

Abstract
This paper reports a reversible switching of the biological activity of an RNA molecule, packaging RNA (pRNA), which is a central component of the DNA-packaging motor of bacteriophage phi29. The switching mechanism contains two components: (1) inhibiting pRNA by a short antisense DNA (asDNA) that can bind to the 3′-end of the pRNA and inactivate the packaging motor; and (2) reactivating pRNA by isothermally removing asDNA from pRNA through a strand displacement strategy. The switching process can be repeated for multiple cycles and has been demonstrated by gel electrophoresis and a virion assembly assay.
Introduction
The genomic DNA of bacteriophage phi29 translocates into a preformed protein shell (procapsid) during viral reproduction.1 This energetically unfavorable process is driven by an ATP-powered DNA packaging motor. The motor has been studied extensively and reconstructed in vitro.1,2 Recently the DNA-packaging motor and its components have also been explored as drug delivery vehicles for the therapy of cancers and viral infection,3,4 and the motor components have been used as a target for developing new approaches in viral therapy.5 The packaging motor contains an RNA component (pRNA), which is indispensable for the motor activity.6 The pRNA molecules fold into helix-rich secondary structures and can further interact with each other through loop-kissing interactions (Figure 1).7, 8 In the absence of viral procapsids, pRNA exists as monomers and dimers. With the native interacting loop sequences, pRNA exists dominantly as monomers and to a small fraction as dimers, but not any higher molecular weight oligomer (Figure 1b). In the presence of viral procapsids, pRNAs bind to procapsids (at the packaging porter) and associate with each other into hexameric rings (Figure 1b). The pRNA hexameric complexes are templated and stabilized by the procapsids. In the final viral structures, the 5′/3′ helical domains of the pRNA molecules are at the outside of the viral particles and accessible to other molecules.
Figure 1.

Reversible switching of the biological activity of the hexameric pRNA complex of bacteriophage phi29 DNA packaging motor. (a) The secondary structure of the native pRNA molecule. The 3′ end of pRNA has been elongated in the current study to facilitate the asDNA binding. The two interacting loops are colored red. pRNA: packaging RNA; asDNA: antisense DNA; rDNA: removal DNA. (b) pRNA self-association. In the absence of viral procapsids, pRNAs reversibly associate with each other into dimers. The equilibrium prefers pRNA monomer when the interacting loop sequences are as shown in (a). No higher oligomeric species can form. In the presence of procapsids, pRNA interacts with procapsids and forms hexameric rings around the packaging porters. (c) Regulation of the pRNA function by reversible and isothermal binding/removal of asDNA. Note that it is not necessary for asDNA to bind to all pRNA to inhibit the packaging motor.
Being able to reversibly control the function of the DNA-packaging motor is desirable for both fundamental biological studies on the DNA packaging mechanism and applications of the DNA packaging motor as nanodevices to actively transport DNA, RNA, and drugs into targeted cells.9 In our previous studies, antisense oligonucleotides were used to target pRNA molecules, thereby inhibiting DNA packaging of the phi29 virus.5 However, this inhibition was irreversible. The DNA-pRNA complex persisted. To effectively utilize phi29 DNA-packaging motor in nanodevices, it is desirable to restore the motor activity. Herein, we report a strategy for reversible switching on/off the biological activity of the phi29 DNA-packaging motor (Figure 1c).
A key to the reversible switching in this study is the isothermal DNA strand displacement.10, 11 It is driven by the maximization of DNA base-pairing. If there is a DNA duplex with single-stranded overhang and a free single-stranded DNA (ssDNA) that is complementary to the long strand of the DNA duplex, the free ssDNA will displace the short strand in the original DNA duplex to form a longer DNA duplex to release the short ssDNA. The process is sequence-specific and takes place under isothermal conditions. Thus, other non-related DNA/RNA interactions will not be affected. This strategy has been extensively used for DNA/RNA nanodevices12-15 and DNA-based computations16 in the last decade. However, it has not yet been explored to use this strategy to reversibly regulate RNA molecules in the content of complex biological systems. Here we apply this strategy to controllably inhibit/activate the RNA-containing phi29 DNA packaging motor (Figure 1c). This strategy involves an antisense (asDNA) and a removal DNA (rDNA). The pRNA is elongated at its 3′ end to contain a 3′-overhang and the asDNA is complementary to the 3′ end of the pRNA. When the asDNA is bound to pRNA, there is a single-stranded tail in asDNA (Figure 1c). This tail will facilitate the removal of the asDNA from the pRNA by the addition of an rDNA, which is complementary to the asDNA. The formation of a longer DNA duplex promoted by a perfect match between asDNA and rDNA drives the dissociation of asDNA from pRNA and reactivates the packaging motor.
Materials and Methods
Oligonucleotides
All DNA oligonucleotides were purchased from Integrated DNA Technologies, Inc (IDT) and used without purification. The sequence of asDNA is: 5′-GTCAGATGTGGTAGGTTAGGA AAGTAGCGTGCACTTTTG-3′; rDNA: 5′-CAAAAGTGCACGCTACTTTCCTAACCTACCACATC TGAC-3′; the shorter asDNA sequences: 5′-GTCA GATGTGGTAGGTTAGGAAAGTAG-3′ and 5′-GTCAGATGTGGTAGGTTAGGAAAG-3′.
Formation of asDNA-pRNA Complexes
For inhibition of DNA packaging activity of pRNA complex, unless otherwise stated, 1μl pRNA stock solution (10 μM), 1.5 μl asDNA stock solution (10 μM), 1 μl 10 × TBM stock buffer, and 6.5 μl DEPC-treated water were mixed to give a final concentration of 1 μM pRNA and 1.5 μM asDNA. TBM buffer consisted of tris(hydroxymethyl)amino methane (Tris, 89 mM), boric acid (89 mM) and MgAc2 (3 mM), pH 7.5. A molar ratio of 1:1.5 between pRNA and asDNA was used to ensure that all pRNA molecules are bound to asDNA. The mixture was incubated at 22 °C for 2 hours for hybridization. To remove asDNA from asDNA-pRNA complexes, 1.5 μl rDNA stock solution (10 μM) was added to the solution, followed by incubation at 22 °C for 2 hours. For the second and third inhibition/activation cycles, asDNA and rDNA stock solution were added to the resulting solution from the first cycle following the same procedure as the first cycle. The molar ratio and incubation condition were the same as the first cycle. For agarose gel electrophoresis, 10 μl sample solution was loaded into each well.
Native Gel Electrophoresis
2% agarose gel was run on a FisherBiotech mini-horizontal electrophoresis unit at 4 °C with a constant voltage of 60 V (electric field strength: 6 V/cm). TBM buffer was used as running buffer. After electrophoresis, the gel was stained by soaking in ethidium bromide (EB) aqueous solution (2.5 μg/ml) for 30 minutes, then destained by soaking in water for 30 minutes to reduce the background, and finally visualized under UV (λ = 365 nm) illumination (FisherBiotech Ultraviolet Transilluminator, FB-TIV-816A).
Preparation of pRNA
The synthesis and purification of pRNA were performed as reported previously.17 The DNA duplex for the transcription of pRNA was prepared by PCR using a DNA template containing the pRNA coding sequence and two primers, 5′-TAA TAC GAC TCA CTA TAG GAA TGG TAC GGT ACT TCC-3′, which contained the T7 promoter (underlined), and 5′-CTT GCC AGG CAC CAT CGT AGG TTA GGA AAG TAG CGT GCA CTT TTG C-3′. The reaction solution contained 2.5 mM MgCl2, 0.2 mM of each dNTP, 2 μM primer each and ∼1 μg DNA template in GoTaq Flex buffer (Promega). After Taq DNA polymerase (Promega) was added, the solution went through 22 thermal cycles (1 min at 95 °C, 2 min at 55 °C, and 30 s at 72 °C), then stayed at 72 °C for 5 min, and finally was cooled down to 4 °C. The pRNA was generated by in vitro transcription with T7 RNA polymerase. The reaction solution contained 5 mM rNTP each, ∼50 ng/μl DNA template from PCR and T7 polymerase in transcription buffer (80 mM HEPES-KOH, pH7.5, 24 mM MgCl2, 2 mM Spermidine, 40 mM DTT). After the solution was incubated at 37 °C for 3 hours, RNase free-DNase I (New England BioLabs) was added and the solution was incubated at 37 °C for 10 min. to digest the DNA template. pRNA was purified by 8% denaturing PAGE in TBE buffer (89 mM Tris-borate 2.5 mM EDTA, pH 8.3) and 8 M urea. The sequence of resulting pRNA is: 5′-GGAAUGGUACGGUACUUCCAUUGUCAUGUGUAU GUUGGGGAUUAAACCCUGAUUGAGUUCAGCCCACAUACUUUGUUGAUUGGUUGUCAAU CAUGGCAAAAGUGCACGCUACUUUCCUAACCUACGAUGGUGCCUGGCAAG-3′.
Assay of Virion Assembly Activity of pRNA Complexes
The activity assay of pRNA via the in vitro phi29 assembly system was performed as reported previously.2 After pre-incubation of pRNA with asDNA or rDNA molecules as indicated, pRNA or pRNA-DNA complexes were incubated with viral procapsids, protein gp16, and the phi29 genomic DNA-gp3 conjugate. The complexes were further assembled into virions to evaluate their infectivities against Bacillus subtilis. In brief, 1 μl of pRNA (0.1 mg/ml) or pRNA-DNA complexes was mixed with 10 μl of purified procapsids (0.3 mg/ml), dialyzed on a 0.025 μm pore size membrane (“V” Series Membrane; Millipore Corp.) for 15 min. against TBE (89 mM Tris-borate 2.5 mM EDTA, pH 8.3), and the buffer was switched to TMS (50 mM Tris, 100 mM NaCl, 10 mM MgCl2, pH 8.0) for 30 min. further dialysis. The pRNA-procapsid solution was mixed with 6μl gp16 (10 μg/ml), 1μl DNA-gp3 (0.2 mg/ml), and 3 μl reaction buffer (10 mM ATP, 50 mM Tris-HCl, 10 mM MgCl2, 100 mM NaCl, pH 8.0) to complete the DNA packaging reaction. After 30 min. at ambient temperature, excess amount of neck, tail, and morphogenic proteins were added to the DNA packaging reaction to complete assembly of infectious virions. The assembled virions were plated with Bacillus subtilis su+44(sup+) to calculate their plaque forming units.
Results and Discussion
Reversible Binding of asDNA to pRNA
The reversible binding of pRNA (monomer or dimer) and asDNA was first demonstrated by native agarose gel electrophoresis (Figure 2). In the gel, pRNA itself shows a predominant fast migrating band (monomer) and a minor slow band (dimer). This phenomenon is consistent with previous reports.2,7,8,17 A red box highlights the oscillation pattern of pRNA mobility in different states. In the presence of asDNA, pRNA (monomer and dimer) associates with asDNA and the resulting pRNA-asDNA complex migrates slower than free pRNA. Upon addition of rDNA, asDNA dissociates from pRNA and forms a perfect asDNA-rDNA duplex, which frees pRNA to resume its original electrophoretic mobility. Such a migration pattern cycles when adding another run of asDNA and rDNA, demonstrating that asDNA can reversibly bind to/dissociate from pRNA. To achieve reversible cycles of binding between asDNA and pRNA, addition of asDNA or rDNA dilutes the sample and causes serial decay of pRNA band intensity in gel for each successive cycle.
Figure 2.
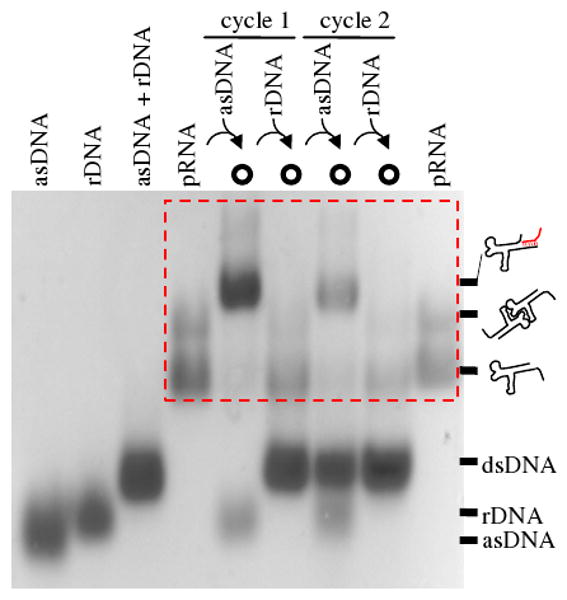
Electrophoretical assay of two cycles of reversible binding/dissociation between asDNA and pRNA by native agarose gel electrophoresis. Sample compositions are indicated above the gel and chemical identities are indicated at the right. Note that the pRNA or pRNA-asDNA complexes are located in the red box.
Biological Assay of Viral Assembly Activity of asDNA-regulated pRNA
Binding of asDNA to pRNA reversibly switches the biological function of the pRNA, demonstated by an in vitro phi29 virion assembly system. Briefly, pRNA was incubated first with DNA to form pRNA/DNA complex. For virion assembly assay, the resulting complex was further incubated with phi29 procapsids, and then with the viral genomic DNA and the viral protein gp16.2 The infectivity of the resulting virions was evaluated by plating on Bacillus subtilis. The virions were counted as plaque-forming units (pfu) per milliliter. When bound to asDNA, pRNA lost its function; the motor could not package its genomic DNA into the procapsid to assemble any infectious virion. However, after the addition of rDNA, asDNA was removed from pRNA and pRNA recovered its biological functions, resulting in the production of infectious viruses. Addition of asDNA decreased the viral yield by ∼ 1000 fold (Figure 3). Addition of rDNA restored the motor function to package the DNA and to produce viruses. The inhibition/activation of the pRNA activity of the motor followed the addition sequence of asDNA and rDNA. In order to check whether random DNA can inhibit the DNA packaging, rDNA alone was used as a control and no inhibition was found on the DNA packaging. When the asDNA was shortened at its 3′ end, its binding capability to the pRNA decreased, and only minor inhibition was found for two slightly different shorter asDNAs. In two parallel experiments, the assembly activity decreases by a factor of 5.4 or 5.5 times for one short asDNA and 15 or 5.4 times for the other short asDNA. These control experiments confirmed that the asDNA worked as designed.
Figure 3.

Virion assembly activity of the asDNA-regulated pRNA. Three cycles of inhibition/reactivation have been shown.
Dependence of the Assembly Activity of pRNA on Relative Ratio of pRNA and asDNA
The pRNA inhibition is efficient and dose dependent. In the DNA-packaging motor, pRNA molecules exist as hexameric ring-like complexes. It has been reported that incorporation of one inactive pRNA mutant into the pRNA complex completely blocks the function of the DNA packaging motor.18 Thus, we reason that it is not necessary for asDNA to bind to all six pRNA molecules to interrupt the biological function of the packaging motor. Instead, only one copy of asDNA binding to one copy of the pRNA in the hexameric pRNA ring is sufficient to disrupt the biological activity of the entire pRNA complex. Therefore, a molecular ratio of 1:1 between asDNA and pRNA is not necessary. To test this hypothesis, we have incubated asDNA and pRNA at various molecular ratios, followed by incubation with procapsids to check the corresponding efficiency of virion assembly (Figure 5). We have found that the efficiency of the virion assembly dramatically decreased by adding 2-3 asDNA molecules per pRNA hexameric complex on average. Further increasing asDNA does not significantly decrease the assembly efficiency.
Conclusions
We have developed an effective strategy to reversibly switch an RNA-containing biological nanomotor by introduction/removal of an antisense DNA. This work helps control biological activities of the phi29 DNA packaging nanomotor and paves a way for its technological applications. We are currently investigating the use of the controllable nanomotor for active drug delivery. It is also conceivable to apply the same strategy for structural and functional control over other RNA-containing biological complexes.
Figure 4.

The effect of relative ratio of asDNA to pRNA hexamer on the activity of pRNA hexameric complex. The columns are experimental data and the thin line is a guide to the eye.
Acknowledgments
This work was supported by the National Institutes of Health through the NIH Roadmap for Medical Research (PN2 EY 018230) and R21EB007472 and the National Science Foundation (CCF-0622093).
References
- 1.Guo P, Grimes S, Anderson D. P Natl Acad Sci USA. 1986;83:3505–3509. doi: 10.1073/pnas.83.10.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee C, Guo P. J Virol. 1995;69:5018–5023. doi: 10.1128/jvi.69.8.5018-5023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoeprich S, et al. Gene Ther. 2003;10:1258–1267. doi: 10.1038/sj.gt.3302002. [DOI] [PubMed] [Google Scholar]
- 4.Guo S, Tschammer N, Mohammed S, Guo P. Hum Gene Ther. 2005;16:1097–1109. doi: 10.1089/hum.2005.16.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C, Garver K, Guo P. Virology. 1995;211:568–576. doi: 10.1006/viro.1995.1439. [DOI] [PubMed] [Google Scholar]
- 6.Guo P, Erickson S, Anderson D. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 7.Guo P, Zhang C, Chen C, Garver K, Trottier M. Mol Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- 8.Chen C, Sheng S, Shao Z, Guo P. J Biol Chem. 2000;275:17510–17516. doi: 10.1074/jbc.M909662199. [DOI] [PubMed] [Google Scholar]
- 9.Guo P, Lee T. J Mol Microbiol. 2007;64:886–903. doi: 10.1111/j.1365-2958.2007.05706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 11.Yan H, Zhang X, Shen Z, Seeman NC. Nature. 2002;415:62–65. doi: 10.1038/415062a. [DOI] [PubMed] [Google Scholar]
- 12.Bath J, Turberfield A. J Nat Nanotechnol. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- 13.Dittmer WU, Kempter S, Radler JO, Simmel FC. Small. 2005;1:709–712. doi: 10.1002/smll.200500074. [DOI] [PubMed] [Google Scholar]
- 14.Zhong H, Seeman NC. Nano Lett. 2006;6:2899–2903. doi: 10.1021/nl062183e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian Y, He Y, Chen Y, Yin P, Mao C. Angew Chem Int Ed. 2005;44:4355–4358. doi: 10.1002/anie.200500703. [DOI] [PubMed] [Google Scholar]
- 16.Seelig G, Soloveichik D, Zhang D, Winfree E. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, Lee C, Guo P. Virology. 1994;201:77–85. doi: 10.1006/viro.1994.1267. [DOI] [PubMed] [Google Scholar]
- 18.Chen C, Guo P. J Virol. 1997;71:3864–3871. doi: 10.1128/jvi.71.5.3864-3871.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]