

Abstract
Wound healing is a dynamic process that involves a coordinated response of many cell types representing distinct tissue compartments and is fundamentally similar among tissue types. Among the many gene products that are essential for restoration normal tissue architecture, several members of the matrix metalloproteinase (MMP) function as positive and, at times, negative regulators of repair processes. MMPs were initially thought to only function in the resolution phase of wound healing, particularly during scar resorption; however, recent evidence suggests that they also influence other wound-healing responses, such as inflammation and re-epithelialization. In this review, we discuss what is currently known about the function of MMPs in wound healing and will provide suggestions for future research directions.
Keywords: Wound Healing, Inflammation, Metalloproteinases, Chemokines, Extracellular Matrix
1. Introduction
1.1 Metalloproteinases
Metalloproteinases are endopeptidases that utilize a Zn2+ or Ca2+ ion in their active site. These include the metzincin family of enzymes that is comprised of the serralysins, astacins, adamalysins (a disintegrin and metalloproteinase domain or ADAMs), and matrixins (matrix metalloproteinases or MMPs) (Huxley-Jones et al., 2007). The majority of research has focused on the matrixins and adamalysins, and thus, in this review, we will highlight the function of these two enzyme families and their inhibitors in wound healing.
MMPs comprise 25 members, 24 of which are found in mammals (Birkedal-Hansen et al., 1993; Parks, Wilson, & Lopez-Boado, 2004). Traditionally, the presumed role for MMPs was confined to catabolism of the extracellular matrix (ECM). In recent years, however, findings from several groups have established that MMPs cleave a wide range of extracellular, bioactive substrates, and regulating the activity of such proteins, typically in a gain-of-function manner, may indeed be the predominant function of MMPs in vivo (Mott & Werb, 2004; Parks et al., 2004). The established functions for MMPs include the release of growth factors from the cell membrane or ECM, cleavage of growth factor receptors from the cell surface, shedding of cell adhesion molecules, and activation of other MMPs, among other, non-catabolic functions (Egeblad & Werb, 2002; Mott & Werb, 2004; Parks et al., 2004).
As their name indicates, ADAMs contain a distintegrin or integrin-binding domain, and a metalloproteinase domain that is similar to the conserved Zn2+-binding catalytic domain in MMPs (Kheradmand & Werb, 2002) They are transmembrane proteases and their primary function is cleavage of extracellular domains of many membrane proteins from the cell surface, a process termed ectodomain shedding (Kheradmand & Werb, 2002; Moss & Bartsch, 2004; Seals & Courtneidge, 2003).
1.2 Inhibitors of Metalloproteinases
Among the endogenous inhibitors of MMPs are TIMPs, of which there are four family members, TIMP-1, -2, -3, and -4 (Baker, Edwards, & Murphy, 2002). TIMPs inhibit MMPs in a 1:1 inhibitor to enzyme ratio through interaction of the N-terminal domain of the TIMP molecule with the active site of the MMP (Brew, Dinakarpandian, & Nagase, 2000). TIMPs co-ordinate the catalytic site Zn2+ and bind to the active site in a similar fashion to an MMP substrate (Brew et al., 2000). Similar to MMPs, ADAMs are inhibited by TIMPs, although this inhibition is regulated primarily by TIMP3 (Amour et al., 2000; Amour et al., 1998; Kashiwagi, Tortorella, Nagase, & Brew, 2001; Loechel, Fox, Murphy, Albrechtsen, & Wewer, 2000). In addition, other proteins and mechanisms can inhibit metalloproteinase activity. These include α2-macroglobulin, a primary inhibitor of metalloproteinases in bodily fluid, such as synovial fluid, and reversion-inducing cysteine-rich protein with Kazal motifs (RECK), the only known membrane-bound MMP inhibitor (Baker et al., 2002; Egeblad & Werb, 2002). Further, modification by reactive oxygen species and internalization have been demonstrated to silence MMP activity (Fu, Parks, & Heinecke, 2007).
1.3 Wound Healing
Wound healing provides a relevant model to illustrate the many functions - mostly beneficial, but some potential deleterious - that metalloproteinases mediate via their ability to dramatically alter the activity of their protein substrates. In response to injury, essentially all cells within the tissue environment and circulation respond concurrently and quickly to stop blood loss, kill microorganisms, and close gaps to the environment. For the purpose of this review, we will focus on three processes of repair: re-epithelialization, which encompasses wound closure and re-differentiation; inflammation; and resolution of scar formation.
Re-epithelialization, which begins immediately after tissue injury (Martin, 1997; Singer & Clark, 1999), requires epithelial cells at the edge of the wounded tissue to loosen their cell-cell and cell-ECM contacts and assume a migratory phenotype. Once the cells at the wound edge begin to migrate, the epithelial cells behind the wound edge begin to proliferate and this continues until a new epithelium covers the damaged tissue (Singer & Clark, 1999; Werner et al., 1994). Although this conversion of epithelial cell behavior is called the Epithelial-to-Mesenchymal Transition (EMT), which is mechanistically similar to early oncogenesis, the epithelial cells do not actually become mesenchymal, i.e., interstitial cells. They remain epithelial cells that are programmed to respond in manner that is highly conserved throughout the Animal Kingdom. Once wound closure is complete, the involved epithelial cells revert to their tissue-specific differentiated state.
The initiation of the inflammatory phase also occurs shortly after injury. The injured cells, which include epithelial and stromal cells as well as platelets from injured blood vessels, become activated and begin to produce chemotactic mediators that culminate in an inflammatory response (Martin & Leibovich, 2005; Singer & Clark, 1999) The primary role of inflammation in wound healing is to control infection, and this task is fulfilled by macrophages and neutrophils (Martin & Leibovich, 2005). It has also been suggested that inflammatory cells are required for other aspects of wound healing (i.e., to release cytokines and growth factors that stimulate cell proliferation and migration); however, this remains to be completely elucidated (Martin & Leibovich, 2005).
A few days after tissue damage occurs, new stromal tissue, called granulation tissue, is formed. This involves the proliferation of fibroblasts surrounding the wound and the subsequent migration of these fibroblasts into the damaged area. Fibroblasts produce ECM structural proteins and lay down a provisional, fibronectin-rich matrix (Clark, Lanigan et al., 1982; Singer & Clark, 1999). Regulation of fibroblast proliferation and migration is thought to occur through the local release of growth factors and cytokines from macrophages as well as through cues from the provisional matrix (Singer & Clark, 1999; Xu & Clark, 1996). The provisional matrix is replaced by a collagen-rich scar, which is, ideally, a temporary scaffold to provide tissue strength. Formation of granulation tissue also requires the formation of new blood vessels to supply the newly formed tissue. Angiogenic signals are released from both macrophages and injured epithelial and endothelial cells (Brown et al., 1992). This process is also regulated by the provisional matrix, which provides a substrate for the sprouting endothelial cells to migrate across (Brooks, Clark, & Cheresh, 1994; Clark, Quinn et al., 1982; Singer & Clark, 1999).
The final phase of wound healing is resolution, which involves ECM remodeling and contraction of the wound. Contraction of the wound occurs after formation of the granulation tissue and requires the fibroblasts to assume a myofibroblast phenotype (Welch, Odland, & Clark, 1990). Remodeling of the ECM, which leads to the conversion of granulation tissue to scar tissue, follows this, and is dependent on the continual synthesis and degradation of collagen fibrils (Parks, 1999; Singer & Clark, 1999).
Metalloproteinases participate in regulating mechanisms in all of these repair processes (Fig. 1). For example, inflammation is shaped by cytokines and chemokines, which arise largely from resident cells (epithelium, endothelium, fibroblasts, etc.). MMPs can activate these mediators, by cleaving them from the cell surface or processing them to increase their activity, or degrade them, thereby inhibiting inflammatory signals (Fig. 1). As well, MMPs are able to cleave components of cell-cell junctions and cell-matrix contacts within the epithelium to promote re-epithelialization (Fig. 1). Furthermore, MMPs are involved in remodeling the scar ECM either directly by proteolytic degradation of proteins, such as collagens, or indirectly via their ability to affect cell behavior. Alteration of the ECM is integral to the resolution of wound healing but also has implications in regulation of inflammation (Fig. 1). Thus, MMPs are key regulators of multiple aspects of tissue repair and further study of these enzymes and their interaction with their substrates will not only advance our basic knowledge of wound healing, but also provide insight into possible therapies.
Figure 1.

Varied metalloproteinase functions during wound healing. Shown in this cartoon are some the many functions that MMPs regulate concurrently during tissue repair. A. Cell Migration: MMPs facilitate epithelial cell migration by affecting the interaction between cells and ECM proteins, either by direct proteolysis of the ECM or processing of cell-ECM adhesion proteins on the cell surface. B. Re-epithelialization: Junctional proteins, like E-cadherin, are shed by MMPs, thereby loosening cell-cell adhesions and subsequently, promoting a shift to a repair phenotype, characterized by dedifferentiation, proliferation, and migration. C. Leukocyte Influx: Proteoglycan/chemokine complexes are shed by MMPs from the basolateral cell surface to establish a chemotactic gradient that guides the transepithelial migration of neutrophils. D. Inflammation: MMPs process multiple chemokines with varied results, as shown in Table 1. Among the consequences of chemokine processing are complete degradation, production of a receptor antagonist, inhibition, or activation.
2. Metalloproteinases in Wound Healing
2.1 Metalloproteinases in Inflammation
Inflammation (i.e., the influx and activation of leukocytes) is influenced by antimicrobial peptides, lipid mediators, homing receptors, chemokines and cytokines, ECM fragments, and more. The production and activity of these factors is controlled by effector proteins, which include metalloproteinases. Findings from our lab and others demonstrate that epithelial-derived MMPs regulate numerous aspects of inflammation, such as the transepithelial migration of leukocytes and the activity and compartmentalization of chemokines (Parks et al., 2004; Yong, 2005).
Inflammatory cells are well known to express MMPs; however, epithelial and stromal cells in wounded tissue have also been demonstrated to express multiple MMPs including MMP1, 2, 3, 7, 9, 10, and 28 (Corry et al., 2004; Corry et al., 2002; Dunsmore et al., 1998; Inoue, Kratz, Haegerstrand, & Ståhle-Bäckdahl, 1995; Pilcher, Sudbeck, Dumin, Welgus, & Parks, 1998; Pilcher et al., 1999; Saarialho-Kere et al., 2002; Saarialho-Kere, Crouch, & Parks, 1995; Salmela et al., 2004; Warner et al., 2004). Many of these MMPs can regulate chemokine activity, either by direct proteolysis or by affecting the formation of chemokine gradients.
Chemokines are divided into subfamilies based on their amino (N)-terminal cysteine residues and have distinct roles in attracting the influx of specific types of leukocytes. The CC chemokines, which have a significant role in monocyte chemotaxis, have the first two N-terminal cysteine residues adjacent to each other (Clark-Lewis et al., 1995; Zlotnik, Yoshie, & Nomiyama, 2006). The other main subfamily of chemokines, the CXC chemokines, primarily regulate neutrophil chemotaxis and as their designation implies, have an extra amino acid residue between the first two N-terminal cysteines (Clark-Lewis et al., 1995; Zlotnik et al., 2006). Chemokines mediate their effect by binding to 7-transmembrane G-protein coupled receptors that are named based on the subfamily of chemokines with which they interact (Zlotnik et al., 2006).
Chemokine cleavage by MMPs most often results in a reduction in chemokine activity (Table 1). This is especially true of CC chemokines, which upon processing by MMPs form receptor antagonists that inhibit downstream signaling (McQuibban et al., 2000; McQuibban et al., 2002; Van den Steen, Proost, Wuyts, Van Damme, & Opdenakker, 2000). To date, as suggested by defined in vitro assays, processing of CCL2, 5, 7, 8, and 13 (MCP1, RANTES, MCP3, MCP2, and MCP4 respectively) is performed most efficiently by MMP1 and 3 (McQuibban et al., 2000; McQuibban et al., 2002; Van den Steen et al., 2000).
Table 1.
Summary of chemokines that are proteolytically processed by metalloproteinases and the subsequent modification to chemokine function.
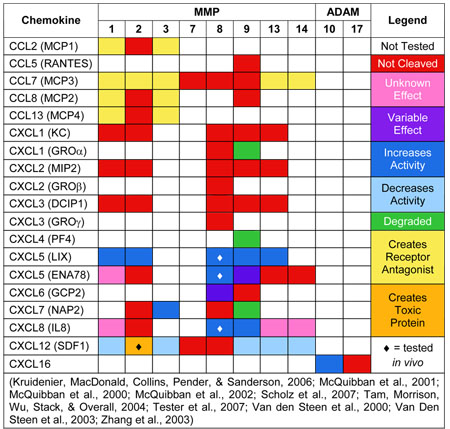 |
CXC chemokines have a far more varied response to MMP cleavage. Some CXC chemokines are completely resistant to processing (CXCL1 or KC, CXCL2 or MIP2, and CXCL3 or DCIP1), while others are readily processed by multiple MMPs (CXCL5 or LIX, CXCL8 or IL8, and CXCL12 or SDF1; see Table 1). This distinction highlights a potential functional difference between humans and mice. Human CXCL8 is processed by multiple MMPs, including MMP8 and 9, leading to a considerable increase in CXCL8 activity (Tester et al., 2007; Van den Steen et al., 2000). Mice lack a CXCL8 homologue; however, CXCL1 and CXCL2 (KC and MIP2 respectively), both of which are resistant to MMPs, are considered to be functional orthologues (Tester et al., 2007). This suggests that if KC and MIP2 are truly functional orthologues of CXCL8 in mice, MMPs may perform different functions in the regulation of inflammatory cell migration by CXCL8 in humans than they do in mice. This potential difference between MMP function in humans and mice underscores the importance of further research into the exact role metalloproteinases have in regulating chemokine signaling, and the mechanism through which they perform this role in both mice and humans. Such knowledge is needed before attempts are made to use metalloproteinases or their inhibitors as therapeutic tools in wound healing and inflammation.
Another CXC chemokine, CXCL5 (LIX), is processed by multiple MMPs, including MMP1, 2, 8, 9, and 13 (Tester et al., 2007; Van Den Steen et al., 2003). Interestingly, as is observed with CXCL8, MMP processing of CXCL5 (LIX) leads to a consequent potentiation of chemokine activity that ultimately increases inflammatory cell recruitment (Tester et al., 2007; Van Den Steen et al., 2003). CXCL12 (SDF1) is also processed by multiple MMPs (MMP1, 2, 3, 9, 13, and 14); however, in this case, MMP processing results in a decrease in chemokine activity or the production of a neurotoxic protein (McQuibban et al., 2001; Zhang et al., 2003).
Of all MMPs examined, MMP1, 3 and 9 appear to have the most widespread ability to regulate chemokine signaling, with effects ranging from complete degradation of the chemokine or creating receptor antagonists to a dramatic increase in chemokine activity (Table 1). MMPs, however, are not the only metalloproteinases capable of processing chemokines. ADAM10 is capable of cleaving CXCL16 from the cell surface and thereby allowing it to bind to its receptor and regulate T-cell activation in the wounded skin (Scholz et al., 2007). Thus, metalloproteinases, including both MMPs and ADAMs, are involved in both positive and negative regulation of chemokine activity.
MMPs not only directly regulate chemokine activity, but they are also required for the establishment of the chemotactic gradient. Work in our lab has established MMP7 (matrilysin) as a key regulator of transepithelial neutrophil migration following lung injury (Li, Park, Wilson, & Parks, 2002). Lung alveolar epithelium has syndecan-1 on its basolateral surface and once injured, expresses CXCL1 (KC) that binds to the heparan sulfate glycosaminoglycan chains on syndecan-1. Wounded epithelium also expresses MMP7, which cleaves the KC/syndecan-1 complex from the epithelium and thereby allows the complex to move across the epithelium into the alveolar space and establish a chemotactic gradient for responding neutrophils (Dunsmore et al., 1998; Li et al., 2002). In the absence of MMP7, no KC is present within the alveolar space (Li et al., 2002). As a result, neutrophils fail to migrate into the alveolar lumen and instead, are sequestered in the interstitial space (Li et al., 2002).
In addition, establishment of chemokine gradients in other forms of inflammation, namely allergen induced inflammation, requires multiple MMPs. Mice lacking MMP2, MMP9, or both have fewer neutrophils and eosinophils present in bronchiole alveolar lavage (BAL) after allergen challenge with Aspergillus fumigatus (Corry et al., 2004; Corry et al., 2002). This is due to an altered CCL11 (eotaxin) transepithelial gradient in Mmp2−/− mice and an altered CCL7 (MARC), CCL11, and CCL17 (TARC) transepithelial gradient in Mmp9−/− or Mmp2/9−/− mice (Corry et al., 2004).
Interestingly, the use of alternative allergens in Mmp9−/− mice has opposite results. Mice lacking MMP9 that were sensitized to OVA and then challenged with aerosolized OVA had increased eosinophils and Th2 cells in both their BAL and tissue compartments (McMillan et al., 2004). This was accompanied by an increase in CCL11 and CCL22, as well as an increase in the Th2 cytokines IL4 and IL13 (McMillan et al., 2004). These findings confirm the importance of MMP9 in regulating inflammation, but also suggest that the role MMP9 has in regulating inflammatory cell migration is dependent on the inflammatory stimulus. Thus, although their exact roles remain to be determined, MMPs have important functions in establishing transepithelial gradients of chemokines and cytokines in response to multiple different inflammatory insults.
Furthermore, metalloproteinases, including both MMPs and ADAMs, have been suggested to control diapedesis. ADAM28, which is expressed by peripheral blood lymphocytes, is found as both a membrane-bound and secreted enzyme (Roberts, Tani, Bridges, Laszik, & Bowditch, 1999). Recent work by Shimoda and co-workers has demonstrated that ADAM28 is capable of binding to P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes and that this enhances binding to P-selectin on endothelial cells (Shimoda et al., 2007). Additionally, MMPs are capable of cleaving occludin and VE-cadherin, components of the tight and adherens junctions, and therefore, are potentially involved in the regulation of endothelial permeability (Alexander & Elrod, 2002).
Inflammation is a key phase of wound healing that is required to protect against infection. Metalloproteinases have many responsibilities in the regulation of inflammation and this includes control of chemokine activity, the establishment of chemotactic gradients, and extravasation of leukocytes out of the blood into the injured tissue. Additional support for this is provided by mice deficient for various MMPs. Challenge of these mice in many cases results in an altered inflammatory response, and conclusions from many studies leads to the inevitable conclusion that a major function of MMPs is the regulation of inflammation (Parks et al., 2004).
2.2 Metalloproteinases in Epithelial Repair
One major component of the repair phase of wound healing is re-epithelialization, which is the re-growth of epithelia over a denuded surface. Re-epithelialization requires the cells at the wound margin to loosen their cell-cell and cell-ECM contacts and begin to migrate across the wound (Singer & Clark, 1999). Multiple MMPs have been associated with this aspect of wound repair, and these include MMP1, 3, 7, 9, 10, 14, and 28 (Atkinson, Toennies, Holmbeck, & Senior, 2007; Dunsmore et al., 1998; McCawley, O'Brien, & Hudson, 1998; Pilcher et al., 1999; Saarialho-Kere et al., 2002).
MMP1 (collagenase-1) is present in human cutaneous wounds during re-epithelialization but turns off once wound closure is complete (Inoue et al., 1995; Saarialho-Kere, Vaalamo et al., 1995). In wounded skin, as keratinocytes migrate off of the basal lamina, they encounter a dermal matrix rich in type I collagen, and ligation to this ECM component via the α2β1 integrin stimulates expression of MMP1, as well as many other genes (Pilcher et al., 1998; Spradling, McDaniel, Lohi, & Pilcher, 2001). Work from our lab indicates that MMP1 facilitates keratinocyte migration over the dermal matrix by lessening the affinity of collagen-integrin contacts (Pilcher et al., 1997). MMP10 (stromelysin-2) is co-localized with MMP1 while MMP3 (stromelysin-1) is localized in cells behind the migrating front. The non-overlapping localization of MMP3 and 10, which are quite similar proteinases, suggests that these two MMPs serve distinct functions in re-epithelialization. Further evidence suggesting a role for MMP1 in re-epithelialization in other tissues includes its expression by migrating enterocytes and its identification as a key regulator of epithelial cell migration in corneal wounds (Mulholland, Tuft, & Khaw, 2005; Salmela et al., 2004).
Another MMP with an established, integral role in re-epithelialization is MMP7. MMP7 is expressed by injured lung epithelium and is required for re-epithelialization after wounding of tracheal explants (Dunsmore et al., 1998). Examination of mice deficient for MMP7 illustrates the importance of MMP7 in this process. In the absence of MMP7, re-epithelialization of tracheal wounds is almost completely abrogated (Dunsmore et al., 1998; McGuire, Li, & Parks, 2003). This role for MMP7 in wound repair also potentially extends to other mucosal epithelia, as MMP7 is also expressed by wound-edge cells in stomach and intestinal ulcers, injured epithelial cells in the kidney, and basal epithelial cells during corneal wound healing (Lu, Ye, Maeda, & Azar, 1999; Salmela et al., 2004; Surendran, Simon, Liapis, & McGuire, 2004).
Work from our lab has suggested that MMP7 regulates re-epithelialization by cleavage of E-cadherin within the adherens junction, which facilitates the migration of epithelial cells away from the wound edge (McGuire et al., 2003). Over-expression of constitutively active MMP7 in lung epithelial cells in vitro leads to E-cadherin shedding and increased epithelial cell migration (McGuire et al., 2003). Additionally, loss of MMP7 results in impaired E-cadherin cleavage in vivo following bleomycin injury (McGuire et al., 2003). MMP7, however, may not be the only MMP involved in cleaving cell junctional proteins. MMP28 (epilysin) is expressed by proliferating keratinocytes distal to the wound edge and is not expressed by migrating keratinocytes. This suggests a possible role in restructuring the basement membrane or cleaving cellular adhesion molecules and thereby, providing further cells for the migrating front (Saarialho-Kere et al., 2002).
MMP9, or gelatinase B, has also been implicated in re-epithelialization after injury. The growth factors epidermal growth factor (EGF) and hepatocyte growth factor (HGF) both stimulate keratinocyte migration in wound assays in vitro (McCawley et al., 1998). This cell migration is dependent on induction of MMP9 activity and is impaired by the presence of either function blocking MMP9 antibodies or a general MMP inhibitor (McCawley et al., 1998). Bove and co-workers have also recently demonstrated that MMP9 is essential to bronchiole epithelial cell migration in culture (Bove et al., 2007). Stimulation of human bronchiole epithelial cells with low concentrations of nitric oxide or with tumor necrosis factor (TNF)α increases MMP9 expression and activation, and this leads to an increase in wound closure (Bove et al., 2007). A few cell culture studies have implicated a role for MMP9 in airway epithelial migration, and indeed, MMP9 is expressed by damaged epithelium in bleomycin-instilled mice (Betsuyaku, Fukuda, Parks, Shipley, & Senior, 2000; d'Ortho et al., 1997; Yao et al., 1998). No defect in airway re-epithelialization, however, is observed in mice lacking MMP9, though cellular differentiation is altered (Betsuyaku et al., 2000).
MMP10 is expressed by epithelium at the edge of the migrating front in all tissues studied, suggesting that this proteinase functions in cell migration. Experiments in vitro and in transgenic mice, however, demonstrate that excess MMP10 only modestly increases cell migration (Krampert et al., 2004; Madlener et al., 1996; Madlener, Parks, & Werner, 1998). Over-expression of a constitutively active MMP10 mutant by wounded epithelium results in somewhat aberrant cell migration at the wound edge and reduced abundance of laminin-5, potentially due to direct cleavage by MMP10 (Krampert et al., 2004). The decrease in laminin-5 deposition is accompanied by altered ECM-cell signaling and increased keratinocyte apoptosis, and as the authors suggest, this highlights the importance of regulated MMP10 activity during re-epithelialization (Krampert et al., 2004). These functions for MMP10 in re-epithelialization have not yet been verified in knock-out mice.
Another aspect of re-epithelialization is proliferation of epithelial cells behind the migrating wound front. MMP14 expression is increased early in lung epithelium within the terminal airways following naphthalene injury, and it appears that MMP14 is involved in the regulation of epithelial cell proliferation after injury (Atkinson et al., 2007). This role is illustrated by decreased Clara cell proliferation in mice lacking MMP14, ultimately resulting in impaired wound healing (Atkinson et al., 2007). One possible mechanism through which this regulation occurs is through the control of keratinocyte growth factor (KGF) receptor expression, which is also increased after lung injury in wild type mice, but not in their MMP14 deficient counterparts (Atkinson et al., 2007).
MMPs may also have negative effects on cell proliferation. MMP2 and 9 are produced by cartilage from the trachea and bronchus, and cartilage conditioned medium decreases respiratory epithelial cell proliferation in vitro (Sigurdson et al., 2003). Addition of a broad, spectrum MMP inhibitor, GM6001, to this culture medium rescues epithelial cell proliferation suggesting that at least some of the MMPs produced by the cartilage repress respiratory cell proliferation (Sigurdson et al., 2003).
In addition to cell proliferation, MMPs are involved in airway epithelial cell differentiation. Experiments employing a human tracheal xenograft model in nude mice found MMP7 and 9 expression and activity to be highest in the later stages of wound healing when epithelial cell differentiation occurs (Coraux et al., 2005). In addition, inhibition of MMP7 or 9 in these xenograft models led to impaired epithelial cell differentiation, confirming that these MMPs are likely required in the airways for appropriate cell differentiation during wound healing (Coraux et al., 2005).
The repair phase of wound healing, much like tissue morphogenesis, is comprised of multiple cellular processes including cell migration, proliferation, differentiation, and death. Although it has been evident for a number of years that MMPs are expressed during all of these processes, the mechanisms through which these proteinases function are only now being discovered. To date, it appears that almost all MMPs that have been examined are positive regulators of these processes; however, it has been suggested that both MMP2 and 9 may have inhibitory effects on cell proliferation.
2.3 Metalloproteinases in Resolution
Wound healing is slowed in mice lacking MMP3 (stromelysin-1). This was not due to altered re-epithelialization as keratinocyte migration was not affected; however, defective wound contraction was observed in these mice compared to their wild type counterparts (Bullard et al., 1999). There were no differences between wild type and Mmp3−/−mice in the number of α-smooth muscle actin positive myofibroblasts but the formation of organized actin bundles in the dermal fibroblasts, which is responsible for early wound contraction, was severely impaired in the mice deficient for MMP3 (Bullard et al., 1999). Thus, the decreased wound contraction resulted in increased wound size and greater distance for epithelial cells to migrate, which ultimately led to the slowed wound healing (Bullard et al., 1999).
Another function of MMPs is the degradation of the ECM and this was originally thought to be their primary function. Although it is now believed that the major role of most MMPs is to process bioactive molecules such as growth factors, cytokines and chemokines, as well as their respective receptors, the ability to degrade ECM proteins has been demonstrated for some members of the MMP family using gain- or loss-of-function approaches (McCawley & Matrisian, 2001; Parks et al., 2004). These include MMP1, 3 and 13 as well as MMP14, which are capable of cleaving collagen, and MMP7, which can process both syndecan-1 and elastin (Parks et al., 2004).
Remodeling of collagen, which includes the degradation of existing collagen fibrils and the synthesis of new ones, is a key part of the resolution phase of wound healing (Singer & Clark, 1999). Because MMPs are capable of this process, it seems plausible that they would have a key role in collagen remodeling during wound resolution. Surprisingly, conclusive evidence of this has yet to be presented in vivo. The localization of certain MMPs, however, does suggest a possible role for them. In anterior keratectomy corneal wounds, MMP2 and 9, and to a lesser extent, MMP3 are localized to the epithelial-stromal interface behind the migrating epithelial cells, which suggests that they may be involved in remodeling of the stroma and reformation of the basement membrane (Mulholland et al., 2005). In culture, it has been demonstrated that keratinocytes express both MMP2 and 9 while fibroblasts express only MMP2 (Sawicki, Marcoux, Sarkhosh, Tredget, & Ghahary, 2005). More importantly, a co-culture of both keratinocytes and fibroblasts leads to increased MMP2 and 9 abundance suggesting that interaction between keratinocytes and fibroblasts, which occurs during wound healing, regulates MMP2 and 9 expression by these cells (Sawicki et al., 2005). The ability of keratinocyte-derived MMP1 to act on dermal type I collagen during wound closure in skin, as discussed above, is spatially restrained and would not affect overall remodeling.
Thus, work to date has provided evidence that MMP3 is required during wound contraction. It has also provided support, through localization of multiple MMPs during wound healing and demonstration of their ability to remodel the ECM in vivo, for continued studies to examine the role of metalloproteinases during the resolution of wound healing. Further support for this has been provided by the examination of metalloproteinase inhibitors, which will be discussed in the subsequent sections.
3. Inhibitors of Metalloproteinases in Wound Healing
3.1 Metalloproteinase Inhibitors in Inflammation
The inflammatory response is mediated by multiple cytokines and chemokines. One of the predominant cytokines involved in acute inflammation is TNFα, which is activated via cleavage from the cell membrane by TNFα Converting Enzyme (TACE) or ADAM17 (Black et al., 1997; Black & White, 1998). One effect that TNFα has on inflammatory cells, like monocytes, is that it stimulates expression of MMP9 through activation of the NFκB and p38 MAP kinase pathways (Nguyen, Gogusev, Knapnougel, & Bauvois, 2006; Zhou, Zhang, Ardans, & Wahl, 2003). Upregulation of MMP9, as outlined above, can alter many aspects of the wound healing process.
TIMP3 is one of the primary inhibitors of ADAM17, and thus has an integral role in the regulation of inflammation (Amour et al., 1998). In the absence of TIMP3, ADAM17 activity is enhanced resulting in increased constitutive TNFα release and a subsequent increase in inflammatory cell infiltration into the livers of aged mice compared with their wild type counterparts (Mohammed et al., 2004). Furthermore, when Timp3−/− mice were challenged with a partial hepatectomy, there was increased cell death due to the altered TNFα signaling, which ultimately resulted in enhanced mortality (Mohammed et al., 2004).
Shedding of TNFα and its receptor from Timp3−/− macrophages was also augmented following stimulation with LPS (Smookler et al., 2006). Increased TNFα activation in the absence of TIMP3 had physiologically relevant consequences as it resulted in increased IL6 release, a downstream effect of TNFα signaling, as well as increased susceptibility to LPS-induced mortality (Smookler et al., 2006). Both of these phenotypes were rescued by deletion of the TNFα receptor or by addition of a synthetic metalloproteinase inhibitor (Mohammed et al., 2004; Smookler et al., 2006). Thus, TIMP3 has a fundamental role in controlling inflammation through the regulation of TNFα signaling.
Additionally, interaction between TIMP3 and ADAM17 likely plays a role in directing leukocyte recruitment. Vascular cell adhesion molecule (VCAM) 1 is involved in leukocyte recruitment, and during an inflammatory response it is shed from the surface of endothelial cells by ADAM 17 (Singh et al., 2005). Addition of TIMP3 blocked the production of soluble VCAM1, while loss of TIMP3 led to an increase in VCAM1 shed from the cell surface (Singh et al., 2005). These findings suggest that TIMP3 regulates inflammatory cell recruitment by its ability to inhibit ADAM17.
TIMP3 is not the only metalloproteinase inhibitor that may function during inflammation. TIMP1 expression is increased in lung injury due to treatment with bleomycin, and in the absence of TIMP1, there is increased vascular permeability and neutrophil diapedesis into the injured pulmonary tissue (Kim et al., 2005; Madtes, Elston, Kaback, & Clark, 2001). The loss of TIMP2, however, has no effect on acute lung injury due to bleomycin, highlighting the importance of TIMP1 in regulating the inflammatory response following lung injury (Kim et al., 2005). One possible mechanism through which TIMP1 may control leukocyte extravasation and vascular permeability may be through the regulation of endothelial cell apoptosis (Boulday, Fitau, Coupel, Soulillou, & Charreau, 2004). TIMP1 exerts an anti-apoptotic effect on cytokine-stimulated endothelial cells through activation of the phosphatidylinositol-3-kinase-Akt pathway (Boulday et al., 2004).
The importance of metalloproteinase inhibition during lung injury and repair was confirmed by studies using the synthetic metalloproteinase inhibitor, Batimastat. Treatment with Batimastat prevented inflammatory cell influx into the lung following bleomycin-induced injury (Corbel et al., 2001). In addition, Batimastat reduced MMP activity in the bronchoalveolar lavage and also diminished the pulmonary fibrosis that is usually associated with bleomycin treatment (Corbel et al., 2001).
Thus, inhibitors of metalloproteinases, especially TIMP3, are required for the regulation of the inflammatory response through control of cytokine signaling and inflammatory cell adhesion receptor processing. The use of synthetic inhibitors illustrated that this is especially true following lung injury and that treatment with these inhibitors may even serve to reduce the severity of the injury.
3.2 Metalloproteinase Inhibitors in Repair
The ability of metalloproteinase inhibitors to regulate MMP activity suggests that they are important to all aspects of wound repair, especially cell migration. This is supported by experiments using the synthetic metalloproteinase inhibitor, GM6001, to treat cutaneous wounds (Mirastschijski, Haaksma, Tomasek, & Ågren, 2004). Wounds treated with GM6001 had impaired re-epithelialization when compared to wounds from control animals, and this was likely due to altered cell migration as epithelial cell proliferation was unaffected by GM6001 (Mirastschijski et al., 2004).
TIMPs also function to regulate aspects of cell migration, apparently by restraining the activity of specific MMPs. Re-epithelialization occurs following isotopic tracheal transplantation; however, following heterotopic tracheal transplantation, this process is impaired and this impairment is accompanied by infiltration of mesenchymal and inflammatory cells into the tracheal lumen (Chen, Farivar, Mulligan, & Madtes, 2006). TIMP1 is induced during this process, and excess levels of this inhibitor may contribute to the impaired epithelial cell migration, as re-epithelialization is normal in heterotopic transplants into TIMP1 deficient recipient mice (Chen et al., 2006).
Cell migration is also decreased in vitro in the presence of metalloproteinase inhibitors. Addition of TIMP2 or the synthetic MMP inhibitor, BB-94, impairs the migration of HT1080 fibrosarcoma cells stably transfected with syndecan-1 (HT1080/SDC) (Endo et al., 2003). This is likely due to the inhibition of MMP14 cleavage of syndecan-1 from the cell surface, which is suggested by the increase in cell-surface associated syndecan-1 (Endo et al., 2003). Interestingly, although TIMP1 has been implicated in the regulation of cell migration following tracheal transplantation, it had no effect in regulating HT1080/SDC cell migration (Endo et al., 2003). The inability of TIMP1 to alter cell migration in this system is possibly due to its low affinity for MMP14 (Baker et al., 2002; Brew et al., 2000). Importantly, this highlights a potential difference between epithelial and stromal cell migration during re-epithelialization and formation of the granulation tissue, and the possible role that TIMPs play in regulating this process.
The exact role of TIMPs in the regulation of cell migration remains to be determined. In fact, and somewhat counterintuitive, other studies have demonstrated that TIMP2 accelerates keratinocyte migration in culture and in vivo (Terasaki, Kanzaki, Aoki, Iwata, & Saiki, 2003) (Terasaki et al., 2003). Thus, although the literature suggests that TIMPs are involved in the regulation of cell migration, much work remains to be done to completely elucidate the specific mechanism through which they fulfill this role.
In addition to their ability to inhibit MMPs, it has been demonstrated that TIMPs have MMP-independent functions (Baker et al., 2002), as exemplified by TIMP3 and its potent inhibition of angiogenesis (Qi et al., 2003). Qi and co-workers demonstrated that TIMP3 binds directly to vascular endothelial growth factor receptor (VEGFR) 2 and inhibits its interaction with VEGF and the subsequent downstream signaling that is required for endothelial cell proliferation and angiogenesis (Qi et al., 2003). During wound repair, angiogenesis is required following the formation of granulation tissue to provide vascular support for the newly formed tissue. Thus, although it remains to be demonstrated during wound healing, it seems likely that the ability of TIMP3 to regulate angiogenesis may be important to successful wound repair.
3.3 Metalloproteinase Inhibitors in Resolution
Wound contraction, an important component of the resolution phase of wound healing, is susceptible to regulation by inhibitors of metalloproteinases. Treatment of cutaneous wounds with GM6001 leads to impaired re-epithelialization, as discussed above, but it also decreases wound contraction and alters the differentiation of cells within the granulation tissue (Mirastschijski et al., 2004). GM6001 treated wounds had a significant reduction in the number of α-smooth muscle actin-positive myofibroblasts in the granulation tissue and this was associated with an impairment of wound contraction when these wounds were evaluated against control treated wounds (Mirastschijski et al., 2004).
Another important phase of wound resolution is remodeling of the ECM, especially collagen, which is required for the transition from granulation tissue to scar tissue (Singer & Clark, 1999). Collagen remodeling may be partially regulated by TIMP3. Mice lacking TIMP3 develop enlarged airspaces and these airspaces continue to increase in size as the animal ages (Leco et al., 2001). This increase in airspace size appears to be due to increased MMP activity in the lungs and is accompanied by altered collagen remodeling (increased degradation and altered deposition) and reduced fibronectin abundance (Leco et al., 2001; Martin et al., 2003). In addition, Timp3−/− mice also have reduced fibronectin abundance in their lungs during bronchiole branching morphogenesis and this is rescued in vivo by treatment with GM6001 (Gill, Pape, Khokha, Watson, & Leco, 2003; Gill, Pape, & Leco, 2006). That both collagen and fibronectin remodeling is altered in the absence of TIMP3 provides further support that TIMP3 and its ability to inhibit metalloproteinases is integral to the maintenance of appropriate ECM composition and that alteration of metalloproteinases or their inhibitors leads to inappropriate remodeling of the ECM.
Experiments using the over-expression of TIMP1 by mammary epithelial cells also provide strong evidence that TIMPs are involved in regulating ECM remodeling. Over-expression of autoactivating MMP3 in mammary epithelial cells led to enhanced degradation of the basement membrane protein, entactin, and a consequent increase in epithelial cell apoptosis (Alexander, Howard, Bissell, & Werb, 1996). When these mice were crossed with mice over-expressing TIMP1, both entactin degradation and apoptosis were reduced to below the levels observed in control animals (Alexander et al., 1996).
Therefore, evidence from both loss- and gain-of-function experiments illustrates the important role that inhibitors of metalloproteinases, especially TIMPs, have in regulating ECM remodeling. This in combination with the evidence suggesting that GM6001 affects wound contraction suggest that inhibitors of metalloproteinases may be fundamental to wound resolution.
4. Summary
Wound healing is a complex process requiring the appropriate temporal and spatial expression of signaling molecules and their receptors, cellular adhesion molecules, and ECM proteins. Together, these molecules regulate the many processes involved in wound healing. Metalloproteinases and their inhibitors are capable of processing many of these signaling molecules, adhesion molecules, and ECM proteins and thus, are likely involved in the control of all aspects of wound healing (Fig. 1).
Metalloproteinase cleavage of signaling molecules and ECM proteins occurs during the inflammatory phase, where MMPs, especially MMP1, 3, 7, and 9, are capable of regulating both chemokine activity (Table 1) and the establishment of chemotactic gradients (Fig. 1). Degradation of cell adhesion molecules as well as ECM proteins by MMPs is also necessary during the repair phase, where MMP1, 7, 9, and 10 are required for cell migration, while the addition of a synthetic metalloproteinase inhibitor leads to impaired cell migration (Fig. 1). MMPs are also required for cell proliferation during this phase and the demonstration that TIMP3 can regulate angiogenesis suggests that it may also be integral to the maintenance of granulation tissue once it has formed. Finally, wound contraction during the resolution phase requires appropriate expression of MMPs and their inhibitors. MMP3 is required for wound contraction to occur and either the loss of MMP3 or the addition of a synthetic inhibitor of metalloproteinases results in slowed wound contraction.
The current literature offers a great deal of evidence that metalloproteinases and their inhibitors are required for wound healing, and this evidence provides a strong foundation for the continuation of this work. Based on the literature, one of the most plausible roles for MMPs and ADAMS, as well as TIMPs, in wound healing is in control of the inflammatory response. Metalloproteinases and their inhibitors play an integral role in regulating inflammation through both the regulation of chemokine and cytokine activity and signaling gradients, and through regulation of inflammatory-endothelial cell adhesion receptor interaction. One well established way that metalloproteinases are involved in inflammation is through direct interaction with chemokines or their receptors; however, much work still remains to examine the rest of the chemokines involved in the inflammatory response. Another focus of the research should be to examine the interaction between metalloproteinases and mediators of chemokine signaling, such as the syndecans. Syndecans are involved in the regulation of chemokine signaling through sequestration of chemokines within the ECM and by presentation of the chemokine to its receptor on the inflammatory cell surface. Processing of syndecan by metalloproteinases provides an indirect method of regulating chemokine activity. Importantly, further understanding of the role of metalloproteinases and their inhibitors in inflammation will not only further our understanding of wound healing, but also of many other pathological processes, including cancer and inflammatory diseases such as arthritis.
Although the goal of medical research is to identify therapeutic interventions for the disease and molecule of interest, much remains to be understood before this should be considered with metalloproteinases and wound healing. Further work utilizing different models of wound healing in mice with gain- and loss-of-function mutations for MMPs, ADAMs, or TIMPs must occur. These experiments will undoubtedly further our knowledge of how metalloproteinases and their inhibitors control the cellular processes involved in wound repair and help to determine the conditions that are required for them to be either positive or negative regulators of these processes.
Thus, although a great deal of work remains to be done, the ability of MMPs and ADAMs to regulate the activity of bioactive molecules, such as growth factors, cytokines and chemokines, as well as the role of MMPs in digestion of both cell adhesion molecules and ECM proteins in the basement membrane suggests an integral role for these enzymes in all phases of wound healing.
Abbreviations
- ADAM
a disintegrin and metalloproteinase
- MMP
matrix metalloproteinase
- ECM
extracellular matrix
- TIMP
tissue inhibitor of metalloproteinase
- RECK
reversion-inducing cysteine-rich protein with Kazal motifs
- EMT
Epithelial-to-Mesenchymal Transition
- BAL
bronchiole alveolar lavage
- PSGL-1
P-selectin glycoprotein ligand-1
- VE-cadherin
vascular endothelial cadherin
- EGF
epidermal growth factor
- HGF
hepatocyte growth factor
- TNFα
tumor necrosis factor α
- KGF
keratinocyte growth factor
- TACE
TNFα Converting Enzyme
- VCAM
Vascular cell adhesion molecule
- SDC
syndecan
- VEGFR
vascular endothelial growth factor receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander CM, Howard EW, Bissell MJ, Werb Z. Rescue of mammary epithelial cell apoptosis and entactin degradation by a tissue inhibitor of metalloproteinases-1 transgene. J. Cell Biol. 1996;135(6 Pt 1):1669–1677. doi: 10.1083/jcb.135.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JS, Elrod JW. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J Anat. 2002;200(6):561–574. doi: 10.1046/j.1469-7580.2002.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, et al. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000;473(3):275–279. doi: 10.1016/s0014-5793(00)01528-3. [DOI] [PubMed] [Google Scholar]
- Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435(1):39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- Atkinson JJ, Toennies HM, Holmbeck K, Senior RM. Membrane-type 1 Matrix Metalloproteinase Is Necessary for Distal Airway Epithelial Repair and Keratinocyte Growth Factor Receptor Expression after Acute Injury. Am J Physiol Lung Cell Mol Physiol. 2007 doi: 10.1152/ajplung.00028.2007. [DOI] [PubMed] [Google Scholar]
- Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115(Pt 19):3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- Betsuyaku T, Fukuda Y, Parks WC, Shipley JM, Senior RM. Gelatinase B is required for alveolar bronchiolization after intratracheal bleomycin. Am. J. Pathol. 2000;157(2):525–535. doi: 10.1016/S0002-9440(10)64563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, et al. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4(2):197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Black RA, White JM. ADAMs: focus on the protease domain. Curr Opin Cell Biol. 1998;10(5):654–659. doi: 10.1016/s0955-0674(98)80042-2. [DOI] [PubMed] [Google Scholar]
- Boulday G, Fitau J, Coupel S, Soulillou JP, Charreau B. Exogenous tissue inhibitor of metalloproteinase-1 promotes endothelial cell survival through activation of the phosphatidylinositol 3-kinase/Akt pathway. Ann N Y Acad Sci. 2004;1030:28–36. doi: 10.1196/annals.1329.004. [DOI] [PubMed] [Google Scholar]
- Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van der Vliet A. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36(2):138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim.Biophys.Acta. 2000;1477(1–2):267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264(5158):569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, et al. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med. 1992;176(5):1375–1379. doi: 10.1084/jem.176.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard KM, Lund L, Mudgett JS, Mellin TN, Hunt TK, Murphy B, et al. Impaired wound contraction in stromelysin-1-deficient mice. Ann. Surg. 1999;230(2):260–265. doi: 10.1097/00000658-199908000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Farivar AS, Mulligan MS, Madtes DK. Tissue inhibitor of metalloproteinase-1 deficiency abrogates obliterative airway disease after heterotopic tracheal transplantation. Am J Respir Cell Mol Biol. 2006;34(4):464–472. doi: 10.1165/rcmb.2005-0344OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, Quinn JH, Winn HJ, Lanigan JM, Dellepella P, Colvin RB. Fibronectin is produced by blood vessels in response to injury. J Exp Med. 1982;156(2):646–651. doi: 10.1084/jem.156.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RAF, Lanigan JM, DellaPelle P, Manseau E, Dvorak HF, Colvin RB. Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound re-epithelization. J. Invest. Dermatol. 1982;79:264–269. doi: 10.1111/1523-1747.ep12500075. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I, Kim KS, Rajarathnam K, Gong JH, Dewald B, Moser B, et al. Structure-activity relationships of chemokines. J Leukoc Biol. 1995;57(5):703–711. doi: 10.1002/jlb.57.5.703. [DOI] [PubMed] [Google Scholar]
- Coraux C, Martinella-Catusse C, Nawrocki-Raby B, Hajj R, Burlet H, Escotte S, et al. Differential expression of matrix metalloproteinases and interleukin-8 during regeneration of human airway epithelium in vivo. J Pathol. 2005;206(2):160–169. doi: 10.1002/path.1757. [DOI] [PubMed] [Google Scholar]
- Corbel M, Caulet-Maugendre S, Germain N, Molet S, Lagente V, Boichot E. Inhibition of bleomycin-induced pulmonary fibrosis in mice by the matrix metalloproteinase inhibitor batimastat. J Pathol. 2001;193(4):538–545. doi: 10.1002/path.826. [DOI] [PubMed] [Google Scholar]
- Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, et al. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry DB, Rishi K, Kanellis J, Kiss A, Song LZ, Xu J, et al. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat. Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Ortho MP, Clerici C, Yao PM, Delacourt C, Delclaux C, Franco-Montoya ML, et al. Alveolar epithelial cells in vitro produce gelatinases and tissue inhibitor of matrix metalloproteinase-2. Am J Physiol. 1997;273(3 Pt 1):L663–L675. doi: 10.1152/ajplung.1997.273.3.L663. [DOI] [PubMed] [Google Scholar]
- Dunsmore SE, Saarialho-Kere UK, Roby JD, Wilson CL, Matrisian LM, Welgus HG, et al. Matrilysin expression and function in airway epithelium. J. Clin. Invest. 1998;102(7):1321–1331. doi: 10.1172/JCI1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Endo K, Takino T, Miyamori H, Kinsen H, Yoshizaki T, Furukawa M, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003;278(42):40764–40770. doi: 10.1074/jbc.M306736200. [DOI] [PubMed] [Google Scholar]
- Fu X, Parks WC, Heinecke JW. Activation and silencing of matrix metalloproteinases. Semin Cell Dev Biol. 2007 doi: 10.1016/j.semcdb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Gill SE, Pape MC, Khokha R, Watson AJ, Leco KJ. A null mutation for tissue inhibitor of metalloproteinases-3 (Timp-3) impairs murine bronchiole branching morphogenesis. Dev Biol. 2003;261(2):313–323. doi: 10.1016/s0012-1606(03)00318-x. [DOI] [PubMed] [Google Scholar]
- Gill SE, Pape MC, Leco KJ. Tissue inhibitor of metalloproteinases 3 regulates extracellular matrix--cell signaling during bronchiole branching morphogenesis. Dev Biol. 2006;298(2):540–554. doi: 10.1016/j.ydbio.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Huxley-Jones J, Clarke TK, Beck C, Toubaris G, Robertson DL, Boot-Handford RP. The evolution of the vertebrate metzincins; insights from Ciona intestinalis and Danio rerio. BMC Evol Biol. 2007;7:63. doi: 10.1186/1471-2148-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Kratz G, Haegerstrand A, Ståhle-Bäckdahl M. Collagenase expression is rapidly induced in wound-edge keratinocytes after acute injury in human skin, persists during healing, and stops at re-epithelialization. J. Invest. Dermatol. 1995;104:479–483. doi: 10.1111/1523-1747.ep12605917. [DOI] [PubMed] [Google Scholar]
- Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5) J Biol Chem. 2001;276(16):12501–12504. doi: 10.1074/jbc.C000848200. [DOI] [PubMed] [Google Scholar]
- Kheradmand F, Werb Z. Shedding light on sheddases: role in growth and development. Bioessays. 2002;24(1):8–12. doi: 10.1002/bies.10037. [DOI] [PubMed] [Google Scholar]
- Kim KH, Burkhart K, Chen P, Frevert CW, Randolph-Habecker J, Hackman RC, et al. Tissue inhibitor of metalloproteinase-1 deficiency amplifies acute lung injury in bleomycin-exposed mice. Am J Respir Cell Mol Biol. 2005;33(3):271–279. doi: 10.1165/rcmb.2005-0111OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampert M, Bloch W, Sasaki T, Bugnon P, Rulicke T, Wolf E, et al. Activities of the matrix metalloproteinase stromelysin-2 (MMP-10) in matrix degradation and keratinocyte organization in wounded skin. Mol Biol Cell. 2004;15(12):5242–5254. doi: 10.1091/mbc.E04-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L, MacDonald TT, Collins JE, Pender SL, Sanderson IR. Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology. 2006;130(1):127–136. doi: 10.1053/j.gastro.2005.09.032. [DOI] [PubMed] [Google Scholar]
- Leco KJ, Waterhouse P, Sanchez OH, Gowing KL, Poole AR, Wakeham A, et al. Spontaneous air space enlargement in the lungs of mice lacking tissue inhibitor of metalloproteinases-3 (TIMP-3) J Clin Invest. 2001;108(6):817–829. doi: 10.1172/JCI12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111(5):635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- Loechel F, Fox JW, Murphy G, Albrechtsen R, Wewer UM. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278(3):511–515. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- Lu PC, Ye H, Maeda M, Azar DT. Immunolocalization and gene expression of matrilysin during corneal wound healing. Invest Ophthalmol Vis Sci. 1999;40(1):20–27. [PubMed] [Google Scholar]
- Madlener M, Mauch C, Conca W, Brauchle M, Parks WC, Werner S. Growth factor regulation of stromelysin-2 expression by keratinocytes: implications for normal and imparied wound healing. Biochemistry J. 1996;320:659–664. doi: 10.1042/bj3200659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlener M, Parks WC, Werner S. Matrix metalloproteinases (MMPs) and their physiological inhibitors (TIMPs) are differentially regulated during excisional skin wound repair in mice. Exp. Cell Res. 1998;242:201–211. doi: 10.1006/excr.1998.4049. [DOI] [PubMed] [Google Scholar]
- Madtes DK, Elston AL, Kaback LA, Clark JG. Selective induction of tissue inhibitor of metalloproteinase-1 in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2001;24(5):599–607. doi: 10.1165/ajrcmb.24.5.4192. [DOI] [PubMed] [Google Scholar]
- Martin EL, Moyer BZ, Pape MC, Starcher B, Leco KJ, Veldhuizen RA. Negative impact of tissue inhibitor of metalloproteinase-3 null mutation on lung structure and function in response to sepsis. Am J Physiol Lung Cell Mol Physiol. 2003;285(6):L1222–L1232. doi: 10.1152/ajplung.00141.2003. [DOI] [PubMed] [Google Scholar]
- Martin P. Wound healing - aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15(11):599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM. Matrix metalloproteinases: they're not just for matrix anymore! Curr. Opin. Cell Biol. 2001;13:534–540. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- McCawley LJ, O'Brien P, Hudson LG. Epidermal growth factor (EGF)- and scatter factor/hepatocyte growth factor (SF/HGF)- mediated keratinocyte migration is coincident with induction of matrix metalloproteinase (MMP)-9. J. Cell Physiol. 1998;176(2):255–265. doi: 10.1002/(SICI)1097-4652(199808)176:2<255::AID-JCP4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol. 2003;162(6):1831–1843. doi: 10.1016/S0002-9440(10)64318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, et al. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J. Immunol. 2004;172(4):2586–2594. doi: 10.4049/jimmunol.172.4.2586. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Butler GS, Gong JH, Bendall L, Power C, Clark-Lewis I, et al. Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J. Biol. Chem. 2001;276(47):43503–43508. doi: 10.1074/jbc.M107736200. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289(5482):1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100(4):1160–1167. [PubMed] [Google Scholar]
- Mirastschijski U, Haaksma CJ, Tomasek JJ, Ågren MS. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Exp Cell Res. 2004;299(2):465–475. doi: 10.1016/j.yexcr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36(9):969–977. doi: 10.1038/ng1413. [DOI] [PubMed] [Google Scholar]
- Moss ML, Bartsch JW. Therapeutic benefits from targeting of ADAM family members. Biochemistry. 2004;43(23):7227–7235. doi: 10.1021/bi049677f. [DOI] [PubMed] [Google Scholar]
- Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr.Opin.Cell Biol. 2004;16(5):558–564. doi: 10.1016/j.ceb.2004.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland B, Tuft SJ, Khaw PT. Matrix metalloproteinase distribution during early corneal wound healing. Eye. 2005;19(5):584–588. doi: 10.1038/sj.eye.6701557. [DOI] [PubMed] [Google Scholar]
- Nguyen J, Gogusev J, Knapnougel P, Bauvois B. Protein tyrosine kinase and p38 MAP kinase pathways are involved in stimulation of matrix metalloproteinase-9 by TNF-alpha in human monocytes. Immunol Lett. 2006 doi: 10.1016/j.imlet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Parks WC. Matrix metalloproteinases in repair. Wound Repair Regen. 1999;7:423–432. doi: 10.1046/j.1524-475x.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4(8):617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG, Parks WC. The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J. Cell Biol. 1997;137:1445–1457. doi: 10.1083/jcb.137.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilcher BK, Sudbeck BD, Dumin JA, Welgus HG, Parks WC. Collagenase-1 and collagen in epidermal repair. Arch Dermatol Res. 1998;290(Suppl):S37–S46. doi: 10.1007/pl00007452. [DOI] [PubMed] [Google Scholar]
- Pilcher BK, Wang M, Qin XJ, Parks WC, Senior RM, Welgus HG. Role of matrix metalloproteinases and their inhibition in cutaneous wound healing and allergic contact hypersensitivity. Ann N Y Acad Sci. 1999;878:12–24. doi: 10.1111/j.1749-6632.1999.tb07671.x. [DOI] [PubMed] [Google Scholar]
- Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003;9(4):407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- Roberts CM, Tani PH, Bridges LC, Laszik Z, Bowditch RD. MDC-L, a novel metalloprotease disintegrin cysteine-rich protein family member expressed by human lymphocytes. J Biol Chem. 1999;274(41):29251–29259. doi: 10.1074/jbc.274.41.29251. [DOI] [PubMed] [Google Scholar]
- Saarialho-Kere U, Kerkela E, Jahkola T, Suomela S, Keski-Oja J, Lohi J. Epilysin (MMP-28) expression is associated with cell proliferation during epithelial repair. J Invest Dermatol. 2002;119(1):14–21. doi: 10.1046/j.1523-1747.2002.01790.x. [DOI] [PubMed] [Google Scholar]
- Saarialho-Kere UK, Crouch EC, Parks WC. Matrix metalloproteinase matrilysin is constitutively expressed in adult human exocrine epithelium. J Invest Dermatol. 1995;105(2):190–196. doi: 10.1111/1523-1747.ep12317104. [DOI] [PubMed] [Google Scholar]
- Saarialho-Kere UK, Vaalamo M, Airola K, Niemi K-M, Oikarinen AI, Parks WC. Interstitial collagenase is expressed by keratinocytes which are actively involved in re-epithelialization in blistering skin diseases. J. Invest. Dermatol. 1995;104:982–988. doi: 10.1111/1523-1747.ep12606231. [DOI] [PubMed] [Google Scholar]
- Salmela MT, Pender SL, Karjalainen-Lindsberg ML, Puolakkainen P, Macdonald TT, Saarialho-Kere U. Collagenase-1 (MMP-1), matrilysin-1 (MMP-7), and stromelysin-2 (MMP-10) are expressed by migrating enterocytes during intestinal wound healing. Scand J Gastroenterol. 2004;39(11):1095–1104. doi: 10.1080/00365520410003470. [DOI] [PubMed] [Google Scholar]
- Sawicki G, Marcoux Y, Sarkhosh K, Tredget EE, Ghahary A. Interaction of keratinocytes and fibroblasts modulates the expression of matrix metalloproteinases-2 and -9 and their inhibitors. Mol Cell Biochem. 2005;269(1–2):209–216. doi: 10.1007/s11010-005-3178-x. [DOI] [PubMed] [Google Scholar]
- Scholz F, Schulte A, Adamski F, Hundhausen C, Mittag J, Schwarz A, et al. Constitutive expression and regulated release of the transmembrane chemokine CXCL16 in human and murine skin. J Invest Dermatol. 2007;127(6):1444–1455. doi: 10.1038/sj.jid.5700751. [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes & Development. 2003;17(1):7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- Shimoda M, Hashimoto G, Mochizuki S, Ikeda E, Nagai N, Ishida S, et al. Binding of ADAM28 to P-selectin glycoprotein ligand-1 enhances P-selectin-mediated leukocyte adhesion to endothelial cells. J Biol Chem. 2007 doi: 10.1074/jbc.M702414200. [DOI] [PubMed] [Google Scholar]
- Sigurdson L, Sen T, Hall L, 3rd, Rubenfeld A, Hard R, Gardella J, et al. Possible impedance of luminal reepithelialization by tracheal cartilage metalloproteinases. Arch Otolaryngol Head Neck Surg. 2003;129(2):197–200. doi: 10.1001/archotol.129.2.197. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341(10):738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Singh RJ, Mason JC, Lidington EA, Edwards DR, Nuttall RK, Khokha R, et al. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. 2005;67(1):39–49. doi: 10.1016/j.cardiores.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol. 2006;176(2):721–725. doi: 10.4049/jimmunol.176.2.721. [DOI] [PubMed] [Google Scholar]
- Spradling KD, McDaniel AE, Lohi J, Pilcher BK. Epsin 3 is a novel extracellular matrix-induced transcript specific to wounded epithelia. J Biol Chem. 2001;276(31):29257–29267. doi: 10.1074/jbc.M101663200. [DOI] [PubMed] [Google Scholar]
- Surendran K, Simon TC, Liapis H, McGuire JK. Matrilysin (MMP-7) expression in renal tubular damage: association with Wnt4. Kidney Int. 2004;65(6):2212–2222. doi: 10.1111/j.1523-1755.2004.00641.x. [DOI] [PubMed] [Google Scholar]
- Tam EM, Morrison CJ, Wu YI, Stack MS, Overall CM. Membrane protease proteomics: Isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc Natl Acad Sci U S A. 2004;101(18):6917–6922. doi: 10.1073/pnas.0305862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki K, Kanzaki T, Aoki T, Iwata K, Saiki I. Effects of recombinant human tissue inhibitor of metalloproteinases-2 (rh-TIMP-2) on migration of epidermal keratinocytes in vitro and wound healing in vivo. J Dermatol. 2003;30(3):165–172. doi: 10.1111/j.1346-8138.2003.tb00367.x. [DOI] [PubMed] [Google Scholar]
- Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, et al. LPS Responsiveness and Neutrophil Chemotaxis In Vivo Require PMN MMP-8 Activity. PLoS ONE. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96(8):2673–2681. [PubMed] [Google Scholar]
- Van Den Steen PE, Wuyts A, Husson SJ, Proost P, Van Damme J, Opdenakker G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur. J. Biochem. 2003;270(18):3739–3749. doi: 10.1046/j.1432-1033.2003.03760.x. [DOI] [PubMed] [Google Scholar]
- Warner RL, Bhagavathula N, Nerusu KC, Lateef H, Younkin E, Johnson KJ, et al. Matrix metalloproteinases in acute inflammation: induction of MMP-3 and MMP-9 in fibroblasts and epithelial cells following exposure to pro-inflammatory mediators in vitro. Exp Mol Pathol. 2004;76(3):189–195. doi: 10.1016/j.yexmp.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Welch MP, Odland GF, Clark RA. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J Cell Biol. 1990;110(1):133–145. doi: 10.1083/jcb.110.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Smola H, Liao X, Longaker MT, Krieg T, Hofschneider PH, et al. The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science. 1994;266:819–822. doi: 10.1126/science.7973639. [DOI] [PubMed] [Google Scholar]
- Xu J, Clark RA. Extracellular matrix alters PDGF regulation of fibroblast integrins. J Cell Biol. 1996;132(1–2):239–249. doi: 10.1083/jcb.132.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao PM, Delclaux C, D'Ortho MP, Maitre B, Harf A, Lafuma C. Cell-matrix interactions modulate 92-kD gelatinase expression by human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1998;18:812–822. doi: 10.1165/ajrcmb.18.6.2984. [DOI] [PubMed] [Google Scholar]
- Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6(12):931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 2003;6(10):1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- Zhou M, Zhang Y, Ardans JA, Wahl LM. Interferon-gamma differentially regulates monocyte matrix metalloproteinase-1 and -9 through tumor necrosis factor-alpha and caspase 8. J Biol Chem. 2003;278(46):45406–45413. doi: 10.1074/jbc.M309075200. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O, Nomiyama H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006;7(12):243. doi: 10.1186/gb-2006-7-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]