

Abstract
Perinatal hypoxia-ischemia (H-I) often manifests as cognitive and/or motor disturbances that appear early in development. Growing evidence indicates that neuroinflammation may exacerbate H-I injury. Resident microglia release proinflammatory cytokines and proteases in response to ischemia. Matrix metalloproteinases (MMPs), in particular, activate cytokines and degrade basement membrane proteins. These actions ultimately permit entry of peripheral leukocytes into the central nervous system neuropil, enhancing neuroinflammation and cell death. Currently, the relative contributions of resident and peripheral immune cells to ischemic brain injury are unclear. The present study employed an ex-vivo model of H-I through oxygen glucose deprivation (OGD) to identify the cellular localization of MMP-9 in organotypic hippocampal slices, and to determine whether inhibiting gelatin-degrading MMPs affords neuroprotection in the absence of peripheral immune cells. Immunohistochemistry revealed ubiquitous neuronal MMP-9 expression in both normoxic and hypoxic slices. Increased MMP-9 expression was detected in CD11b – positive microglia after 48 hrs exposure to OGD relative to normoxic controls. Consistent with these data, in situ zymography showed increased gelatinolytic activity after OGD. Gelatin-cleaved fluorescence localized to astrocytic processes and somata of various cellular morphologies. Treatment with either the MMP inhibitor AG3340 (prinomastat) or minocycline dampened OGD – induced gelatinolytic activity and neural injury, as measured by Fluoro-Jade staining, relative to vehicle controls. These results show that resident microglia, in the absence of peripheral immune cells, were sufficient to enhance neural injury after OGD in the organotypic hippocampal slice. Additionally, these effects were associated with upregulation or secretion of MMP-9, and were blocked after treatment with either the gelatinase-selective compound AG3340 or the anti-inflammatory compound minocycline. These data, coupled with the effectiveness of these compounds previously shown in vivo, support the selective targeting of gelatin-degrading MMPs and activated microglia as potential therapeutic approaches to combat neonatal H-I injury.
Keywords: inflammation, neuroprotection, central nervous system, matrix metalloproteinase, microglia, oxygen glucose deprivation
Perinatal hypoxia-ischemia (H-I) produces cerebral white matter injury (Back et al., 2002, McQuillen and Ferriero, 2004) and neurodegeneration (Vannucci et al., 1999), both of which result in loss of function (Back and Rivkees, 2004, Folkerth, 2005). Similarly, neonatal rodents exposed to H-I exhibit cognitive deficits (Young et al., 1986, Ikeda et al., 2001, Arteni et al., 2003) and impaired motor function (Jansen and Low, 1996, Bona et al., 1997, Ten et al., 2003). Previous work demonstrated that oxidative stress (Back et al., 1998, Bernardo et al., 2003) and NMDA receptor activation (Kaur et al., 2006) contribute to white matter injury, while glutamatergic blockade immediately following H-I reduced white matter injury (Follett et al., 2000) and cerebral infarction (Dingley et al., 2006). However, selective targeting of glutamate receptors and free radicals have failed in clinical trials (Hoyte et al., 2004, Shuaib et al., 2007, Lo, 2008).
The lack of effective therapies underscores the need for novel approaches. The proinflammatory roles of resident microglia are well-documented in the developing brain (Chew et al., 2006). Following ischemia, activated microglia upregulate the expression of cytokines and chemokines (Fukui et al., 2006, Zhao et al., 2006, Van Lint and Libert, 2007). Blood brain barrier degradation further enhances microglial activation and macrophage recruitment (Alvarez-Diaz et al., 2007). Numerous data suggest that matrix metalloproteinases (MMPs) may be instrumental in the production and maintenance of a proinflammatory microenvironment (del Zoppo et al., 2007). Astrocytes and microglia become activated within hours after ischemia. In vitro, MMP-9 expression was induced in astrocytes after stimulation with either TNF-α or IL-1β (Gottschall and Yu, 1995), and in microglia after stimulation with LPS (Rosenberg et al., 2001). Conversely, MMPs have been shown to process both TNF-α (Gearing et al., 1994) and IL-1β (Schonbeck et al., 1998) to their biologically active forms, while other data demonstrated a role for MMPs in monocyte chemoattractant protein-1 (MCP-1) chemokine signaling (Overall et al., 2002). Additionally, MMP-mediated basement membrane proteolysis occurs after ischemia (Cunningham et al., 2005, Manicone and McGuire, 2007), linking MMP activity to peripheral monocyte infiltration. MMP-9 knockout mice showed improved outcomes that were associated with attenuated blood brain barrier degradation (Asahi et al., 2001, Gidday et al., 2005) and reduced microglial activation (Svedin et al., 2007). Other in vivo experiments revealed increased MMP-9 expression in neutrophils and endothelial cells after middle cerebral artery occlusion (MCAO) and elevated MMP-2 expression in astrocytic endfeet (Rosenberg et al., 2001). Cortical gelatinase activity also increased after MCAO, and neuronal laminin degradation was associated with upregulated MMP-9 expression (Gu et al., 2005).
These data suggest a complex regulatory system driven by the concerted actions of gelatin-degrading MMPs (MMP-2, MMP-9) and proinflammatory molecules. Minocycline, a second generation tetracycline with anti-inflammatory properties, is neuroprotective in several rat injury models (Cai et al., 2006, Fan et al., 2006, Wasserman and Schlichter, 2007) and inhibits MMP activity both in vitro and in vivo (Machado et al., 2006). AG3340 (prinomastat), a small molecule hydroxamate-based MMP inhibitor with high selectivity for gelatinases (Santos et al., 1997, Shalinsky et al., 1998), showed efficacy in limiting tumor growth in rodent cancer models (Price et al., 1999, Alves et al., 2001) and was neuroprotective in adult rodents exposed to chronic ischemia (Nakaji et al., 2006). Both compounds demonstrate good oral bioavailability. Previous work in our laboratory showed that both AG3340 and minocycline were efficacious in reducing H-I – induced neuroinflammation and infarction in the neonatal rat (Leonardo et al., 2008a), yet the relative contributions of peripheral monocytes/macrophages and resident microglia, and their association with MMP activity, are not clear. The present study utilized an ex-vivo model that lacks peripheral immune cell involvement to assess gelatinase activity, to identify the cellular localization of MMP-9, and to determine whether inhibition of gelatin-degrading MMPs protects organotypic hippocampal slices from injury after oxygen glucose deprivation (OGD).
Experimental Procedures
Animal Care
All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals with a protocol approved by the Institutional Animal Care and Use Committee at the University of South Florida. Experiments were designed to limit the number of animals required. Timed pregnant Sprague-Dawley dams were purchased from Harlan Labs (Indianapolis, IN), maintained on a 12 hr light/dark cycle (7 am – 7 pm) and given access to food and water ad libitum. Neonatal rats birthed from time-pregnant dams were used for all organotypic slice culture experiments. Postnatal day 28 (P28) rats were used for the Lipopolysaccharide infusion experiments.
Organotypic Slice Culture
Organotypic slice cultures were prepared using a method previously described (Stoppini et al., 1991), with slight adaptations. P8-P10 rat pups were decapitated, brains were removed, and intact hippocampi were dissected in cold isotonic buffer (136.89 mM NaCl, 5.37 mM KCl, 169 nM Na2HPO4, 22.04 nM KH2PO4, 27.52 nM glucose, 59.01 mM sucrose). Whole hippocampi were sliced sagitally at 400 μm thickness using a McIlwain Tissue Chopper (Mickle Laboratory Engineering Co. Ltd. Gomshall, Surrey, England). Only slices that appeared thin and translucent were selected for culture. Slices were then incubated for 90 min at 4°C. Cultures were maintained on Millicell CM (Millipore Corp., Billerica, MA) inserts and placed in 6-well plates containing Neurobasal media supplemented with B27 and 5 mM L-glutamine. Slices were cultured for 14 days in a standard tissue culture incubator at 37°C, receiving partial media changes every 3-4 days prior to experimentation. Each experiment utilized 2 pups for slice preparation. A total of 6 slices per treatment group were used for each experiment, and data were collected from 3 separate experiments.
Oxygen Glucose Deprivation
Organotypic slices were subjected to 48 hrs of normoxia or OGD to determine first, whether gelatinase activity is associated with neural injury, and second, whether selective gelatinase inhibition could reduce cell death after OGD in the absence of peripheral immune cell effects. Slices were assigned to 1 of 2 exposures (normoxia or OGD) and 1 of 3 treatments (vehicle, AG3340, or minocycline). Immediately prior to exposure, inserts were transferred into new 6-well plates containing vehicle, AG3340 (AG3340 was kindly provided by Dr. Peter Baciu, Allergan, Irvine, CA), or minocycline (Sigma, St. Louis, MO). Vehicle consisted of 0.1% DMSO in Dulbecco's Modified Eagles Medium (DMEM; Mediatech, Herndon, VA) for normoxia or DMEM without glucose (Invitrogen, Carlsbad, CA) for OGD. Stock solutions of minocycline and AG3340 were prepared, aliquoted and stored at -80°C prior to experiments. Fresh working solutions of AG3340 (3 μM) and minocycline (30 μM) were prepared for each experiment. Drug concentrations were selected based on doses that reduced neuroinflammation and neural injury in vivo (Leonardo et al., 2008a). Cultures were maintained in a standard tissue culture incubator during the normoxia exposure. For the OGD exposure, a chamber (CBS Scientific Co. Inc., Del Mar, CA) was flushed with a gas mixture containing 1% O2 and 5% CO2 (N2 balanced) and held at 37°C.
Lipopolysaccharide Infusions
Lipopolysaccharide (LPS), a well-known activator of MMPs, was infused unilaterally into P28 rats (N = 2) to verify the selectivity of anti-MMP-9. Animals were anesthetized with 2.5% isofluorane, placed on a heating pad (37°C) and maintained at 350 ml/min of oxygen and 1.5% isofluorane with an interfaced scavenging system for the duration of the surgery. Rats were secured into the stereotaxic apparatus (David Kopf Instruments, Tujunga, CA), the skull was exposed, and a hole was drilled through the skull. A Hamilton syringe, containing a 5 μg/μl solution of LPS (Sigma) dissolved in phosphate-buffered saline (PBS, pH 7.4), was carefully lowered into the right frontoparietal motor cortex. After a 5 min equilibration period, 2 μl of LPS was infused at a rate of 1 μl/min to yield a total concentration of 10 μg. Following a 5 min equilibration period, the needle was carefully removed, the skull hole was filled in with bone wax and the wound was sutured closed. The cerebral cortex was targeted using the following Bregma coordinates: AP = 1.2 mm; ML = 2.0 mm; DV = 1.5 mm.
Histology and Immunohistochemistry
Immunohistochemistry was performed as previously described (Leonardo et al., 2008b) to verify the selectivity of MMP-9 and to determine the cellular localization of MMP-9 in organotypic hippocampal slices after exposure to normoxia or OGD. For the LPS infusion experiments, rats (N = 2) were anesthetized with pentobarbital (60 mg/kg) and intracardially perfused with PBS followed by 4% paraformaldehyde. Brains were removed and cryopreserved with increasing sucrose concentrations (15%, 30%), sectioned coronally on a cryostat at 30 μm thickness and thaw-mounted onto Superfrost slides (Fisher Scientific, Suwane, GA). For organotypic slices, inserts were transferred into 12 well plates containing 1.1 mL of 4% paraformaldehyde immediately following the 48 hr exposure. After a 24 hr fixation at 4°C, inserts were submerged in PBS. Slices were carefully dissociated from the inserts using a small paint brush, mounted onto Superfrost slides and dried overnight at room temperature.
For immunohistochemistry, slices and brain sections were rinsed with PBS, permeabilized and blocked for 60 min at room temperature (3% Triton X-100, 3% 1M Lysine, 10% Normal Goat Serum in PBS), and incubated overnight with primary antibody at 4°C (3% Triton X-100, 2% Normal Goat Serum in PBS). On day 2, slices were washed with PBS, incubated 60 min with secondary antibody at room temperature, washed again and coverslipped using Vectashield aqueous mounting media (Vector Labs, Burlingame, CA). Double-label immunohistochemistry was achieved by co-incubation with anti-mouse and anti-rabbit primary antibodies, and subsequent co-incubation with secondary antibodies conjugated to distinct fluorophores for each respective species of primary antibody.
Primary antibodies used in these studies were mouse anti-glial fibrillary acidic protein (GFAP; Roche Applied Science, Indianapolis, IN; 1:1000), mouse anti-CD11b (OX-42; Serotec, Raleigh, NC; 1:3000), rabbit anti-neuron specific enolase (NSE; Polysciences, Inc., Warrington, PA; 1:1000), mouse anti-CD-45 (Serotec; 1:200), rabbit anti-MMP-9 (Abcam, Cambridge, MA; 1:200) and mouse anti-MMP-9 (Chemicon, Temecula, CA, 1:1000). Primary antibodies were visualized using either Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies (Molecular Probes, Eugene, OR). Working concentrations for secondary antibodies were as follows: 1:300 for OX-42 and CD-45; 1:1000 for all other primary antibodies. The specificities of all antibodies were verified both in vitro and in vivo.
Fluoro-Jade Staining
Fluoro-Jade staining was performed to determine whether treatment with AG3340 or minocycline affords neural protection in organotypic hippocampal slices exposed to OGD. The Fluoro-Jade stain identifies dead and degenerating cells, thus providing a positive quantitative marker as opposed to the absence of labeling that is observed when using Nissl stain (Leonardo et al., 2008b). Fluoro-Jade was previously shown to be a more sensitive measure of cell death when compared to triphenyltetrazolium chloride in a murine model of stroke (Duckworth et al., 2005). This method was adapted from Schmued and colleagues (Schmued et al., 1997) and subsequently detailed (Duckworth et al., 2005). Slices were prepared and mounted on Superfrost slides as described for histology. Slides were sequentially placed in 100% ethanol for 3 min, 70% ethanol for 1 min and ddH2O for 1 min. Slices were oxidized for 15 min using 0.06% KMnO4 solution followed by 3 brief rinses in ddH2O. Slides were then immersed in a 0.001% solution of Fluoro-Jade (Histo-Chem, Jefferson, AR) in 0.1% acetic acid for 30 min, rinsed with ddH2O, dried for 60 min at 37°C, cleared with xylene and coverslipped using DPX medium (Electron Microscopy Sciences, Ft. Washington, PA).
Thionin and Propidium Iodide Staining
The viability of cultured organotypic slices was verified using the thionin stain. Thionin labels Nissl bodies and dark staining demonstrates indicates intact, viable slices. For staining, slices were dried for 1 hr at 50°C and sequentially placed in 100% EtOH, 95% EtOH, 70% EtOH, and 50% EtOH for 2 min each. Slices were then placed in dH20 for 1 min and stained with thionin (Sigma) in a glacial acetic acid/ I N NaOH solution for 50 sec. After staining, slices were dehydrated in reverse order, cleared with xylene and coverslipped with Permount.
Propidium iodide (PI) staining was also performed on separate slices during the normoxia and hypoxia exposures to verify the specificity of the Fluoro-Jade stain for neurodegenerative tissue. PI intercalates into cells through compromised plasma membranes and binds to nucleic acids, thus revealing cellular injury. For staining, a 2 mM working solution was prepared in dH20 containing 0.1% sodium azide. The working solution was added to each media-containing well (1:1000) just prior to the exposure to yield a final concentration of 2 μM.
In Situ Zymography
In situ zymography was performed using a method previously described (Oh et al., 1999) to assess gelatinase activity in both LPS – infused rat brain sections and organotypic hippocampal slices exposed to normoxia or OGD. For the LPS infusion experiments, rats were anesthetized with pentobarbital (60 mg/kg), brains were quickly removed on ice and snap-frozen in dry ice. Brains were then sectioned coronally on a cryostat at 12 μm thickness, thaw-mounted onto Superfrost slides and stored at -80°C. Organotypic slices were dissociated from the inserts immediately following the 48 hr exposure and mounted onto Superfrost slides. Slices were dried briefly prior to incubation.
For detection of gelatinase activity, slices and brain sections were coated with in situ zymography buffer (0.05 M Tris HCl, 0.15 M NaCl, 5 mM CaCl, 0.2 mM NaN3) containing 20 μg of DQ gelatin (Invitrogen), coverslipped and incubated 90 min at 37°C to allow for enzymatic cleavage of the gelatin. Quenched FITC molecules contained within the gelatin emit fluorescence when the gelatin is enzymatically cleaved by MMP-2 or MMP-9. Following incubation, fluorescence was visualized using a Zeiss Axioscope 2 equipped with a BP 450-490 fluorescent filter (450-490 nm excitation; 515-565 nm emission).
Gelatin Zymography
AG3340 has been previously shown to be a selective inhibitor of gelatin-degrading MMPs. Gelatin zymography was performed to verify that this compound inhibits the activity of MMP-9. Gelatinase zymography standards (Chemicon) were loaded and separated by SDS PAGE using a 10% Tris-Glycine gel containing 0.1% gelatin. Following electrophoresis, the gel was bisected for subsequent incubations with either developing buffer alone or developing buffer containing 3 mM AG3340. Gels were then incubated for 30 min at room temperature in renaturing buffer, followed by 30 min equilibration in developing buffer. Developing buffer was then decanted, fresh developing buffer was added and gels were incubated for 24 hrs at 37°C. Following incubation, gels were stained 25 min with 0.5% Coomassie Blue B-7920 (Sigma). Renaturing and developing was performed using Novex® zymogram reagents (Invitrogen).
Western Blotting
Western blotting was performed as previously described (Ajmo et al., 2008), with minor adaptations. Animals were euthanatized by exposure to excess CO2 until death and immediately decapitated. Cerebral cortices from ischemic rat brain were rapidly dissected, extracted with a teflon-glass homogenizer in 5 volumes of Triton-X-100-containing buffer (20 mM Tris-HCl at pH 7.4, 10 mM EDTA, 1% Triton-X-100, and 1:100 protease inhibitor cocktail [Calbiochem type III, LaJolla, CA]) and homogenized for 2 min. Homogenates were centrifuged in a microcentrifuge at 6800 × g for 5 min. Supernatants were collected and stored at -80°C prior to SDS-PAGE.
Tissue extracts were loaded (20 μg/well total protein) onto pre-cast, 1.5 mm, 4–20% gradient SDS-PAGE gels (Novex Tris-Glycine, Invitrogen, Carlsbad, CA). Separated proteins were electrophoretically transferred to a polyvinylidine difluoride membrane (PVDF, Immobilon, Millipore, Billerica, MA). For immunoblotting, the membrane was washed with Buffer B (10 mM phosphate buffered saline, pH 7.4 containing 0.05% Tween 20) for 5 min, blocked for 60 min in 5% non-fat dry milk diluted in Buffer B, and incubated for 2 hrs in mouse anti-MMP-9 (Chemicon, Temecula, CA, 1:500). The membrane was then washed 3 × 5 min in Buffer B and incubated 60 min in anti-mouse secondary antibody conjugated to horseradish peroxidase (Chemicon, Temecula, CA). Antigens were visualized using a chemiluminescence developing system (SuperSignal, Pierce, Rockford, IL).
Image Analyses and Quantification
For quantification, images were acquired using a Zeiss Axioscope 2 microscope controlled by Openlab (Improvision Ltd, Lexington, MA) software, and photomicrographs were captured with a Zeiss Axicam Color camera. All images subjected to direct comparisons were captured at the same exposure and digital gain settings to eliminate confounds of differential background intensity or false-positive fluorescent signal across sections. Fluorescence was quantified using NIH ImageJ software. Photomicrographs of organotypic hippocampal slices were imported into ImageJ and background subtraction was achieved by enhancing contrast until background particles were eliminated from the images. To control for size differences between slices, fluorescence values were calculated by dividing the total fluorescence by the total area of the slice, thus yielding percent of total area occupied by fluorescent signal. This method of quantification has been used in previous studies and was shown to correlate with reductions in both NeuN and cresyl violet staining (Leonardo et al., 2008b).
Additional high-power micrographs were captured for MMP-9 immunohistochemistry and in situ zymography to verify cell-specific expression of MMP and to better determine the cellular localization of gelatinase activity. These images were obtained using a Leica SP-2 confocal microscope and Leica LCS software.
Statistical Analyses
Data from all treatment groups were expressed as mean ± SEM of the percent area occupied by fluorescent signal. Group means were then subjected to two-way ANOVAs with “p values” set at 0.05. Pair-wise comparisons of group means were made using Bonferroni post-hoc tests.
Results
Oxygen Glucose Deprivation in the Organotypic Hippocampal Slice
Organotypic slices were cultured for 14 days and exposed to 48 hrs of normoxia or OGD. Slices exposed to normoxia retained viability and three-dimensional structure (Fig. 1). Thionin staining showed dense, intact pyramidal and dentate neuronal layers (Fig. 1A). Exposure to OGD resulted in severe cellular injury. Fluoro-Jade staining was intense in slices exposed to OGD and was predominantly localized to the neuronal layers (Fig. 1B), demonstrating high selectivity for injured neurons. Fluoro-Jade staining was verified with PI, which showed robust staining throughout slices exposed to OGD (Fig. 1D) when compared to the low basal PI staining present in normoxic controls (Fig. 1C). While PI staining identifies compromised plasma membranes of all cell types, Fluoro-Jade stain is a more sensitive and reliable marker for neuronal injury.
Figure 1. OGD in the organotypic hippocampal slice.

Organotypic slices were cultured for 14 days and exposed to 48 hrs of normoxia or OGD. Thionin staining revealed intact hippocampal slices (A, inset) with dense neuronal layers (A, dark labeling) after exposure to normoxia. Fluoro-Jade staining revealed neural injury that was predominantly localized to the neuronal layers after OGD (B, white labeling), demonstrating selectivity for neuronal cell death. OGD – induced cell death was validated with propidium iodide (PI) staining (C,D, white labeling), showing marked PI elevations in slices exposed to OGD (D) compared to normoxic controls (C). Scale bars = 100 μm.
Validation of Anti-MMP-9 Antibody Selectivity and Gelatinase Inhibition
LPS is a known activator of gelatin-degrading MMPs. To verify the specificity of anti-MMP-9, in situ zymography and immunohistochemistry were performed on rat brain tissues following unilateral cortical infusion of 10 μg LPS (Fig. 2) Gelatinase activity, as indicated by gelatin-cleaved fluorescent signal, was prominent throughout the frontoparietal motor cortex surrounding the infusion site (Fig. 2B,E). Fluorescent signal was nearly absent from contralateral control hemispheres with the exception of faint labeling that was restricted to blood vessels (Fig. 2A,D). Immunohistochemistry revealed dense populations of CD-45 – expressing microglia/macrophages (Fig. 2C) surrounding the infusion site. Many of these cells also displayed intense immunoreactivity for MMP-9 (Fig. 2F), consistent with LPS – induced gelatinase activity.
Figure 2. Verification of gelatinase inhibition and MMP-9 specificity.
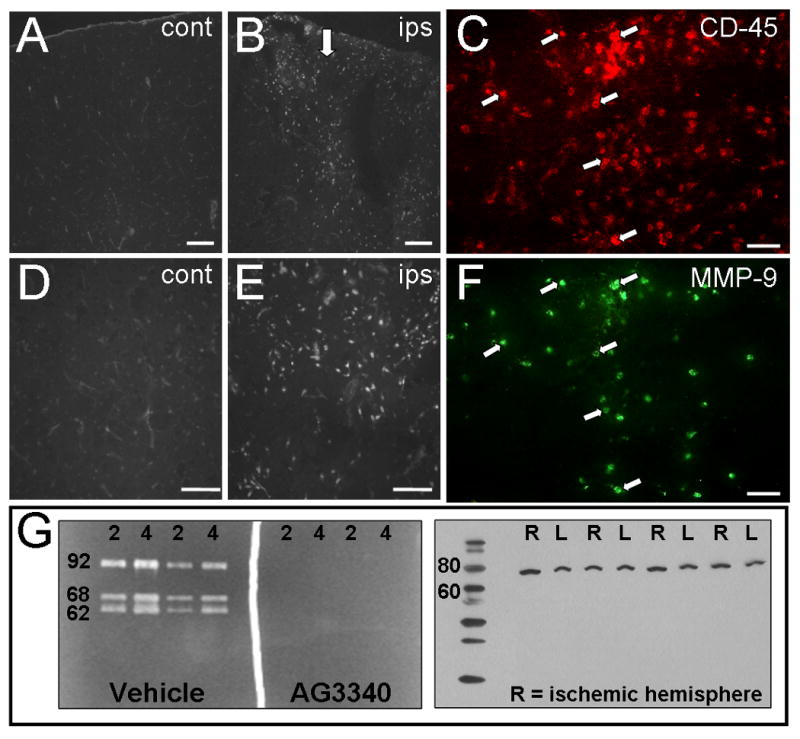
In situ zymography (A,B,D,E) was performed in tissues from rats that received 10 μg cortical infusions of LPS (B, arrow). Gelatin-cleaved fluorescence was prominent in cortical regions adjacent to the LPS infusion site (B,D). Fluorescent signal in contralateral control hemispheres was faint by comparison and was detected only in blood vessels (A,D). Consistent with increased gelatinase activity, LPS induced elevations in CD-45 (C) and MMP-9 (F) immunoreactivity. Gelatin zymography was performed using 2 ng and 4 ng of purified gelatinase zymography standards (G, left panel). Intense bands of cleaved gelatin were detected at the predicted positions on SDS-PAGE for MMP-9 (92 kDa) and MMP-2 (68, 62 kDa). Incubation with 3 mM AG3340 showed complete inhibition of gelatinase activity. Western Blot of cortical homogenates from ischemic rat brain (G, right panel) shows a single band migrated at the predicted position on SDS-PAGE for activated MMP-9 (∼78 kDa). N = 2 (in situ zymography), N = 4 (Western Blot; R = ischemic hemisphere). Scale bars = 100 μm. Arrows indicate cells expressing both antigens.
To determine whether AG3340 could inhibit MMP-9 activity, gelatin zymography was performed using purified standards containing both MMP-2 and MMP-9 (Fig. 2G, left panel). The proteins migrated at the predicted positions on SDS-PAGE for the MMP-9 proenzyme (92 kDa), the MMP-2 proenzyme (68 kDa) and activated MMP-2 (62 kDa). The addition of 3 mM AG3340 to the developing buffer abolished all activity of MMP-2 and MMP-9, demonstrating the efficacy of this compound in conferring gelatinase inhibition. The specificity of anti-MMP-9 was additionally confirmed by loading 20 μg/well of total protein from ischemic rat cortex onto SDS-PAGE and performing a Western Blot. Results showed a single band that migrated at approximately 80 kDa (Fig. 2G, right panel), consistent with the predicted position for activated MMP-9 on SDS-PAGE (78 kDa).
Cellular Localization of MMP-9
MMPs are known to be involved in the neuropathological response to ischemia. While MMP-2 activity contributes to peripheral monocyte infiltration through blood brain barrier disruption, MMP-9 activity may contribute to neural injury by enhancing inflammation. Therefore, immunohistochemistry was employed to characterize the cellular localization of MMP-9 in an ex-vivo model of H-I that lacks peripheral immune cell infiltration. Organotypic hippocampal slices were cultured for 14 days and exposed to normoxia or OGD. MMP-9 expression was ubiquitous in slices exposed to normoxia (Fig. 3). GFAP – positive astrocytes were prevalent throughout the slices and displayed the typical morphology associated with the quiescent state (Fig. 3B). The majority of MMP-9 expression did not colocalize with astrocytes (Fig. 3C), though MMP-9 did localize to some astrocytic processes (Fig. 3A-C, arrows). To determine whether basal MMP-9 expression occurs in resident immune cells, slices were double-stained with anti-MMP-9 and anti-CD11b, an antigen that is commonly expressed on the cell surfaces of microglia. CD11b immunopositive cells displayed the classic amoeboid morphology associated with activated microglia (Fig. 3E), consistent with previous data indicating that these cells exhibit a heightened activation state under culture conditions. Interestingly, little MMP-9 immunoreactivity was associated with microglia (Fig. 3D-F). Instead, the majority of MMP-9 immunoreactivity colocalized with NSE-positive neurons (Fig. 3G-I, arrows) and was predominantly cell surface-associated (Fig. 3J, arrows).
Figure 3. Basal MMP-9 expression.

Micrographs show cellular expression of MMP-9 (A,D,H), GFAP (B), CD11b (E) and NSE (I) after 48 hrs exposure to normoxia. (A-C) Basal MMP-9 expression colocalized with astrocytic processes (C). (D-F) CD11b – expressing microglia displayed the activated phenotype in normoxic slices (E) but did not colocalize with MMP-9 (F). (G-J) MMP-9 expression was prominent on cell surfaces of NSE-immunopositive neurons (J). Scale bars = 100 μm. Arrows indicate cells expressing both antigens.
MMP-9 immunoreactivity was elevated in slices exposed to OGD relative to normoxic controls. GFAP immunoreactivity was also intense, localizing to astrocytic cell bodies and processes throughout the slices (Fig. 4). In contrast to normoxic slices, hypertrophic astrocytes, a hallmark of H-I injury, were prevalent in slices subjected to OGD (Fig. 4A). Similar to the expression profile observed in normoxic slices, MMP-9 immunoreactivity did not colocalize with GFAP – positive cell bodies (Fig. 4C). However, exposure to OGD resulted in increased CD11b – expressing microglia (Fig. 4D) that were generally detected adjacent to the hippocampal pyramidal cell layers, and MMP-9 immunoreactivity colocalized with these cells (Fig. 4D-F, arrows). As with normoxic slices, MMP-9 was also expressed in NSE-positive neurons (Fig. 4G-I, arrows) and showed abundant colocalization (Fig. 4J, arrows).
Figure 4. MMP-9 expression after OGD.

Micrographs show cellular expression of MMP-9 (B,E,I), GFAP (A), CD11b (D) and NSE (H) after 48 hrs exposure to OGD. (A-C) Hypertrophic astrocytes showed intense GFAP immunoreactivity (A) but little colocalization with MMP-9 (C). (D-F) CD11b – expressing microglia were ubiquitous (D) and colocalized with MMP-9 (F) after OGD. (G-J) NSE – positive neurons (H) expressed MMP-9 on cell surfaces (J). Scale bars = 100 μm. Arrows indicate cells expressing both antigens.
Gelatinase Activity Increases after Oxygen Glucose Deprivation
To determine whether increased MMP-9 expression after OGD was associated with increased gelatinolytic activity, in situ zymography was performed on slices exposed to normoxia or OGD (Fig. 5). Gelatin-cleaved fluorescence was observed in slices exposed to normoxia (Fig. 5A,C), indicative of basal MMP-2 and/or MMP-9 activity in organotypic slice culture. Gelatinase activity was primarily localized in the nuclei and throughout the cytoplasm. After exposure to OGD, slices showed increased fluorescent labeling (Fig. 5B,D). As in normoxic slices, gelatinase activity was nuclear and cytoplasmic. However, compact, amoeboid cells were ubiquitous after OGD (Fig. 5B), and this morphology occurred rarely in normoxic slices upon comparison. Cells also exhibited greater fluorescent intensity after OGD, which was also localized to cellular processes when viewed at higher magnification (Fig. 5D). Gelatin-cleaved fluorescence in normoxic slices was faint by comparison and was not detected in cellular processes (Fig. 5C).
Figure 5. Gelatinase activity is elevated after OGD.

Confocal micrographs show in situ zymography in organotypic hippocampal slices after 48 hrs exposure to normoxia or OGD. Basal gelatinase activity was detected in normoxic cells (A,C) and was increased after exposure to OGD (B,D). Gelatinase activity localized to cellular processes and cells with amoeboid morphology after OGD (D) when compared to normoxia (C). Scale bars = 100 μm (A,B), 50 μm (C,D).
Treatment with AG3340 or Minocycline Attenuates Gelatinase Activity
In situ zymography was performed on organotypic slices that were exposed to 48 hrs of normoxia or OGD in media containing vehicle, AG3340, or minocycline to assess the activity of gelatin-degrading MMPs in response to OGD, and to test the MMP-inhibitory efficacy of these compounds. Quantification of gelatinase activity was performed by analyzing photomicrographs (3 slices per group; N = 2 experiments) for percent of total hippocampal area occupied by fluorescent signal (Fig. 6). Gelatin-cleaved fluorescence was increased in slices exposed to OGD (27.6% +/- 2.9%) relative to normoxia (15.5% +/- 4.1%; p<0.01) after treatment with vehicle alone. Activity was significantly attenuated in slices exposed to OGD and treated with AG3340 (16.1% +/- 2.6%; p<0.01) or minocycline (20.1% +/- 3.4%; p<0.05) relative to those treated with vehicle alone. There were no significant differences between normoxia and OGD for either treatment.
Figure 6. Treatment with AG3340 or minocycline reduces gelatinase activity.

Gelatin-cleaved fluorescence was quantified in organotypic hippocampal slices after exposure to normoxia or OGD in media containing vehicle, AG3340, or minocycline. Slices treated with vehicle alone showed a significant increase in fluorescent signal after exposure to OGD relative to normoxic controls (p<0.01). Gelatinase activity was significantly reduced in slices exposed to OGD and treated with either AG3340 (p<0.01) or minocycline (p<0.05) relative to those treated with vehicle alone. N = 6. * indicates significant from normoxia; # indicates significant from OGD + vehicle.
Treatment with AG3340 or Minocycline Reduces Neural Injury
To determine whether gelatinase activity was directly associated with neural injury after OGD, organotypic slices were exposed to 48 hrs of normoxia or OGD in media containing vehicle, AG3340, or minocycline and were stained with Fluoro-Jade (Fig. 7). Basal levels of Fluoro-Jade stain were detected in normoxic slices after all treatments. Staining was most prominent in the hippocampal neuronal cell layers but was also present in surrounding tissue. In general, Fluoro-Jade stain was more intense and occupied larger areas in slices exposed to OGD and treated with vehicle alone (Fig. 7D) relative to normoxic controls (Fig. 7A). Fluoro-Jade was also elevated in slices exposed to OGD and treated with AG3340 (Fig. 7E) when compared to normoxic controls (Fig. 7B). Slices treated with minocycline and exposed to OGD showed staining that occupied smaller areas of total tissue (Fig. 7F), resembling normoxic controls (Fig. 7C). Quantification of Fluoro-Jade staining was performed by analyzing photomicrographs (6 slices per group; N = 2 experiments) for percent of total hippocampal area occupied by fluorescent signal (Fig. 7G). Fluoro-Jade staining was significantly elevated in slices exposed to OGD (21.5% +/- 2.4%) relative to normoxia (8.0% +/- 1.0%) after treatment with vehicle alone (p<0.001). Quantification also revealed a significant increase in staining after OGD (10.7% +/- 1.2%) relative to normoxia (5.8% +/- 0.3%) in slices treated with AG3340 (p<0.05), yet the degree of staining was significantly lower when compared to those exposed to OGD and treated with vehicle alone (p<0.001). Fluoro-Jade staining was also significantly reduced after OGD in slices treated with minocycline (7.7% +/- 1.1%) relative to those treated with vehicle alone (p<0.001), and the degree of staining was not statistically different from normoxic controls (5.1% +/- 1.1%).
Figure 7. Treatment with AG3340 or minocycline reduces neural injury.

Organotypic hippocampal slices exposed to normoxia or OGD in media containing vehicle, AG3340, or minocycline were stained with Fluoro-Jade. Basal Fluoro-Jade staining was detected in slices exposed to normoxia after all treatments (A-C). Staining was markedly elevated in slices exposed to OGD and treated with vehicle alone (D) compared to normoxic controls (A), and this effect was attenuated in slices exposed to OGD and treated with either AG3340 (E) or minocycline (F) (scale bars = 100 μm). Quantification (G) showed a significant increase in slices exposed to OGD and treated with vehicle alone relative to normoxic controls (p<0.001). Treatment with either AG3340 or minocycline attenuated the OGD – induced increase in Fluoro-Jade stain (p<0.001), and minocycline reduced staining to normoxic control levels. N = 6. * indicates significant from normoxia; # indicates significant from OGD + vehicle.
Discussion
H-I insults produce neural sequelae characterized by substantial white matter injury and neurodegeneration. Inflammation mediates the delayed injury response in ischemic brain. Both peripheral and resident immune cells may promote proinflammatory signaling, yet the relative contributions of infiltrating leukocytes and resident microglia have yet to be elucidated. Gelatin-degrading MMPs proteolytically process endothelial cell junction proteins (Reijerkerk et al., 2006, Rosenberg and Yang, 2007) and inflammatory cytokines (Ito et al., 1996, Schonbeck et al., 1998). Previous reports have demonstrated that gelatinases modulate neuroinflammation (Gidday et al., 2005, Svedin et al., 2007), blood brain barrier degradation (Back et al., 1998, Asahi et al., 2001) and cell death (Asahi et al., 2000, Lee et al., 2004) after ischemia. However, the association of MMPs with neuroinflammation and injury in the absence of peripheral immune cell effects has not been investigated to date. Therefore, the present study was conducted to determine whether selective gelatinase inhibition can confer neuroprotection in an ex vivo model of H-I that lacks peripheral immune cell involvement. Results show that MMP-9 expression was upregulated in organotypic hippocampal slices after 48 hrs exposure to OGD. Furthermore, gelatinase activity and neural injury increased in slices exposed to OGD, effects that were attenuated when slices were incubated with media containing either the selective gelatinase inhibitor AG3340 or minocycline.
To our knowledge, this is the first study utilizing organotypic slice culture methodology to investigate the link between ischemic injury and MMP activity in the absence of peripheral immune cell effects. Thionin staining showed that the slices retained three-dimensional structure with dense neuronal layers after extended exposure to culture conditions, indicating that the in vivo hippocampal morphology was maintained throughout the experiments. Immunohistochemistry revealed ubiquitous MMP-9 expression that was associated with plasma membranes. Normoxic slices contained few microglia labeled with the CD11b antibody. The colocalization of MMP-9 – positive cell bodies with NSE, coupled with the general lack of colocalization with GFAP or CD11b, shows that MMP-9 expression was predominantly neuronal. In contrast, MMP-9 colocalized with CD11b – positive cell bodies in slices exposed to OGD, indicating that the insult induced MMP-9 expression. Additionally, MMP-9/CD11b - expressing cells displayed an amoeboid morphology consistent with activated microglia. These data are in agreement with previous studies that localized gelatinase activity to neurons and glia (Lee et al., 2004, Back et al., 2007, Amantea et al., 2008).
In situ zymography experiments demonstrated that there is basal gelatinase activity in organotypic hippocampal slices. Gelatin-cleaved fluorescence was predominantly limited to cells with neuronal morphology, consistent with the neuronal expression profile of MMP-9 in normoxic slices. Gelatinase activation in normoxic slices likely occurred in response to cellular injury from the slice preparation itself. The presence of Fluoro-Jade and PI staining in normoxic slices supports this explanation. Upon exposure to OGD, gelatin-cleaved fluorescence was markedly increased. Gelatinase activity was not limited to neurons after OGD. Instead, activity appeared in cells with amoeboid morphology and also localized to cellular processes. The immunohistochemical experiments indicate that the amoeboid cells were activated microglia and the cellular processes were of astrocytic origin, as MMP-9 was expressed in CD11b – positive cells and some astrocytic processes, but was not detected in neuronal processes. The latter expression profile is consistent with several previous studies that showed gelatinase expression in activated astrocytes in vitro (Apodaca et al., 1990, Rosenberg et al., 2001, Muir et al., 2002).
Both AG3340 and minocycline were efficacious in attenuating OGD – induced gelatinase activity and Fluoro-Jade staining. Gelatinase activity, therefore, correlated with increased neural injury, and both compounds dampened these effects. Consistent with these data, AG3340 was previously shown to be neuroprotective in the adult brain after ischemia (Nakaji et al., 2006). AG3340 is a small molecule hydroxamate-based MMP inhibitor with high selectivity for gelatinases (Santos et al., 1997, Shalinsky et al., 1998, Price et al., 1999). Minocycline has also been shown to possess MMP-inhibitory properties (Machado et al., 2006) but acts primarily as an anti-inflammatory agent. This compound has also improved outcomes in several rodent injury models (Cai et al., 2006, Fan et al., 2006, Wasserman and Schlichter, 2007). Indeed, in vivo doses equivalent to the concentrations used in the present study were neuroprotective in neonatal rat pups exposed to H-I (Leonardo et al., 2008b). Although the former study did not assess enzyme activity or MMP expression, the cellular expression profile of MMP-9 in organotypic slices suggests a direct link between gelatinase activity and neuroinflammation in response to OGD. While the AG3340 data indicate that selective inhibition of active MMP-9 and/or MMP-2 is sufficient to confer neuroprotection, the fact that minocycline was equally efficacious supports the notion that the resident immune cell response is involved in facilitating MMP expression and activation. Interestingly, neither compound was able to completely abolish gelatinase activity in normoxic slices. One possible explanation is that the 3 μM dose, while efficacious in reducing high levels of activity induced by exposure to OGD, is below the threshold required to inhibit all activity within the slice. It is also possible that the basal gelatin cleavage observed in normoxic slices resulted from the activation of other endogenous enzymes that are not currently known to degrade gelatin.
In summary, the present study exploited a model that lacks the peripheral immune cell response to determine whether gelatinase activity is of resident immune cell origin, and whether heightened MMP activity is associated with neural injury after OGD. Advantages of this model are that it is more conducive to mechanistic experimentation and more closely resembles the microenvironment in brain when compared to primary cell culture or mixed glial culture. Results here indicate that gelatinase activity is linked to the resident microglial response within the first 48 hrs after OGD, and selective targeting of gelatin-degrading MMPs may be an avenue for therapeutic intervention to combat H-I neuropathies.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke (RO1-NS052839 to K.P.); and the University of South Florida Department of Molecular Pharmacology and Physiology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ajmo JM, Eakin AK, Hamel MG, Gottschall PE. Discordant localization of WFA reactivity and brevican/ADAMTS-derived fragment in rodent brain. BMC Neurosci. 2008;9:14. doi: 10.1186/1471-2202-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Diaz A, Hilario E, de Cerio FG, Valls-i-Soler A, Alvarez-Diaz FJ. Hypoxic-ischemic injury in the immature brain--key vascular and cellular players. Neonatology. 2007;92:227–235. doi: 10.1159/000103741. [DOI] [PubMed] [Google Scholar]
- Alves F, Borchers U, Padge B, Augustin H, Nebendahl K, Kloppel G, Tietze LF. Inhibitory effect of a matrix metalloproteinase inhibitor on growth and spread of human pancreatic ductal adenocarcinoma evaluated in an orthotopic severe combined immunodeficient (SCID) mouse model. Cancer Lett. 2001;165:161–170. doi: 10.1016/s0304-3835(01)00420-7. [DOI] [PubMed] [Google Scholar]
- Amantea D, Corasaniti MT, Mercuri NB, Bernardi G, Bagetta G. Brain regional and cellular localization of gelatinase activity in rat that have undergone transient middle cerebral artery occlusion. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2007.12.030. [DOI] [PubMed] [Google Scholar]
- Apodaca G, Rutka JT, Bouhana K, Berens ME, Giblin JR, Rosenblum ML, McKerrow JH, Banda MJ. Expression of metalloproteinases and metalloproteinase inhibitors by fetal astrocytes and glioma cells. Cancer Res. 1990;50:2322–2329. [PubMed] [Google Scholar]
- Arteni NS, Salgueiro J, Torres I, Achaval M, Netto CA. Neonatal cerebral hypoxia-ischemia causes lateralized memory impairments in the adult rat. Brain Res. 2003;973:171–178. doi: 10.1016/s0006-8993(03)02436-3. [DOI] [PubMed] [Google Scholar]
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Craig A, Kayton RJ, Luo NL, Meshul CK, Allcock N, Fern R. Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab. 2007;27:334–347. doi: 10.1038/sj.jcbfm.9600344. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405–414. doi: 10.1053/j.semperi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Bernardo A, Greco A, Levi G, Minghetti L. Differential lipid peroxidation, Mn superoxide, and bcl-2 expression contribute to the maturation-dependent vulnerability of oligodendrocytes to oxidative stress. J Neuropathol Exp Neurol. 2003;62:509–519. doi: 10.1093/jnen/62.5.509. [DOI] [PubMed] [Google Scholar]
- Bona E, Johansson BB, Hagberg H. Sensorimotor function and neuropathology five to six weeks after hypoxia-ischemia in seven-day-old rats. Pediatr Res. 1997;42:678–683. doi: 10.1203/00006450-199711000-00021. [DOI] [PubMed] [Google Scholar]
- Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137:425–435. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Chew LJ, Takanohashi A, Bell M. Microglia and inflammation: impact on developmental brain injuries. Ment Retard Dev Disabil Res Rev. 2006;12:105–112. doi: 10.1002/mrdd.20102. [DOI] [PubMed] [Google Scholar]
- Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI, Koziol JA. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007;38:646–651. doi: 10.1161/01.STR.0000254477.34231.cb. [DOI] [PubMed] [Google Scholar]
- Dingley J, Tooley J, Porter H, Thoresen M. Xenon provides short-term neuroprotection in neonatal rats when administered after hypoxia-ischemia. Stroke. 2006;37:501–506. doi: 10.1161/01.STR.0000198867.31134.ac. [DOI] [PubMed] [Google Scholar]
- Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR. Temporary focal ischemia in the mouse: technical aspects and patterns of Fluoro-Jade evident neurodegeneration. Brain Res. 2005;1042:29–36. doi: 10.1016/j.brainres.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Fan LW, Lin S, Pang Y, Rhodes PG, Cai Z. Minocycline attenuates hypoxia-ischemia-induced neurological dysfunction and brain injury in the juvenile rat. Eur J Neurosci. 2006;24:341–350. doi: 10.1111/j.1460-9568.2006.04918.x. [DOI] [PubMed] [Google Scholar]
- Folkerth RD. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005;20:940–949. doi: 10.1177/08830738050200120301. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui O, Kinugasa Y, Fukuda A, Fukuda H, Tskitishvili E, Hayashi S, Song M, Kanagawa T, Hosono T, Shimoya K, Murata Y. Post-ischemic hypothermia reduced IL-18 expression and suppressed microglial activation in the immature brain. Brain Res. 2006;1121:35–45. doi: 10.1016/j.brainres.2006.08.121. [DOI] [PubMed] [Google Scholar]
- Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, Drummond AH, Galloway WA, Gilbert R, Gordon JL, et al. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gottschall PE, Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J Neurochem. 1995;64:1513–1520. doi: 10.1046/j.1471-4159.1995.64041513.x. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyte L, Kaur J, Buchan AM. Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol. 2004;188:200–204. doi: 10.1016/j.expneurol.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Mishima K, Yoshikawa T, Iwasaki K, Fujiwara M, Xia YX, Ikenoue T. Selective and long-term learning impairment following neonatal hypoxic-ischemic brain insult in rats. Behav Brain Res. 2001;118:17–25. doi: 10.1016/s0166-4328(00)00287-4. [DOI] [PubMed] [Google Scholar]
- Ito A, Mukaiyama A, Itoh Y, Nagase H, Thogersen IB, Enghild JJ, Sasaguri Y, Mori Y. Degradation of interleukin 1beta by matrix metalloproteinases. J Biol Chem. 1996;271:14657–14660. doi: 10.1074/jbc.271.25.14657. [DOI] [PubMed] [Google Scholar]
- Jansen EM, Low WC. Long-term effects of neonatal ischemic-hypoxic brain injury on sensorimotor and locomotor tasks in rats. Behav Brain Res. 1996;78:189–194. doi: 10.1016/0166-4328(95)00248-0. [DOI] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Ang LS, Sundaresan A. Hypoxic damage to the periventricular white matter in neonatal brain: role of vascular endothelial growth factor, nitric oxide and excitotoxicity. J Neurochem. 2006;98:1200–1216. doi: 10.1111/j.1471-4159.2006.03964.x. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo CC, Eakin AK, Ajmo JM, Collier LA, Pennypacker KR, Strongin AY, Gottschall PE. Delayed administration of a matrix metalloproteinase inhibitor limits progressive brain injury after hypoxia-ischemia in the neonatal rat. J Neuroinflammation. 2008a;5:34. doi: 10.1186/1742-2094-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo CC, Eakin AK, Ajmo JM, Gottschall PE. Versican and brevican are expressed with distinct pathology in neonatal hypoxic-ischemic injury. J Neurosci Res. 2008b;86:1106–1114. doi: 10.1002/jnr.21553. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- Machado LS, Kozak A, Ergul A, Hess DC, Borlongan CV, Fagan SC. Delayed minocycline inhibits ischemia-activated matrix metalloproteinases 2 and 9 after experimental stroke. BMC Neurosci. 2006;7:56. doi: 10.1186/1471-2202-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2007 doi: 10.1016/j.semcdb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillen PS, Ferriero DM. Selective vulnerability in the developing central nervous system. Pediatr Neurol. 2004;30:227–235. doi: 10.1016/j.pediatrneurol.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Muir EM, Adcock KH, Morgenstern DA, Clayton R, von Stillfried N, Rhodes K, Ellis C, Fawcett JW, Rogers JH. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Brain Res Mol Brain Res. 2002;100:103–117. doi: 10.1016/s0169-328x(02)00132-8. [DOI] [PubMed] [Google Scholar]
- Nakaji K, Ihara M, Takahashi C, Itohara S, Noda M, Takahashi R, Tomimoto H. Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke. 2006;37:2816–2823. doi: 10.1161/01.STR.0000244808.17972.55. [DOI] [PubMed] [Google Scholar]
- Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM, McQuibban GA, Clark-Lewis I. Discovery of chemokine substrates for matrix metalloproteinases by exosite scanning: a new tool for degradomics. Biol Chem. 2002;383:1059–1066. doi: 10.1515/BC.2002.114. [DOI] [PubMed] [Google Scholar]
- Price A, Shi Q, Morris D, Wilcox ME, Brasher PM, Rewcastle NB, Shalinsky D, Zou H, Appelt K, Johnston RN, Yong VW, Edwards D, Forsyth P. Marked inhibition of tumor growth in a malignant glioma tumor model by a novel synthetic matrix metalloproteinase inhibitor AG3340. Clin Cancer Res. 1999;5:845–854. [PubMed] [Google Scholar]
- Reijerkerk A, Kooij G, van der Pol SM, Khazen S, Dijkstra CD, de Vries HE. Diapedesis of monocytes is associated with MMP-mediated occludin disappearance in brain endothelial cells. Faseb J. 2006;20:2550–2552. doi: 10.1096/fj.06-6099fje. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, Miller K, Gearing A. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007;22:E4. doi: 10.3171/foc.2007.22.5.5. [DOI] [PubMed] [Google Scholar]
- Santos O, McDermott CD, Daniels RG, Appelt K. Rodent pharmacokinetic and anti-tumor efficacy studies with a series of synthetic inhibitors of matrix metalloproteinases. Clin Exp Metastasis. 1997;15:499–508. doi: 10.1023/a:1018418725453. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Mach F, Libby P. Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol. 1998;161:3340–3346. [PubMed] [Google Scholar]
- Shalinsky DR, Brekken J, Zou H, Kolis S, Wood A, Webber S, Appelt K. Antitumor efficacy of AG3340 associated with maintenance of minimum effective plasma concentrations and not total daily dose, exposure or peak plasma concentrations. Invest New Drugs. 1998;16:303–313. doi: 10.1023/a:1006204901140. [DOI] [PubMed] [Google Scholar]
- Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357:562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Svedin P, Hagberg H, Savman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007;27:1511–1518. doi: 10.1523/JNEUROSCI.4391-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten VS, Bradley-Moore M, Gingrich JA, Stark RI, Pinsky DJ. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav Brain Res. 2003;145:209–219. doi: 10.1016/s0166-4328(03)00146-3. [DOI] [PubMed] [Google Scholar]
- Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J Leukoc Biol. 2007;82:1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, Vannucci SJ. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res. 1999;55:158–163. doi: 10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Wasserman JK, Schlichter LC. Minocycline protects the blood-brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol. 2007 doi: 10.1016/j.expneurol.2007.06.025. [DOI] [PubMed] [Google Scholar]
- Young RS, Kolonich J, Woods CL, Yagel SK. Behavioral performance of rats following neonatal hypoxia-ischemia. Stroke. 1986;17:1313–1316. doi: 10.1161/01.str.17.6.1313. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, Wang X, Lo EH. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]