

Abstract
Rationale
Endothelial function and dysfunction are central to the focal origin and regional development of atherosclerosis; however, an in vivo endothelial phenotypic footprint of susceptibility to atherosclerosis preceding pathological change remains elusive.
Objective
To conduct a comparative multi-site genomics study of arterial endothelial phenotype in athero-susceptible and athero-protected regions.
Methods and Results
Transcript profiles of freshly isolated endothelial cells from 7 discrete arterial regions in normal swine were analyzed to determine the steady state in vivo endothelial phenotypes in regions of varying susceptibilities to atherosclerosis. The most abundant common feature of the endothelium of all athero-susceptible regions was the upregulation of genes associated with endoplasmic reticulum (ER) stress. The unfolded protein response (UPR) pathway, induced by ER stress, was therefore investigated in detail in endothelium of the athero-susceptible aortic arch and was found to be partially activated. ER transmembrane signal transducers IRE1α and ATF6α and their downstream effectors, but not PERK, were activated concomitant with a higher transcript expression of protein folding enzymes and chaperones, indicative of ER stress in vivo.
Conclusions
The findings demonstrate the prevalence of chronic endothelial ER stress and activated UPR in vivo at athero-susceptible arterial sites. We propose that chronic localized biological stress is linked to spatial susceptibility of the endothelium to the initiation of atherosclerosis.
Keywords: hemodynamics, DNA microarrays, gene expression
INTRODUCTION
Atherosclerosis originates as focal arterial lesions having a predictable distribution to regions of bifurcations, branches and inner curvatures 1. Here, blood flow separates from unidirectional laminar flow to create complex laminar and occasional turbulent flow collectively referred to as disturbed flow 2, 3. Functional and structural endothelial responses to systemic atherosclerotic risk factors within large arteries alter endothelial permeability, coagulation balance, and vasoactive properties of the arterial wall4. However, the initiation of atherogenesis is localized to specific susceptible sites. Endothelial cells display phenotype heterogeneity reflecting the various functions they perform in different regions of the circulation5 and they play a key role in the initiation and progression of lesions3 but the mechanisms that link regional heterogeneity and atherosusceptibility in vivo are poorly understood. Previous in vitro 6 and in vivo 7 studies have established a link between complex hemodynamics and athero-susceptible endothelial phenotypes. However, a molecular signature for spatial susceptibility in vivo has not yet been established, principally because few in vivo phenotyping studies have been performed.
The effects of the local hemodynamic environment on EC phenotypic heterogeneity have been implicated through multiple in vitro studies that provide extensive information on flow-induced endothelial responses 6, 8–10. However, most in vitro flow devices designed to recapitulate in vivo flow conditions fail to simulate one or more components of the complex arterial flow fields 11. An in vivo EC profiling study showed complex gene expression with the coexistence of both pro- and anti-atherosclerotic pathways in the athero-susceptible aortic arch region of normal swine 7, and the dominance of site specificity over gender or diet as a determinant of phenotype was demonstrated in a follow-up study 12. Overall, these studies suggest that the presence of disturbed flow is associated with a steady-state EC phenotype that is primed for the initiation of atherosclerosis, but a common mechanism of athero-susceptibility at multiple sites in vivo has not been identified.
Here, we profile endothelial phenotypes of normal adult swine arteries at multiple regions (Fig. 1) that are susceptible or resistant to atherosclerosis in similar locations to those of humans 13. Each susceptible site is associated with complex disturbed flow characteristics. Using unbiased genomic analyses we identify ER-stress-related protein biosynthesis as the prevalent endothelial genomic signature in all athero-susceptible locations. Biochemical measurements in endothelium of the athero-susceptible aortic arch demonstrated activation of two of the three UPR signaling pathways. Together the studies demonstrate that chronic ER stress characterizes the pre-pathological state of athero-susceptible endothelial phenotypes in vivo.
Figure 1.
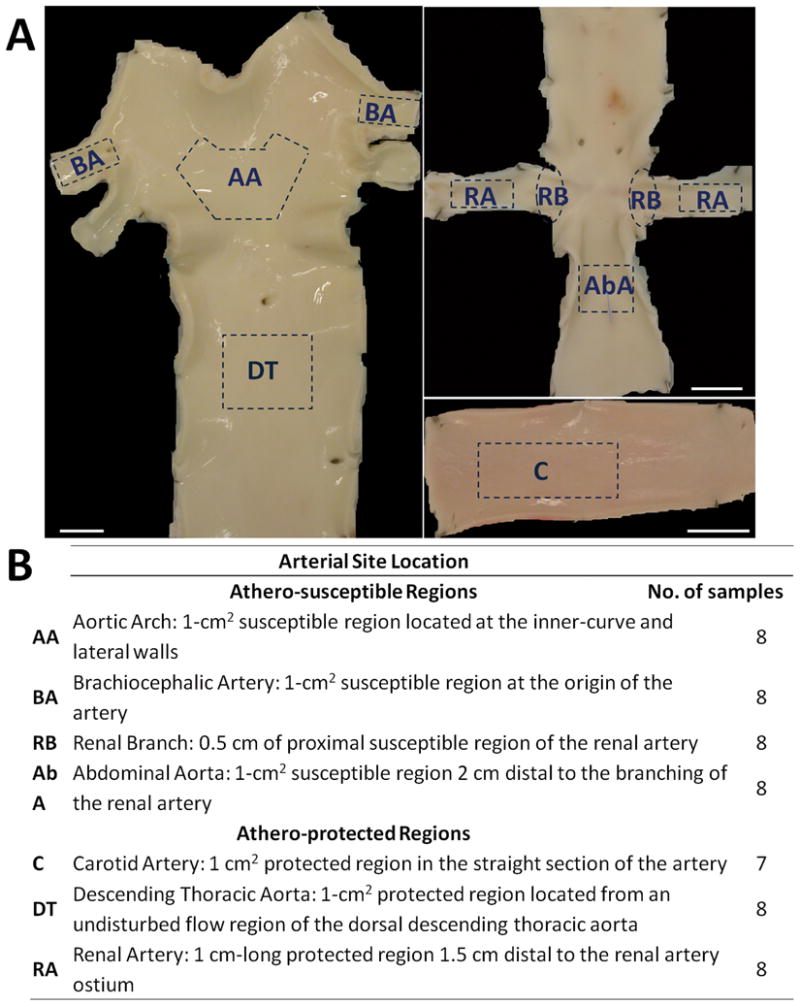
Arterial regions of endothelial isolation. ECs were gently scraped from multiple athero-susceptible and athero-protected regions for transcript and protein analysis. Representative images showing regions of isolation. Scale bar = 1cm.
METHODS
The complete MIAME compliant annotated study has been deposited into the public repository ArrayExpress (accession number E-CBIL-42). Detailed MIAME compliant annotation and data for this study are also available for user-friendly querying at www.cbil.upenn.edu/RAD (RAD study_id=3265). Supplementary information and files are available online.
Sample collection
Fifty five endothelial cell (EC) samples, 7–8 from each of 7 distinct arterial regions (Fig. 1a,b), were collected by gentle scraping from 45 different adult swine shortly after sacrifice at a local abattoir (6 months-old, ~250 lbs, Hatfield Industries, Hatfield, PA). EC purity was 96.5% with 2.8% smooth muscle cell and 0.7% leukocyte contamination (Fig. S1). Regions showed normal histology without inflammation or lipid insudation. Cells were transferred to lysis buffer for RNA and protein extraction.
Microarray hybridization
High quality total RNA was linearly amplified and hybridized to custom-printed porcine microarrays (ArrayExpress A-CBIL-16). Cy5-labelled sample mRNA was combined with Cy3-labelled common reference RNA that consisted of aRNA amplified from pooled total RNA from all regions. Microarrays were scanned with an Agilent DNA Microarray Scanner and the images were analyzed with Agilent Feature Extraction Software (version 9.1).
Bioinformatics
Data pre-processing is described in the Supplementary Information. Differential expression analysis of genes was performed using Patterns of Gene Expression (PaGE v5.1.6) 14. Genes with less than 25% false discovery rate (FDR) were considered to be differentially expressed. The list of differentially expressed genes was interrogated for statistically significant over-represented biological themes using DAVID 15 and for network formation and direct interactions using Ingenuity Pathway Analysis (IPA) 16. Differential expression analysis of gene sets was performed using Gene Set Enrichment Analysis (GSEA)17.
Quantitative real time PCR and western blots
A separate cohort of 10 animals were used to isolate ECs from aortic arch (AA), thoracic aorta (DT), renal branch (RB) and renal artery (RA) to study gene expression. An additional cohort of 12 animals was used to isolate ECs from AA and DT, where sufficient material was obtained to study protein expression. PCR primers for genes of interest are listed in Table S1. Quantitative real time PCR was performed using LightCycler FastStart DNA Master SYBR Green I on a LightCycler System (Roche Applied Science, Indianapolis, IN). Primary and secondary antibodies used for Western blotting are listed in the expanded methods.
Statistical significance was assessed for the gene and protein expression ratios using one sided one-sample Wilcoxon test.
RESULTS
Differential gene expression analysis identified 133 genes in athero-susceptible regions consistent with, and extending, our previously published work7; they included Connexin 43 (GJA1), identified as the most significantly upregulated gene in athero-susceptible ECs in agreement with previous work that showed higher Connexin 43 protein expression in flow dividers in rat arteries18 and upregulated Connexin 43 mRNA expression in cultured endothelial cells that were subjected to disturbed flow in vitro19. VCAM1 was also upregulated in athero-susceptible ECs, in agreement with previously published work20 and its upregulation was confirmed by QRT PCR (Fig S2).
ER stress-related gene expression is upregulated in ECs of athero-susceptible arterial regions
Three independent and unbiased bioinformatics analyses indicated the presence of ER stress in regions of athero-susceptibility. Gene Ontology terms of protein folding, endoplasmic reticulum and unfolded protein binding were identified by the analysis tool DAVID15 to be over-represented functional categories of the 133 differentially expressed genes (Fisher’s Exact p value = 10−6−10−3; Table 1). Furthermore, Gene Set Enrichment Analysis (GSEA) identified upregulation of gene sets related to ER metabolism, unfolded protein binding, ubiquitin conjugation and proteasome degradation in susceptible regions (Table S2). By Ingenuity Pathway Analysis, 73% of the upregulated genes formed a tightly connected network of interactions with highly significant enrichment scores (Fisher’s exact p value = 10−69 to 10−17) based on known gene-protein and protein-protein direct relations (Fig. S3). This network contained multiple genes that function in protein synthesis, protein folding and post-translational modification as well as inflammation and apoptosis, cellular processes that have been linked to endoplasmic reticulum (ER) stress 21. Collectively, the genomic analysis indicated prominent differences in endothelial ER stress in regions of athero-susceptibility.
A subset of the differentially expressed genes was functionally categorized into protein biosynthesis and related pathways (Table 2 and Table S3). Each category showed elevated expression in the endothelium of athero-susceptible regions:
mRNA processing
Genes that code for proteins of the spliceosome complex, which processes hetero-nuclear RNA to mRNA, were upregulated in the endothelium from athero-susceptible regions. These included heterogeneous nuclear ribonucleoproteins (hnRP) A/B, D, M and U, PTBP1 (hnRNP I) and splicing factor PQ (SFPQ), genes that have been shown to function in mRNA splicing to remove introns from pre-mRNA 22; expression of hnRNP A1 has been determined to increase with stress 23. Translation initiation (eIF3S2) and elongation (eEF1E1) genes, DEAD box polypeptide (DDX3) which interacts with eIF3 to promote translation 24 as well as deoxyhypusine synthase (DHPS) which encodes hypusine, an unusual amino acid only found in eukaryotic translation initiation factor 5A, were also upregulated 25. Collectively, mRNA processing activity of ECs in susceptible arterial regions was increased.
Protein folding
Numerous genes that have crucial functions in protein folding and post-translational modifications had higher expression in ECs from susceptible regions. Transcripts for two critical protein folding enzymes were upregulated: Peptidylprolyl isomerase (PPID) catalyzes the cis-trans isomerization of peptide bonds and protein disulphide isomerase (PDIA4 and PDIA5) catalyzes the formation of disulfide bonds 26. Molecular chaperones, which aid protein folding, were more highly expressed in susceptible regions. Hsp70 (HSPA4, HSPA5) and Hsp40 (DNAJB6, DNAJB9) families of molecular chaperones bind to the growing polypeptide chains to increase the efficiency of folding 27. Chaperonins (CCT4A, CCT6, CCT8, TCP1) recognize misfolded proteins in the cytosol and because of their barrel-like structure, create a favorable environment for protein refolding 27. Observed increased folding capacity in susceptible ECs was consistent with an adaptive response to higher protein synthesis and ER stress in cells isolated from athero-susceptible sites.
Protein transport and quality control
Proteins that are destined for secretion are transported into the ER through the SEC61 protein translocator (SEC61A1, SEC61B) for glycosylation and proper folding 28. Calnexin (CANX) and calreticulin (CALR) are two lectins that are part of the ER quality control system that ensures proteins are properly folded before they exit. Higher expression of SEC61 and CANX/CALR genes indicated increased ER quality control machinery in susceptible ECs consistent with higher expression of protein folding genes in these regions.
Protein degradation
Accumulation of polypeptides that fail to acquire their native conformation can jeopardize cellular function; therefore, misfolded proteins are removed by ER-associated degradation (ERAD) in conjunction with the ubiquitin/proteasome system (POMP, PSMD12, UFD1L) 29; the latter genes were upregulated in susceptible ECs providing evidence for a protective mechanism against the accumulation of misfolded proteins in these regions.
ER lipid synthesis
ER stress results in an increase in the size of the ER membrane in stressed cells (ER dilation) 30. Sterol-C4-methyl oxidase (SC4MOL) and squalene epoxidase (SQLE), which catalyze the intermediate reactions of cholesterol synthesis from squalene 31, were more highly expressed in susceptible ECs, consistent with their rate-limiting role in the synthesis of cholesterol 32, an important component of the ER membrane.
Collectively, these data show a coordinated and significant upregulation of endothelial genes related to protein synthesis, folding, quality control and degradation indicative of the presence of chronic ER stress in susceptible regions.
Overload of misfolded proteins triggers the heat shock response in the cytosol and the unfolded protein response in the ER (Fig 2A), also known as ER stress 33. A hallmark of ER stress is the upregulation of HSPA5, known as Binding Protein or GRP78 33. HSPA5 transcript was upregulated in susceptible regions (Table 2). This result was confirmed by QRT-PCR and Western blot (Fig 2B) in a comparison of ECs from aortic arch (AA; susceptible) and descending thoracic aorta (DT; protected). The ER stress marker, SERP1 34, was also more highly expressed in susceptible regions.
Figure 2.

ER stress and UPR. (A) UPR signaling outline (figure adapted from 30). (B) ER stress marker HSPA5 gene and protein (78 kDa) expression in aortic arch, AA, normalized to descending thoracic aorta, DT, for each paired sample based on their animal origin. Gene (n=6 paired samples) and protein (n=12 paired samples) expression was normalized to GAPDH and β-actin, respectively. Values > 1.0 indicate higher expression in AA. Data represent mean±SEM.*p≤0.05 one-sample, one sided, paired Wilcoxon test.
Unfolded protein response pathway is partially upregulated in athero-susceptible endothelium of the aorta
ER stress triggers UPR signaling 30. Further analysis of UPR was focused on comparisons of gene and protein expression in the endothelium from the athero-susceptible AA and athero-protected DT where sufficient cells were accessible to allow protein measurements.
UPR is an adaptive response that is regulated by three ER membrane-localized signal transducers: inositol requiring kinase 1 (IRE1α), protein kinase-like ER kinase (PERK) and activating transcription factor 6α (ATF6α) (Fig 2A)30. IRE1α and ATF6α gene expressions were upregulated in AA compared to DT (Fig 3A, 3B); PERK expression, however, was unchanged (Fig 3C). Protein expression of the three transducers in their inactive form was not different between the two regions (Fig 3). Activation of the UPR occurs when HSPA5 dissociates from the signal transducers to bind unfolded proteins. ATF6α 90 kDa protein translocates to the Golgi where it is cleaved, and subsequently to the nucleus where the active 50 kDa form binds ER stress element (ERSE) and induces the transcription of molecular chaperones and apoptosis-related genes. Significantly higher expression of the 50 kDa ATF6α (Fig 3A) in AA was consistent with increased chaperone gene expression in susceptible ECs (Table 2).
Figure 3.
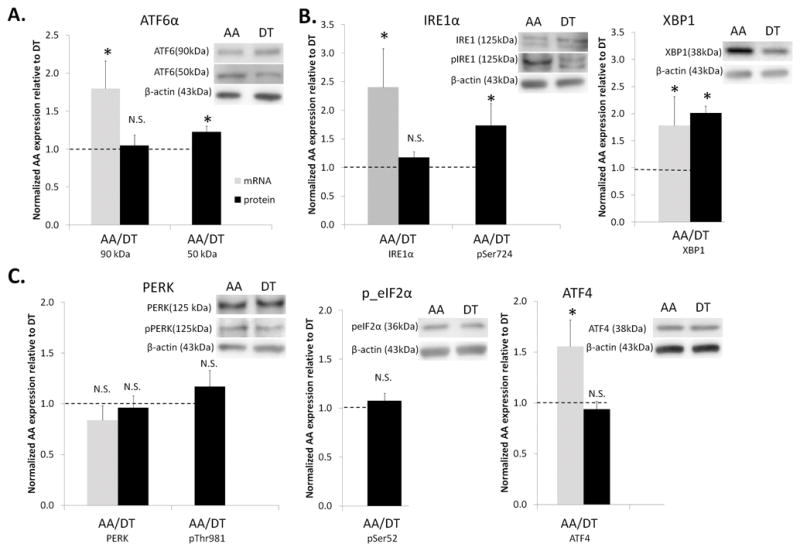
UPR expression in arterial endothelium. Gene and protein expression of tripartite branches of UPR signaling: (A) ATF6α branch (B) IRE1α branch (C) PERK branch in aortic arch, AA, normalized to descending thoracic aorta, DT, for each paired sample based on their animal origin. Gene (n=6–10 paired samples) and protein (n=10–12 paired samples) expression was normalized to GAPDH and β-actin, respectively. Values > 1.0 indicate higher expression in AA. Data represent mean±SEM. N.S.: non-significant. *p≤0.05 one-sample, one sided, paired Wilcoxon test.
The release of HSPA5 from IRE1α leads to homodimerization and auto-phosphorylation. Phosphorylated IRE1α had 70% higher expression in AA compared to DT (Fig 3B). Both phosphorylated IRE1α via its endoribonuclease activity, and ATF6α via its ERSE binding, upregulate the transcription of the spliced form of X-box binding protein 1 (XBP1) which is translated into XBP1 transcription factor. XBP1 gene and protein expression was elevated in AA compared to DT (Fig 3B). XBP1 nuclear translocation induces its binding to the UPR response element (UPRE) which leads to the transcription of ER associated degradation (ERAD) genes that include transcripts of ubiquitin/proteasome, glycosylation, disulphide bond formation and lipid synthesis consistent with the endothelial transcript profile in the susceptible regions (Table 2).
Similar to IRE1α, PERK activation also involves homodimerization and auto-phosphorylation comprising the third UPR signaling pathway. In contrast to the other 2 branches, however, PERK gene and protein expression and phospho-PERK expression were unchanged in AA compared to DT (Fig 3C). Phospho-PERK phosphorylates serine 52 of the eukaryotic translation initiation factor 2α (eIF2α) causing translational attenuation of most proteins with the exception of activating transcription factor 4 (ATF4). Translated ATF4 protein translocates to the nucleus where it binds to the UPRE. Neither phospho-eIF2α nor ATF4 protein expression was different between AA and DT, a finding consistent with inactivity of the PERK branch of UPR (Fig 3C). However, ATF4 mRNA expression was higher in AA compared to DT (Fig 3C) indicating translational control of ATF4 expression.
The presence of ER stress was demonstrated in another athero-susceptible arterial region. ECs from athero-susceptible renal branch (RB) had significantly higher expression of HSPA5, ATF4 and XBP1 transcripts compared to ECs from athero-protected renal artery (RA) (Fig 4), providing further evidence for the presence of chronic ER stress in multiple athero-susceptible regions.
Figure 4.
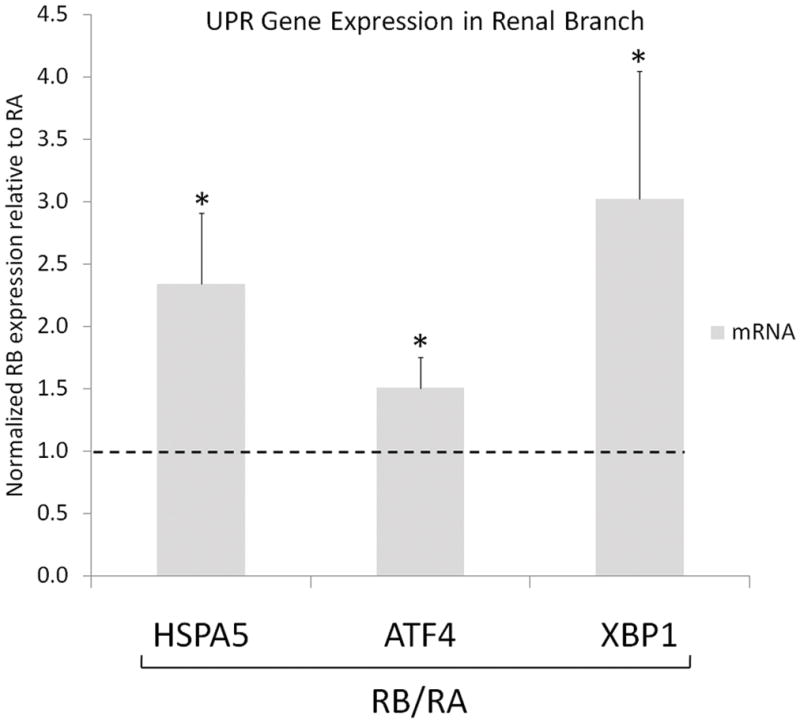
UPR gene expression at renal branch and renal artery. HSPA5, ATF4 and XBP1 transcript in renal branch, RB, normalized to renal artery, RA, for each paired sample based on their animal origin. Gene expression (n= 6 paired samples) was normalized to GAPDH. Values >1.0 indicate higher expression in RB. Data represent mean±SEM. *p≤0.05 one-sample, one sided, paired Wilcoxon test.
Activation of both ATF6α and PERK branches of UPR induces the expression of pro-apoptotic transcription factor CHOP. PERK induces the expression of anti-oxidative stress response genes 30, 35 by phosphorylating nuclear factor erythroid 2-like 2 (NRF2), a transcription factor that upregulates anti-oxidative genes 35. Although CHOP transcript expression was more than two fold increased in AA compared to DT, NRF2 transcript expression was not changed (Fig 5), a finding in agreement with the lack of activation in the PERK branch of UPR. CHOP protein expression, however, was unchanged in AA compared to DT. Increased ATF4 and CHOP gene expression but not protein expression indicates an endothelial phenotype that is primed for further activation of the PERK branch of UPR. Furthermore, although CHOP transcript had higher expression in susceptible ECs, several anti-apoptotic genes that may mitigate apoptosis were also upregulated (Table S4). For example, ARMET, which inhibits ER-stress induced cell death 36, had significantly higher transcript expression in susceptible ECs (Table S4). Similarly, API537, BIRC238 and NIFL339, which have been shown to inhibit apoptosis in other cells as well as ECs, were also upregulated in susceptible regions. In addition to apoptotic balance, pro- and anti-inflammatory genes were also co-expressed in susceptible ECs (Table S4) providing further evidence for a steady-state phenotype of susceptible ECs primed for pathological change 7.
Figure 5.
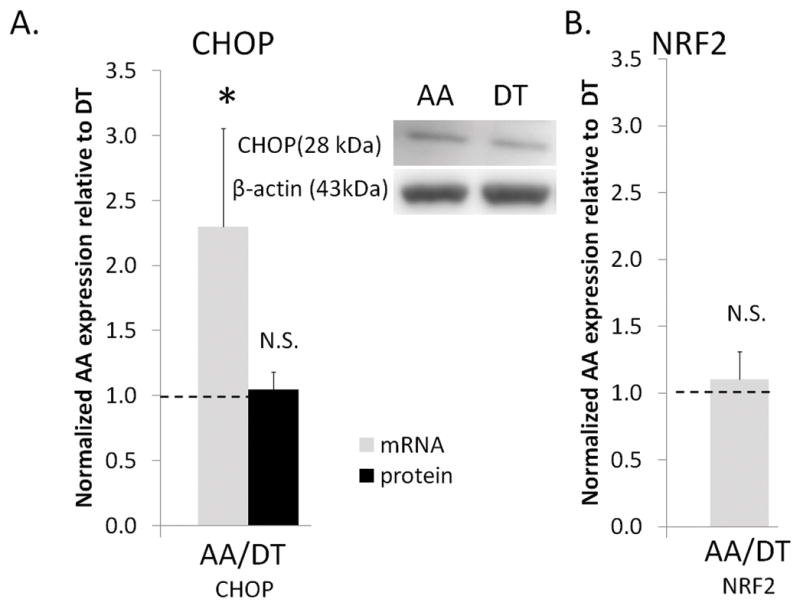
Pro-apoptotic and anti-oxidative transcription factor expression in arterial endothelium. (A) Pro-apoptotic transcription factor CHOP gene (n=6 paired) and protein (n=12 paired) expression. (B) Anti-oxidative transcription factor NRF2 gene (n=5 paired) expression in aortic arch, AA, normalized to thoracic aorta, DT. Values > 1.0 indicate higher expression in AA. Data represent mean±SEM. N.S.: non-significant.*p≤0.05 one-sample, one sided, paired Wilcoxon test.
In summary, these data demonstrate the activation of adaptive UPR through ATF6α and IRE1α in response to chronic ER stress in susceptible ECs. In contrast, PERK, the third regulatory branch of UPR appears not to be involved in this adaptive response. Adaptive UPR signaling provides protection from the accumulation of misfolded proteins by selectively upregulating the protein folding and quality control mechanisms to cope with the protein overload in susceptible ECs.
DISCUSSION
Heterogeneity in the functional state of the endothelium likely plays a key role in the spatial arterial susceptibility to atherosclerosis. Here, we have demonstrated the presence of ER stress as a signature for athero-susceptible endothelial phenotype in multiple arterial regions from a large cohort of animals from different backgrounds. While inter-animal variation is amplified in this approach, emergent findings are likely to reflect those in a general population. We report that endothelial phenotypes exhibiting chronic ER stress-related gene and protein expression characterize regions more susceptible to atherogenesis. Adaptive UPR signaling at these sites may mitigate localized stress effects by preventing the accumulation of pathological misfolded proteins.
UPR is a three pronged signaling network designed to maintain ER homeostasis by relieving the cells from accumulation of unfolded proteins. If the unfolded protein overload is not cleared, terminal UPR activates apoptosis. In acute UPR, eIF2α is rapidly phosphorylated to inhibit translation. On the other hand, adaptive UPR occurring over an extended period in vivo increases the protein folding and processing capacity of the ER 40. Rutkowski and Kaufman 40 proposed that suppression of PERK and activation of ATF6α may occur in adaptive UPR. In agreement with their proposal, we present in vivo evidence for the activation of adaptive UPR in susceptible ECs through ATF6α and IRE1α but not PERK. In such a scenario, additional risk factors may be required to trigger PERK activation. Many studies suggest that eIF2α phosphorylation, as a result of PERK activation, leads to increased ATF4 expression30; however, ATF4 mRNA, but not protein, expression was increased in AA compared to DT despite equivalent eIF2α phosphorylation. CHOP mRNA and protein expression had a similar profile to ATF4 in these regions. It is plausible that ATF4 and CHOP protein expression in nuclear protein extracts is different between the two regions; however, small number of ECs isolated from these regions did not allow for enough nuclear protein for quantitative measurements. Nuclear extracts pooled from 10 animals did not provide conclusive results (Fig S4). In response to pharmacologically-induced mild ER stress in mouse embryonic fibroblasts in vitro higher ATF4 and CHOP protein degradation rates have been reported compared to other UPR proteins 41. The same study also demonstrated PERK activation in the apparent absence of eIF2α phosphorylation; similarly we observed a modest (16%) but non-significant increase in PERK phosphorylation (Fig 3C). This agreement between in vivo and in vitro results suggests a role for translational control of ATF4 and CHOP mRNA, the availability of which may provide susceptible ECs with a response mechanism by stabilizing their protein products to cope with acute increases in misfolded protein load as a result of local increases in pathological factors. The coexistence of pro-apoptotic CHOP expression and anti-apoptotic transcripts in susceptible regions suggests endothelial priming of, but not commitment to, apoptosis. A similar mechanism has been reported in macrophages during atherosclerosis where macrophage UPR was activated in the absence of apoptosis in early atherosclerotic lesions. However, apoptotic cells were apparent in advanced lesions in addition to further activation of UPR 42.
The initiation and progression of atherosclerosis involves inflammatory elements 43. Low-grade chronic inflammation, as indicated by leukocyte accumulation in the intima was shown in the athero-susceptible region of the normal mouse aortic arch compared to more resistant regions 44. Growing evidence links ER stress and inflammation 21. Endothelial inflammation was shown to be UPR-mediated via XBP1 and ATF4 upregulation 45 and UPR activation in macrophages has been documented in early and advanced atherosclerotic lesions in apoE-deficient mice42. ROS generation due to protein folding provides the strongest link between inflammation and ER stress. The second link is via the IRE1α signaling molecule. Pro-inflammatory transcription factor NF-κB was shown to be activated by the association of IRE1α and adaptor protein TNF receptor-associated factor 2 (TRAF2) 46, although the details are not well understood. The third link is the phosphorylation of eIF2α, which inhibits translation, thereby repressing the synthesis of NF-κB inhibitor IκB. However, our previous study showed that NF-κB was not activated in swine AA or DT regions 7 suggesting that adaptive UPR does not induce NF-κB activation in normal animals in the absence of additional risk factors. Atherosclerotic risk factors, oxidized phospholipids 45, 47, oxidized LDL 48 and homocysteine 49 induce ER stress and upregulate the UPR pathway in ECs, suggesting that prolonged exposure to systemic risk factors can trigger the terminal UPR.
Athero-susceptible regions in vivo correlate with the presence of complex hemodynamics. Athero-susceptible disturbed flow triggers ER stress in ECs in vitro. Feaver et al.50 provided in vitro evidence for the induction of HSPA5 expression by athero-susceptible flow through p38 activation. HSPA5 upregulation triggered the UPR pathway via ATF6α in cultured ECs. These in vitro studies provide evidence for a direct role of disturbed flow-induced ER stress in ECs that are consistent with the in vivo genomic and protein analyses presented here, although we cannot presume that the hemodynamics are the single most important determinant in vivo. While this manuscript was under review, a study demonstrating the increased expression of XBP1 at branch points of mouse aorta was published 51. The study also showed that the overexpression of spliced XBP1 contributed to neointima formation in an artery isograft model. Taken together, the current study and studies by Feaver et al.50 and Zeng et al.51 indicate a role for ER stress in the susceptibility to atherosclerosis.
Activation of UPR has been observed in disease pathogenesis 33 and in normal immune cells and hepatocytes whose function is to produce and secrete proteins 40. However, this is the first study that demonstrates a steady state link between endothelial ER stress and disease susceptibility in healthy animals under physiological conditions. We propose ER stress/UPR to be an in vivo signature for athero-susceptible endothelial phenotype in which chronic low-grade stress primes the cells for pathological change. It will be important to understand the molecular mechanisms of adaptive UPR and the threshold signaling for terminal UPR to devise effective preventative therapies for atherosclerosis; our model can serve as an experimental system to study adaptive UPR under controlled manipulations. Recent studies identify antioxidants and chemical chaperones that reduce ER stress in vitro 52 and in vivo 53, respectively. A therapeutic role for these molecules to increase the protein folding capacity to reduce ER stress in ECs may be a viable therapy for atherosclerosis occurring in a variety of arterial vessels.
Supplementary Material
Acknowledgments
We thank Dr. Gregory Grant for the microarray annotation, Drs. Scott L. Diamond and Craig A. Simmons for critical reading of the paper, and Drs. Marie Guerraty and Yun Fang for helpful discussions.
Funding Sources: This work was supported by an American Heart Association predoctoral fellowship (0315286U) and NIH Grants HL062250 and HG004521.
Non-standard Abbreviations and Acronyms
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- EC
Endothelial cell
- ER
Endoplasmic reticulum
- ERAD
ER associated degradation
- ERSE
Endoplasmic reticulum stress element
- UPR
Unfolded protein response
- GSEA
Gene Set Enrichment Analysis
- IPA
Ingenuity Pathway Analysis
- MIAME
Minimum Information About a Microarray Experiment
- PaGE
Patterns of Gene Expression
- UPRE
UPR response element
Gene Names
- ACSL4
Acyl-CoA synthetase long-chain family member 4
- ATF4
Activating transcription factor 4
- ATF6α
Activating transcription factor 6 alpha
- ANKRD45
Ankryin repear domain 45
- API5
Apoptosis inhibitor 5
- ARMET
Arginine-rich, mutated in early stage tumors
- ASB8
Ankyrin repeat and SOCS box-containing 8
- BIRC2
Baculoviral IAP repeat-containing 2
- CALR
Calreticulin
- CALX
Calnexin
- CALU
Calumenin
- CCT
Chaperonin containing T-complex polypeptide
- CHOP
C/EBP homologous protein
- CNIH
Cornichon
- DDX3X
DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, X-linked
- DHPS
Deoxyhypusine synthase
- DNAJ
Heat-shock protein 40 kDa
- EEF1E1
Eukaryotic translation elongation factor 1 epsilon 1
- eIF2α
eukaryotic translation initiation factor 2α
- EIF3S2
Eukaryotic translation initiation factor 3, subunit 2 beta
- GJA1
gap junction protein, alpha 1, 43kDa
- GRP78
Glucose-regulated protein 78 kDa
- IRE1α
Inositol-requiring enzyme 1 alpha
- NFIL3
Nuclear factor, interleukin 3 regulated
- NRF2
Nuclear factor erythroid 2-like 2
- HNRN
Heterogeneous nuclear ribonucleoprotein
- HSPA
Heat-shock 70 kDa protein
- HSPH1
Heat-shock 105/110 kDa protein 1
- HUWE1
HECT, UBA and WWE domain containing protein 1
- KPNA2
Karyopherin alpha-2
- NUCB2
Nucleobindin 2
- PERK
Protein kinase-like ER kinase
- POMP
Proteasome maturation protein
- PPAP2A
Phosphatidic acid phosphatase type 2A
- PSMD12
Proteasome 26S subunit, non-ATPase, 12
- PTBP1
Polypyrimidine tract binding protein 1
- PDIA
Protein disulphide isomerase
- PPID
Peptidylprolyl isomerase
- RPN1
Ribophorin I
- SAR1B
SAR1 gene homolog B
- SC4MOL
Sterol-C4-methyl oxidase-like
- SEC61
SEC61 complex
- SERP1
Stress-associated endoplasmic reticulum protein 1
- SFPQ
Splicing factor proline/glutamine-rich
- SPPL2A
Signal peptide peptidase-like 2A
- SQLE
Squalene epoxidase
- SURF4
Surfeit 4
- SYNCRIP
Synaptotagmin binding, cytoplasmic RNA interacting protein
- TPST1
Tyrosylprotein sulfotransferase 1
- TRAF2
TNF receptor-associated factor 2
- UFD1L
Ubiquitin fusion degradation 1 like
- VCAM1
Vascular cell adhesion molecule 1
- XBP1
X-box binding protein 1
Footnotes
Disclosures: Authors do not report any conflict of interest.
References
- 1.Schwartz CJ, Mitchell JR. Observations on localization of arterial plaques. Circ Res. 1962;11:63–73. [PubMed] [Google Scholar]
- 2.Caro CG, Fitz-Gerald JM, Schroter RC. Arterial wall shear and distribution of early atheroma in man. Nature. 1969;223:1159–1160. doi: 10.1038/2231159a0. [DOI] [PubMed] [Google Scholar]
- 3.Gimbrone MA, Jr, Anderson KR, Topper JN, Langille BL, Clowes AW, Bercel S, Davies MG, Stenmark KR, Frid MG, Weiser-Evans MC, Aldashev AA, Nemenoff RA, Majesky MW, Landerholm TE, Lu J, Ito WD, Arras M, Scholz D, Imhof B, Aurrand-Lions M, Schaper W, Nagel TE, Resnick N, Dewey CF, Gimbrone MA, Davies PF. Special communicationthe critical role of mechanical forces in blood vessel development, physiology and pathology. J Vasc Surg. 1999;29:1104–1151. doi: 10.1016/s0741-5214(99)70252-1. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 5.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100:174–190. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 6.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Passerini AG, Polacek DC, Shi C, Francesco NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY, Stoeckert CJ, Jr, Davies PF. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci U S A. 2004;101:2482–2487. doi: 10.1073/pnas.0305938101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berk BC. Atheroprotective signaling mechanisms activated by steady laminar flow in endothelial cells. Circulation. 2008;117:1082–1089. doi: 10.1161/CIRCULATIONAHA.107.720730. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Cardena G, Gimbrone MA., Jr Biomechanical modulation of endothelial phenotype: implications for health and disease. Handb Exp Pharmacol. 2006;176(Pt 2):79–95. doi: 10.1007/3-540-36028-x_3. [DOI] [PubMed] [Google Scholar]
- 10.Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16–26. doi: 10.1038/ncpcardio1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huo Y, Guo X, Kassab GS. The flow field along the entire length of mouse aorta and primary branches. Ann Biomed Eng. 2008;36:685–699. doi: 10.1007/s10439-008-9473-4. [DOI] [PubMed] [Google Scholar]
- 12.Passerini AG, Shi C, Francesco NM, Chuan P, Manduchi E, Grant GR, Stoeckert CJ, Jr, Karanian JW, Wray-Cahen D, Pritchard WF, Davies PF. Regional determinants of arterial endothelial phenotype dominate the impact of gender or short-term exposure to a high-fat diet. Biochem Biophys Res Commun. 2005;332:142–148. doi: 10.1016/j.bbrc.2005.04.103. [DOI] [PubMed] [Google Scholar]
- 13.Skold BH, Getty R, Ramsey FK. Spontaneous atherosclerosis in the arterial system of aging swine. Am J Vet Res. 1966;27:257–273. [PubMed] [Google Scholar]
- 14.Grant GR, Liu J, Stoeckert CJ., Jr A practical false discovery rate approach to identifying patterns of differential expression in microarray data. Bioinformatics. 2005;21:2684–2690. doi: 10.1093/bioinformatics/bti407. [DOI] [PubMed] [Google Scholar]
- 15.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 16.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabriels JE, Paul DL. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ Res. 1998;83:636–643. doi: 10.1161/01.res.83.6.636. [DOI] [PubMed] [Google Scholar]
- 19.DePaola N, Davies PF, Pritchard WF, Jr, Florez L, Harbeck N, Polacek DC. Spatial and temporal regulation of gap junction connexin43 in vascular endothelial cells exposed to controlled disturbed flows in vitro. Proc Natl Acad Sci U S A. 1999;96:3154–3159. doi: 10.1073/pnas.96.6.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dansky HM, Barlow CB, Lominska C, Sikes JL, Kao C, Weinsaft J, Cybulsky MI, Smith JD. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21:1662–1667. doi: 10.1161/hq1001.096625. [DOI] [PubMed] [Google Scholar]
- 21.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 23.Guil S, Long JC, Caceres JF. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol Cell Biol. 2006;26:5744–5758. doi: 10.1128/MCB.00224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CS, Dias AP, Jedrychowski M, Patel AH, Hsu JL, Reed R. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res. 2008;36:4708–4718. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolff EC, Kang KR, Kim YS, Park MH. Posttranslational synthesis of hypusine: evolutionary progression and specificity of the hypusine modification. Amino Acids. 2007;33:341–350. doi: 10.1007/s00726-007-0525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 27.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 28.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 29.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Kaplan J. Characterization of yeast methyl sterol oxidase (ERG25) and identification of a human homologue. J Biol Chem. 1996;271:16927–16933. doi: 10.1074/jbc.271.28.16927. [DOI] [PubMed] [Google Scholar]
- 32.Chugh A, Ray A, Gupta JB. Squalene epoxidase as hypocholesterolemic drug target revisited. Prog Lipid Res. 2003;42:37–50. doi: 10.1016/s0163-7827(02)00029-2. [DOI] [PubMed] [Google Scholar]
- 33.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hori O, Miyazaki M, Tamatani T, Ozawa K, Takano K, Okabe M, Ikawa M, Hartmann E, Mai P, Stern DM, Kitao Y, Ogawa S. Deletion of SERP1/RAMP4, a component of the endoplasmic reticulum (ER) translocation sites, leads to ER stress. Mol Cell Biol. 2006;26:4257–4267. doi: 10.1128/MCB.02055-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Apostolou A, Shen Y, Liang Y, Luo J, Fang S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res. 2008;314:2454–2467. doi: 10.1016/j.yexcr.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tewari M, Yu M, Ross B, Dean C, Giordano A, Rubin R. AAC-11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res. 1997;57:4063–4069. [PubMed] [Google Scholar]
- 38.Santoro MM, Samuel T, Mitchell T, Reed JC, Stainier DY. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat Genet. 2007;39:1397–1402. doi: 10.1038/ng.2007.8. [DOI] [PubMed] [Google Scholar]
- 39.Ikushima S, Inukai T, Inaba T, Nimer SD, Cleveland JL, Look AT. Pivotal role for the NFIL3/E4BP4 transcription factor in interleukin 3-mediated survival of pro-B lymphocytes. Proc Natl Acad Sci U S A. 1997;94:2609–2614. doi: 10.1073/pnas.94.6.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–476. doi: 10.1016/j.tibs.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–1821. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 43.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 44.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006;203:2073–2083. doi: 10.1084/jem.20060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 46.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A. 2006;103:12741–12746. doi: 10.1073/pnas.0605457103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanson M, Auge N, Vindis C, Muller C, Bando Y, Thiers JC, Marachet MA, Zarkovic K, Sawa Y, Salvayre R, Negre-Salvayre A. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ Res. 2009;104:328–336. doi: 10.1161/CIRCRESAHA.108.183749. [DOI] [PubMed] [Google Scholar]
- 49.Outinen PA, Sood SK, Pfeifer SI, Pamidi S, Podor TJ, Li J, Weitz JI, Austin RC. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood. 1999;94:959–967. [PubMed] [Google Scholar]
- 50.Feaver RE, Hastings NE, Pryor A, Blackman BR. GRP78 upregulation by atheroprone shear stress via p38-, alpha2beta1-dependent mechanism in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1534–1541. doi: 10.1161/ATVBAHA.108.167999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng L, Zampetaki A, Margariti A, Pepe AE, Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, Mori K, Li YS, Hu Y, Chien S, Xu Q. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc Natl Acad Sci U S A. 2009;106:8326–8331. doi: 10.1073/pnas.0903197106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.