

Abstract
Background
Adult neurogenesis augments neuronal plasticity, and deficient neurogenesis may contribute to mood disorders and schizophrenia and impede treatment responses. Since these diseases may be associated with inadequately controlled glycogen synthase kinase-3 (GSK3), we tested if blocked inhibitory serine-phosphorylation of GSK3 impairs neurogenesis.
Methods
Neural precursor cell (NPC) proliferation was measured by dentate gyrus BrdU labeling in GSK3alpha/beta21A/21A/9A/9A knockin mice with serine-to-alanine mutations to block inhibitory serine-phosphorylation of GSK3 while it remains within the physiological range, since GSK3 is not overexpressed.
Results
There was a drastic 40% impairment in neurogenesis in vivo in GSK3 knockin mice compared with wild-type mice. Impaired neurogenesis could be due to effects of GSK3 in NPCs or in surrounding cells that modulate NPCs. In vitro proliferation was equivalent for NPCs from GSK3 knockin and wild-type mice, suggesting an in vivo deficiency in GSK3 knockin mice of external support for NPC proliferation. Measurements of two neurotrophins that promote neurogenesis demonstrated less hippocampal vascular endothelial growth factor, but not brain-derived growth factor, in GSK3 knockin mice than wild-type mice, reinforcing the possibility that insufficient environmental support in GSK3 knockin mice may contribute to impaired neurogenesis. In vivo chronic co-administration of lithium and fluoxetine, which each increase inhibitory serine-phosphorylation of wild-type GSK3, increased NPC proliferation in wild-type, but not GSK3 knockin, mice.
Conclusions
Blocked inhibitory control of GSK3 impaired neurogenesis and the capacity of therapeutic drugs to stimulate neurogenesis, likely through deficient environmental factors that support neurogenesis, which may contribute to psychiatric diseases and responses to therapeutic drugs.
Keywords: neurogenesis, mood disorders, glycogen synthase kinase-3, neural precursor cells, lithium, fluoxetine
Introduction
Neurogenesis, the proliferation and differentiation of neural precursor cells (NPCs), provides new neurons that may be integrated into the developing and adult brain. In adults, NPCs appear to be important mediators of plasticity of the CNS, providing a source of new neurons that may supplement functional networks or replace dysfunctional neurons (1, 2). Deficiencies in NPCs have been implicated in the etiology of major depression (3-5), bipolar disorder (6), and schizophrenia (7). Evidence that impaired neurogenesis contributes to these diseases stems in part from findings that neurogenesis is augmented by therapeutic interventions that include antidepressants (8-12), electroconvulsive shock (13-16), mood stabilizers (17-19), and antipsychotics (20-23). Especially compelling is a report that ablation of NPCs in mice blocked behavioral responses to antidepressants (24).
Although there is strong evidence that therapeutic interventions enhance neurogenesis, much less is known about what factors might impair neurogenesis in psychiatric diseases. One factor may be inadequate neurotrophins, since in rodents deficient neurotrophic signaling impairs NPC proliferation (25-28), administration of neurotrophins increases neurogenesis (25, 29, 30), and therapeutic agents increase neurotrophins (12, 31-33). For several reasons we considered the possibility that glycogen synthase kinase-3 (GSK3) also may have roles in impaired neurogenesis and its augmentation by therapeutic interventions. Hyperactive GSK3 may contribute to mood disorders and schizophrenia (34) and GSK3 is inhibited by neurotrophins and by therapeutic drugs used in these diseases, including lithium (35, 36), the classical mood stabilizer used to treat bipolar disorder (37), antidepressants (38), and antipsychotics (39-42). Additionally, activation of 5-HT1A receptors enhances neurogenesis (3, 43) and also causes inactivation of GSK3 (38). Inhibition of GSK3 also bolsters NPC survival from apoptotic conditions (44), which may counteract the elimination by apoptosis of many NPCs before they mature and differentiate (45-47).
These findings raised the possibility that deficient neurogenesis in psychiatric diseases may be linked to GSK3 that is not sufficiently inhibited by serine phosphorylation. A prominent mechanism regulating the activity of the two isoforms of GSK3, GSK3α and GSK3β, is phosphorylation of a regulatory serine, Ser9 in GSK3β and Ser21 in GSK3α, which inhibits GSK3 (48). In homozygous GSK3α/β21A/21A/9A/9A knockin mice the regulatory serines of both GSK3 isoforms are mutated to alanines, forming S9A-GSK3β and S21A-GSK3α (49). These mutations maintain GSK3 unable to be inhibited by this serine-phosphorylation while levels of GSK3 are within the physiological range since both GSK3 isoforms are expressed at normal levels. In the present study we utilized these GSK3 knockin mice to test if lack of inhibitory serine-phosphorylation of GSK3 impacts NPCs in vivo and in vitro.
Methods and Materials
Animals and drugs
Male 8-10 week old homozygous GSK3α/β21A/21A/9A/9A knockin mice (hereafter referred to as GSK3 knockin mice) and matched wild-type mice of C57Bl6, Balb/c, Ba11 mixed background (49) were used. For chronic lithium treatment, mice were given water and saline ad libitum and were fed pelleted chow containing 0.2% lithium carbonate (Teklad, Madison, Wisconsin) for 21 days. Fluoxetine (10 mg/kg/day; from the NIMH Chemical Synthesis and Drug Supply Program) was injected intraperitoneally (ip) for 21 consecutive days. All mice were housed and treated in accordance with NIH and the University of Alabama at Birmingham Institutional Animal Care and Use Committee guidelines and the experiments were approved by the UAB Institutional Animal Care and Use Committee. For in vitro experiments, NPCs were prepared at gestational day 12-13 as described previously (44, 50).
Administration of BrdU, immunohistochemistry, and stereology
To measure proliferation in vivo, BrdU (100 mg/kg; Sigma-Aldrich, St Louis, MO) was administered ip three times at 2 hr intervals and mice were analyzed 24 hr later. Mice were transcardially perfused with phosphate-buffered saline (PBS; pH 7.4), followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were postfixed overnight in 4% paraformaldehyde at 4°C and cryoprotected in 30% sucrose/PBS for 48 hr. Each brain was sliced coronally (40 μm) with a microtome (Leica, Nuβloch, Germany) through the rostrocaudal hippocampus and stored in PBS. Every seventh section was analyzed for BrdU-specific immunohistochemistry as previously described with slight modifications (8, 51) or for active caspase-3 immunostaining. Sections were preincubated in SSC, followed by incubation in 50% formamide-2/SSC at 65°C for 2 hr, and washed in 2×SSC for 10 min. DNA was denatured in 2 N HCl at 37°C for 30 min, followed by neutralization in borate buffer for 10 min at room temperature. After washing in PBS, sections were incubated for 30 min in 3% H2O2 to eliminate endogenous peroxidases. Sections were incubated in PBS-blocking buffer (3% normal goat/donkey serum, 0.3% Triton-X-100, in PBS) for 2 hr at 37°C and incubated with anti-BrdU antibody (1:500; BU1/75; Abcam) or anti-active caspase-3 (1:50; Cell Signaling, Beverly, MA) for 48 hr at 4°C. Sections were washed with PBS and incubated with horseradish peroxidase-conjugated secondary antibody (1:1000; Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 hr at room temperature. Antigen-antibody complexes were detected by tyramide signal amplification (Perkin-Elmer Life Sciences Products, Boston, MA) using cyanine-3-conjugated tyramide according to the manufacturer's instructions. Cell nuclei were stained by incubating sections for 1 hr in 0.2 μg/ml bisbenzimide (Hoechst 33258; Sigma). BrdU or active caspase-3 positive cells in the granule cell layer of the dentate gyrus and the subgranular zone were counted in each section by an investigator blind to the treatment and analyzed by unbiased stereology using the Neurolucida system (MicroBrightField, Williston, VT). To distinguish single cells within clusters, all counts were performed using a 60× oil immersion objective (Olympus BX-51), omitting cells in the outermost focal plane. The total number of BrdU-labeled cells per section was determined and multiplied by 7 to obtain the total number of cells per dentate gyrus.
For triple labeling, brain sections were incubated in PBS-blocking buffer and incubated for 2 days at 4°C with anti-BrdU [1:500; BU1/75 (ICR1); Abcam], anti-neuronal nuclei (NeuN) (1:200; Chemicon, Temecula, CA), and anti-glial fibrillary acidic protein (GFAP) (1:200; DAKO, Glostrup, Denmark). Tyramide signal amplification (1:500; Perkin-Elmer Life Sciences Products, Boston, MA) using cyanine-3-conjugated tyramide, Alexa Fluor 488 goat anti-mouse (1:1000), and Cy5-donkey-anti-rabbit (1:200; Jackson ImmunoResearch) were applied for 1 hr. Slices were analyzed on a confocal microscope (Leica TCS SP5) and at least 50 dentate gyrus BrdU-positive cells per animal were analyzed with z-plane sectioning (1 μm steps) to confirm the colocalization of both BrdU and the phenotypic markers for neurons (NeuN) and glia (GFAP).
In vitro proliferation assays
For in vitro proliferation measurements, NPCs isolated and cultured on poly-L-lysine/laminin-coated glass coverslips as previously described (44) were treated with 10 μM BrdU for 6 hr. Cells were fixed in 4% paraformaldehyde followed by incubation in 2 N HCl at 37°C for 30 min and then permeabilized and blocked in PBS, pH 7.4, containing 0.2% Triton-X-100, 3% BSA, and 0.2% skim milk. Cells were incubated overnight at 4°C with anti-BrdU antibody (BU1/75; Abcam) and diluted in PBS containing 3% BSA and 0.2 % skim milk. The cells were washed three times with PBS and incubated with horseradish peroxidase-conjugated secondary antibody (1:3000; Jackson ImmunoResearch Laboratories) for 1 hr at room temperature. Antigen-antibody complexes were detected by tyramide signal amplification (Perkin-Elmer Life Sciences Products) using cyanine-3-conjugated tyramide according to the manufacturer's instructions. Nuclei were labeled with 1 μg/ml bisbenzimide (Sigma) for 10 min at room temperature. NPCs were then washed three times with PBS, and coverslips were adhered to glass slides in mounting medium (0.1% p-phenylenediamine in 75% glycerol in PBS). Cells were viewed and photographed with an inverted Nikon fluorescence microscope. Positively stained cells were counted in at least 12 fields from a minimum of 3 different NPC preparations, and the total number of cells counted was always greater than 5000. In vitro proliferation was also measured using the CellTiter96Aqueous One Solution Cell Proliferation Assay (MTS assay) according to the manufacturer's instructions (Promega, Madison, Wisconsin) after dissociating neurospheres with Hanks' balanced salt solution with 0.05% trypsin, 0.02% EDTA, 0.2% BSA for 2 min at 37°C, diluting in 2 vol of Dulbecco's modified Eagle's medium with 10% fetal calf serum, and replating into 96-well plates at 2×103 cells/well. Quantitative measurements from at least three independent experiments were tested for statistical significance using analysis of variance.
Tissue extraction and ELISA
The hippocampus from GSK3 knockin and matched wild-type mice was dissected and weighed. Tissue samples were homogenized in lysis buffer containing 137 mM NaCl, 20 mM Tris-HCl, 1% NP-40, 10% glycerol, 0.5 mM sodium vanadate, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin A, and 1 nM okadaic acid, and centrifuged at 14,000g for 20 min at 4°C. The supernatant was diluted 5-fold with Dulbecco's PBS and acidified to pH 2.6. After 15 min of incubation at room temperature, the supernatants were neutralized to pH 7.6 and frozen. The levels of vascular endothelial growth factor (VEGF) (R&D Systems, Minneapolis, MN) and brain-derived neurotrophic factor (BDNF) (Promega Corporation, Madison, WI) were measured by ELISA according to the manufacturer's instructions.
RNA and RT-PCR
Total RNA was extracted from hippocampi of GSK3 knockin and wild-type mice using Trizol (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. cDNA was synthesized using ImProm-II™ Reverse Transcription System (Promega) and semi-quantitative PCR reactions were performed. Briefly, each PCR cycle included the following steps: 94°C for 30 s, 58°C for 30 s (60°C for 30 s for VEGF), 72°C for 30 s. A sequential series of PCR reactions using each primer pair was initially run to determine optimal annealing temperature and cycle number to ensure amplification within the exponential phase of the amplification curve for both the gene under study and for the housekeeping gene β2 microglobulin. The expression of β2 microglobulin was used as an internal standard, to which other PCR amplification products were normalized. The sequences of oligonucleotide primers were VEGF sense, 5′-CAGGCTGCTGTAACGATGAA-3′; VEGF anti-sense, 5′-CAGGAATCCCAGAAACAACC-3′; BDNF exon 8 (coding exon), sense, 5′-GATGCCGCAAACATGTCTATGA-3′ and anti-sense, 5′-TAATACTGTCACACACGCTCAGCTC-3′; BDNF exon 3, sense, 5′-TAAATGAAGTTTATACAGTACAGTGGTTCTACA-3′ and antisense, 5′-AGTTGTGCGCAAATGACTGTTT-3′; β2 microglobulin sense, 5′-TTCTGGTGCTTGTCTCACTGA-3′ and anti-sense, 5′-CAGTATGTTCGGCTTCCCATTC-3′. The values presented are the average of PCR analyses for 4 mice per group, each normalized to β2 microglobulin. The PCR products were electrophoresed in 2% agarose gels (1.5% for VEGF), visualized by ethidium bromide staining, and the signals were quantified using UN-SCAN-IT gel 6.1 (Silk Scientific Corporation).
Immunoblot analysis
Cells were washed twice with PBS and harvested in 100 μl lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin A, 1 nM okadaic acid, and 1% Triton-X-100). The lysates were centrifuged at 14,000 rpm for 20 min at 4°C. Protein concentrations were determined using the Bradford method (52). Samples were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for 5 min. Proteins (20 μg) were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with primary antibodies to phospho-Ser9-GSK3β (1:1000), phospho-Ser21-GSK3α (1:1000), total GSK3α/β (1:2000), and phospho-Tyr279/216-GSK3α/β (1:1000; Cell Signaling). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence.
Cell cycle analysis
NPCs were trypsinized, fixed in ethanol at -20°C for 10 min, and rehydrated by adding PBS at room temperature for 15 min. Cells were stained with 3 μM propidium iodide in staining buffer (100 mM Tris, pH 7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2, 0.1% NP-40) for 15 min at room temperature, and analyzed on a single-laser flow cytometer (Becton-Dickinson LSR II, San Jose, CA). Quantitative measurements from at least three independent experiments were tested for statistical significance using analysis of variance.
Results
Neurogenesis is impaired in GSK3 knockin mice
The lack of serine-phosphorylated GSK3α/β in GSK3 knockin mice was confirmed in immunoblots of hippocampal extracts, which also showed that the tyrosine phosphorylation and total levels of each GSK3 isoform were the same as in age- and sex-matched wild-type mice (Figure 1A). GSK3 knockin mice reproduce and develop apparently normally with no overt phenotype (49) and general morphological features of wild-type and GSK3 knockin brains and staining for neuronal nuclei (NeuN) and glial fibrillary acidic protein (GFAP) were equivalent (Supplemental Figure S1). Immunohistochemical analysis of cell proliferation in the hippocampus measured 24 hr after three injections of BrdU (100 mg/kg) given at 2 hr intervals showed that BrdU-labeled mitotic cells were predominantly located in the subgranular zone of the dentate gyrus in both wild-type and GSK3 knockin mice (Figure 1B). Quantitative unbiased stereology analysis revealed that the number of BrdU-labeled cells within the dentate gyrus in GSK3 knockin mice was significantly 40% lower (n=10 mice/group; p<0.001) than in matched wild-type mice (Figure 1C). Immunohistochemical staining with Ki67 and PCNA to detect changes in number of proliferating cells demonstrated results comparable to the BrdU labeling (Supplemental Figure S1). Impaired neurogenesis was not due to a change in the volume of the dentate gyrus in GSK3 knockin mice, which was not significantly different (F=2.87, p=0.16) from wild-type mice (Figure 1D). Although apoptosis is a rapid event and few cells could be expected to be undergoing apoptosis at any one time, active caspase-3 was measured as an apoptotic marker to determine if differences in apoptosis were evident. Quantitative unbiased stereology analysis of active caspase-3-positive cells within the dentate gyrus showed no significant difference between GSK3 knockin and wild-type mice (Figure 1E).
Figure 1. NPC proliferation is impaired in the dentate gyrus of GSK3 knockin mice.

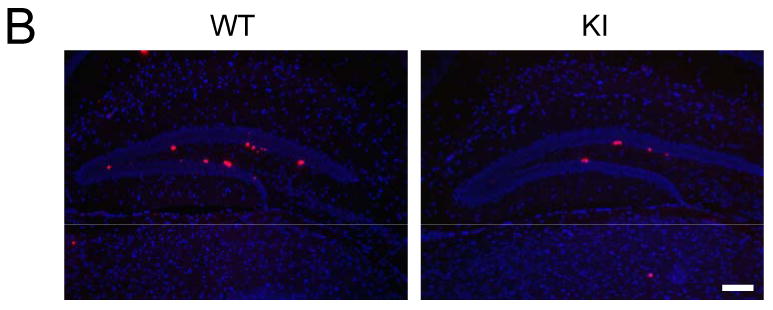

(A) Immunoblots showing that serine phosphorylation of GSK3α and GSK3β is absent in the hippocampus of GSK3 knockin mice, whereas total and tyrosine-phosphorylated levels of GSK3α and GSK3β are equal in GSK3 knockin (KI) and wild-type (WT) mice. (B) Immunohistochemical detection of BrdU-positive cells (red) in the hippocampus of WT and KI mice. Nuclei are labeled with bisbenzimide (blue). Scale bar, 100 μm. (C) Unbiased stereological quantitation of BrdU-positive cells in the hippocampal dentate gyrus (DG). Values are means ± S.E.; n=10/group; *p<0.001. (D) Dentate gyrus volumes of WT and KI mice. Values are means ± S.E.; n=10/group. (E) Unbiased stereological quantitation of active caspase-3-positive cells in the hippocampal dentate gyrus WT and KI mice. Values are means ± S.E.; n=5/group.
We examined if the differentiation of NPCs was altered in GSK3 knockin mice. To test this, BrdU (100 mg/kg) was administrated once daily for 3 days and labeled cells were analyzed 28 days after the last BrdU injection. Representative immunohistochemical analysis of cells co-labeled with BrdU and the neuronal marker NeuN or the astrocyte marker GFAP are shown in Figure 2 (A-H). Most BrdU-positive cells in the dentate gyrus were co-labeled with NeuN in accordance with previous reports (8, 53, 54). A similar percentage of BrdU-positive cells co-expressed NeuN (95±2 % in wild-type mice; 98±2% in GSK3 knockin mice; Figure 2I) or co-expressed GFAP (3±1% in wild-type mice; 2±1% in GSK3 knockin mice; Figure 2J). Thus, the proliferation of NPCs in GSK3 knockin mice was lower than in wild-type mice, but the percentage differentiating to neurons and astrocytes was similar to wild-type mice.
Figure 2. Phenotype of BrdU-positive cells in the dentate gyrus.
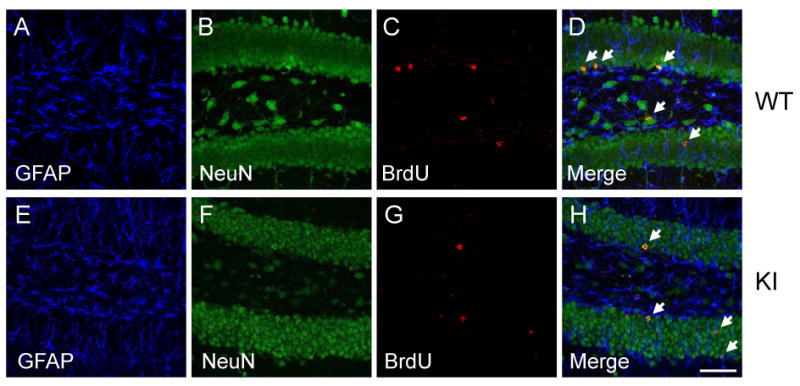

GSK3 knockin (KI) and matched wild-type (WT) mice were administrated BrdU (100 mg/kg) once daily for 3 days and were analyzed 28 days after the last BrdU injection. Sections were labeled with markers for neurons (NeuN) and glia (GFAP). Representative confocal images are shown for (A, E) GFAP (blue), (B, F), NeuN (green), and (C, G) BrdU (red). Labels are merged in (D, H). The arrows indicate representative BrdU(+)/NeuN(+) cells in the subgranular zone. Scale bar, 50 μm. Percentage of (I) BrdU(+)/NeuN(+) cells and (J) BrdU(+)/GFAP(+) cells in the dentate gyrus from WT and KI mice. Values are means ± S.E.; n=4/group.
Effects of blocked inhibitory serine-phosphorylation of GSK3 on NPCs in vitro
Cultured NPCs were used to test if impaired proliferation observed in GSK3 knockin mice in vivo were also evident in vitro. Immunoblots confirmed undetectable levels of serine-phosphorylated GSK3α/β and unaltered tyrosine phosphorylation and total levels of each GSK3 isoform in NPCs from GSK3 knockin mice compared with wild-type mice (Figure 3A). Cell proliferation was measured by immunocytochemical analysis after incubation with 10 μM BrdU for 6 hr (Figures 3B and 3C) or using the MTS assay (Figure 3D). Quantitative analysis showed that there was not a significant difference between proliferation of GSK3 knockin and wild-type NPCs. Flow cytometry measurements of the cell cycle distribution of NPCs from GSK3 knockin and matched wild-type mice showed that they were not different, confirming the equivalent proliferation measurements (Figure 3F). In addition, there were no differences in apoptosis as measured by active caspase-3 between the GSK3 knockin and wild-type NPCs in culture (Figure 3E)
Figure 3. Blocked inhibitory serine-phosphorylation of GSK3 does not impair proliferation of NPCs in vitro.
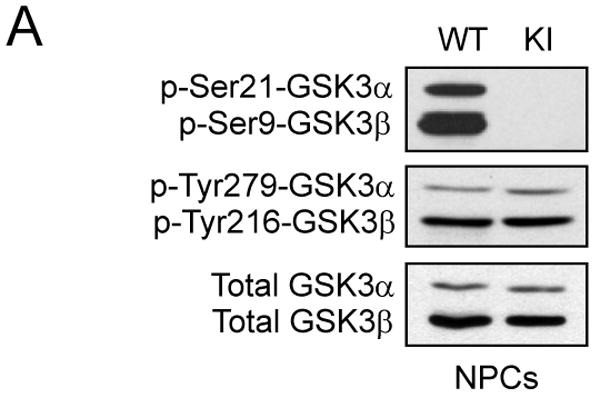
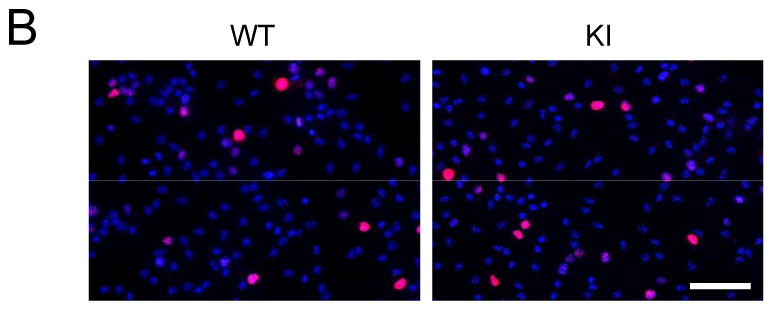


(A) Western blots showing the absence of serine phosphorylation of GSK3α and GSK3β in NPCs from GSK3 knockin mice (KI), whereas tyrosine-phosphorylated and total levels of GSK3α/β are equal in NPCs from wild-type (WT) and KI mice. (B) Immunocytochemical images of BrdU-positive cells (red) in NPCs from WT and KI mice. Nuclei were counterstained with bisbenzimide (blue). Scale bar, 100 μm. (C) Quantitative analysis of BrdU-positive cells in NPCs from WT and KI mice. Values are means ± S.E.; n=8 per group. (D) NPC proliferation for 3 days measured using the MTS assay. Values are means ± S.E.; n=3 per group. (E) Active caspase-3-positive cells in NPCs from WT and KI mice. Values are means ± S.E.; n=3 per group. (F) Cell cycle distribution of NPCs using propidium iodide staining and FACS analysis. Values are means ± S.E.; n=3 per group.
The effect of blocked inhibitory serine-phosphorylation of GSK3 on VEGF and BDNF in the hippocampus
Since in vivo, but not in vitro, NPC proliferation was diminished in GSK3 knockin mice compared with wild-type mice, we tested if the concentrations of two neurotrophins known to support neurogenesis, VEGF and BDNF, were different in hippocampi of GSK3 knockin and matched wild-type mice. The level of VEGF was ∼30% lower in the hippocampus of GSK3 knockin mice compared to the wild-type mice (Figure 4A), whereas the level of BDNF was equivalent in the two groups (Figure 4B). We also examined mRNA levels of three isoforms of VEGF, VEGF188 (a 708-bp product), VEGF164 (a 636-bp product), and VEGF120 (a 522-bp product) and BDNF splice variants containing exon-8 and -3 in the hippocampal extracts from GSK3 knockin and matched wild-type mice. In accordance with protein levels, VEGF mRNA levels were ∼25% lower in GSK3 knockin mice compared with wild-type mice (Figures 4C and 4D), but BDNF mRNA levels were the same in the two groups (Figures 4E-G). After in vitro treatment with VEGF there was significantly increased proliferation of wild-type NPCs by 9%, but only a non-significant 4% increased proliferation of NPCs from GSK3 knockin mice (Supplemental Figure S2).
Figure 4. VEGF and BDNF in the hippocampus of wild-type and GSK3 knockin mice.
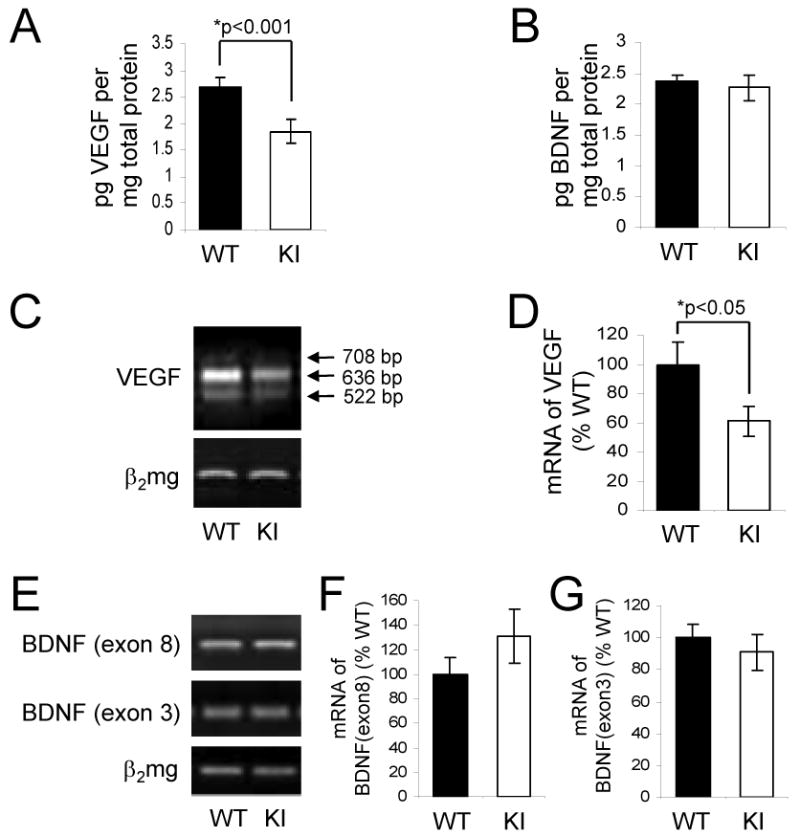
Quantification of (A) VEGF and (B) BDNF protein levels in the hippocampus of GSK3 knockin (KI) and matched wild-type (WT) mice determined by ELISA. Values are means ± S.E.; n=15 per group. (C) Representative result, and (D) quantitation of RT-PCR analysis of VEGF mRNA in the hippocampus of KI and WT mice. PCR products were separated by electrophoresis in 1.5 % agarose and visualized by ethidium bromide staining. VEGF amplification products of 708-, 636-, and 522-bp correspond to VEGF188, VEGF164, and VEGF120, respectively. (E) Representative result, and (F, G) quantitation of RT-PCR analysis of BDNF mRNA in the hippocampus of KI and WT mice. PCR products were separated by electrophoresis in 2% agarose and visualized by ethidium bromide staining. RT-PCR for β2 microglobulin (β2mg) was used as a control for RNA loading. Values for individual data were normalized to β2 microglobulin and expressed as the percentage of the WT values. Quantitative values are means ± S.E.; n=4 per group; *p<0.05.
Co-administration of fluoxetine plus lithium increases proliferation in the dentate gyrus of wild-type but not GSK3 knockin mice
To test if the proliferation of cells in the dentate gyrus in response to fluoxetine and lithium is different in GSK3 knockin and wild-type mice, mice were treated with each drug alone or both together for 21 days. On the last day of drug administration, BrdU (100 mg/kg) was administrated 3 times at 2 hr intervals and the brains were analyzed 24 hr later. In wild-type mice, BrdU-positive cells in the dentate gyrus were not significantly changed after treatment with fluoxetine or lithium individually, but co-administration of both fluoxetine and lithium together induced a 63% increase (Figure 5A). In contrast, co-administration of fluoxetine and lithium did not increase proliferation in GSK3 knockin mice. The levels of VEGF and BDNF in the hippocampus were not increased by administration of fluoxetine or lithium in wild-type or GSK3 knockin mice, but surprisingly treatment with lithium or lithium plus fluoxetine reduced VEGF in wild-type mice (Figure 5B), indicating that activation of neurogenesis by the drugs in wild-type mice was not due to bolstering the production of these neurotrophins. In wild-type mice, co-administration of fluoxetine and lithium significantly increased the inhibitory phosphorylation on serine-21-GSK3α and serine-9-GSK3β in both the hippocampus (Figure 5C) and cerebral cortex (Figure 5D) but did not change the tyrosine-phosphorylation or total levels of either GSK3 isoform, as expected from previous reports (38, 55). These findings suggest that the inability of fluoxetine and lithium to inhibit GSK3 by increasing its serine-phosphorylation in GSK3 knockin mice impairs their capacity to promote neurogenesis.
Figure 5. Fluoxetine and lithium-induced proliferation in the dentate gyrus is impaired in GSK3 knockin mice.

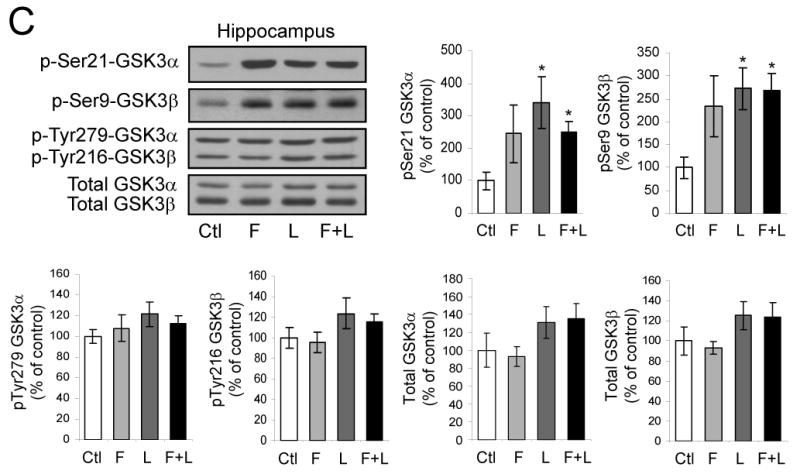
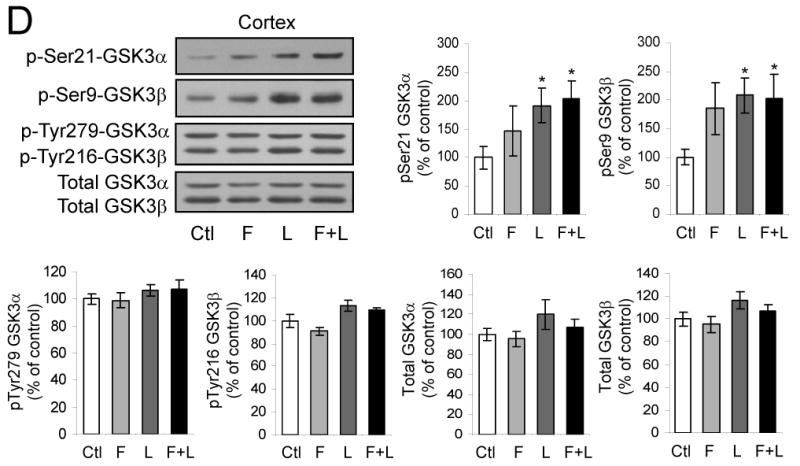
Fluoxetine (F), lithium (L), or both together were administrated to mice for 21 days. (A) BrdU (100 mg/kg) was injected the day before sacrifice followed by unbiased stereological quantitation of BrdU-positive cells in the hippocampal dentate gyrus (DG). Values are means ± S.E.; n=5-10 per group; *p<0.001. (B) Fluoxetine (F), lithium (L), or both together were administrated to mice for 21 days. Hippocampal extracts were evaluated for VEGF and BDNF by ELISA. Quantitative values are means ± S.E.; n=5 per group. *p<0.05 comparing VEGF after drug treatments to untreated wild-type mice, **p<0.05 comparing GSK3 knockin mice to wild-type. (C) Hippocampus and (D) cerebral cortex extracts were immunoblotted for phospho-Ser21-GSK3α, phospho-Ser9-GSK3β, phospho-Tyr279-GSK3α, phospho-Tyr216-GSK3β, and total GSK3α/β. Quantitative values are means ± S.E.; n=5 per group; *p<0.05.
Discussion
Impaired neurogenesis may contribute to certain psychiatric diseases, particularly mood disorders, and enhancement of neurogenesis may be partly responsible for the therapeutic effects of mood stabilizers, antidepressants, and antipsychotics (2-4, 56, 57, 64). However, it is not evident what mechanisms can impair neurogenesis in mood disorders that can be reversed by therapeutic agents. One candidate mechanism is hyperactive GSK3, because accumulating evidence suggests that dysregulated GSK3 may contribute to mood disorders and GSK3 is inhibited by mood stabilizers, antidepressants, and antipsychotics (34). Therefore, we tested if dysregulated GSK3 affected neurogenesis in the mouse dentate gyrus. The results showed that in vivo expression of GSK3 that cannot be inhibited by serine phosphorylation impaired neurogenesis in mice and also blocked the enhancement of neurogenesis induced by coadministration of lithium and fluoxetine. Thus, impaired neurogenesis may result from inadequate regulatory mechanisms that normally inhibit GSK3 through serine-phosphorylation, such as deficient serotonergic signaling associated with depression (38), and these deficiencies may be counteracted by therapeutic agents that increase the inhibitory control of GSK3.
A large impairment in the in vivo proliferation of NPCs was found in the hippocampus of adult mice that express mutations of both GSK3 isoforms that block inhibitory serine phosphorylation of GSK3 (49). Importantly, tyrosine phosphorylation of each GSK3 isoform is unaltered in GSK3 knockin mice and both isoforms of GSK3 are expressed at normal levels, so GSK3 is present at physiologically meaningful levels but cannot be inhibited by serine-phosphorylation. Thus, GSK3 in these mice models conditions of impaired signaling that would otherwise cause inhibitory serine-phosphorylation of GSK3. Regulators that can inhibit GSK3 by increasing serine-phosphorylation include serotonin (38) and BDNF (58), and deficiencies of each of these have been linked to mood disorders. Considering that there is accumulating evidence for both hyperactive GSK3 and impaired neurogenesis in mood disorders, the present findings indicate that the two may be linked in such a way that neurogenesis is disrupted if GSK3 is not properly controlled by inhibitory serine phosphorylation.
Impaired in vivo neurogenesis in GSK3 knockin mice appears not to be due to a direct effect of GSK3 in NPCs because in vitro proliferation was equivalent in NPCs from GSK3 knockin mice and wild-type mice. This suggested that disruption of NPC proliferation in vivo in GSK3 knockin mice may be due to reduced signaling by neurotrophins or other modulators that promote NPC proliferation, although it is important to consider that there may be fundamental differences in the influence of GSK3 on cultured embryonic NPCs and neurogenesis in adults. Since no deficiencies have yet been reported in the brains of GSK3 knockin mice, to test if this may underlie impaired neurogenesis in these mice we examined two neurotrophins well-established to promote neurogenesis, BDNF and VEGF (56, 59-61). Measurements in hippocampal extracts from GSK3 knockin mice and matched wild-type mice showed that BDNF levels were the same but there was a significant deficit in VEGF levels in the GSK3 knockin mice. This represents the first detection of a deficient neurotrophin in GSK3 knockin mouse brain, and raises the likelihood of other deficits. This supports the hypothesis that neurogenesis may be impaired in GSK3 knockin mice because of deficient production of molecules that support neurogenesis, with VEGF identified as one such neurotrophin. Neurogenesis relies on many environmental factors involving multiple types of cells, so the critical insufficient factors stemming from the effects of GSK3 remain to be determined.
We tested if deficient neurogenesis in GSK3 knockin mice could be repaired by three weeks of treatment with fluoxetine, lithium, or both together. Co-administration of fluoxetine and lithium significantly increased neurogenesis in wild-type mice, but neither drug administered alone was effective. As noted in the Introduction, administration of antidepressants or lithium has been reported to increase neurogenesis in vivo, but species and strain differences affect the extent of this response (62, 63), so we expect that accounted for increases in neurogenesis only being evident with co-administration of the two drugs in the mice used in present study. In contrast to wild-type mice, in GSK3 knockin mice co-administration of fluoxetine and lithium did not increase neurogenesis. Both fluoxetine and lithium previously were reported to increase the inhibitory serine-phosphorylation of GSK3 in mouse brain in vivo and it was speculated that this inhibition of GSK3 may contribute to their therapeutic effects (38, 55). The present findings indicate that inhibition of GSK3 by increasing its serine-phosphorylation is crucial for fluoxetine and lithium to increase neurogenesis, lending further support to the hypothesis that inhibition of GSK3 by increasing serine-phosphorylation contributes to their therapeutic actions.
In summary, loss of inhibitory control of GSK3 impaired neurogenesis and VEGF production in mouse hippocampus, and impaired the capacity of fluoxetine and lithium to promote neurogenesis. These findings reinforce the possibility that dysregulated GSK3 contributes to the pathophysiology of mood disorders and is a therapeutic target of antidepressants and mood stabilizers. GSK3 can impair neurogenesis in vivo and promotes NPC apoptosis in vitro (44), indicating that if neurogenesis is important in mood disorders, these actions of GSK3 may contribute to the susceptibility or progression of mood disorders. This prospect is further strengthened by the recent finding that lack of inhibition of GSK3 impairs neurogenesis upon deficient expression of DISC1 (Disrupted in Schizophrenia 1), a gene disrupted in a family with a high incidence of depression, schizophrenia, and bipolar disorder (64). However, there is still much to learn about how neurogenesis is involved in psychiatric diseases and how neurogenesis is regulated by GSK3.
Supplementary Material
Acknowledgments
We are grateful to Dr. Dario Alessi for providing the mice used in this research. The authors acknowledge the valuable support of Dr. K. A. Roth and the Neuroscience Core Facilities at UAB (NS47466, NS57098), the fluoxetine from the NIMH Chemical Synthesis and Drug Supply Program, and Anna Zmijewska for technical assistance. This research was supported by grant MH38752 from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- 2.Abrous DN, Koehl M, Le Moal M. Adult neurogenesis: from precursors to network and physiology. Physiol Rev. 2005;85:523–569. doi: 10.1152/physrev.00055.2003. [DOI] [PubMed] [Google Scholar]
- 3.Jacobs BL, Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- 4.Duman RS, Malberg J, Nakagawa S, D'Sa C. Neuronal plasticity and survival in mood disorders. Biol Psychiatry. 2000;48:732–739. doi: 10.1016/s0006-3223(00)00935-5. [DOI] [PubMed] [Google Scholar]
- 5.Kempermann G. Regulation of adult hippocampal neurogenesis - implications for novel theories of major depression. Bipolar Disord. 2002;4:17–33. doi: 10.1034/j.1399-5618.2002.40101.x. [DOI] [PubMed] [Google Scholar]
- 6.Schloesser RJ, Chen G, Manji HK. Neurogenesis and neuroenhancement in the pathophysiology and treatment of bipolar disorder. Int Rev Neurobiol. 2007;77:143–178. doi: 10.1016/S0074-7742(06)77005-2. [DOI] [PubMed] [Google Scholar]
- 7.Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, Lesch KP. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11:514–522. doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- 8.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czeh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M, Bartolomucci A, Fuchs E. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci USA. 2001;98:12796–12801. doi: 10.1073/pnas.211427898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manev H, Uz T, Smalheiser NR, Manev R. Antidepressants alter cell proliferation in the adult brain in vivo and in neural cultures in vitro. Eur J Pharmacol. 2001;411:67–70. doi: 10.1016/s0014-2999(00)00904-3. [DOI] [PubMed] [Google Scholar]
- 11.Banasr M, Soumier A, Hery M, Mocaer E, Daszuta A. Agomelatine, a new antidepressant, induces regional changes in hippocampal neurogenesis. Biol Psychiatry. 2006;59:1087–1096. doi: 10.1016/j.biopsych.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 12.Warner-Schmidt JL, Duman RS. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA. 2007;104:4647–4652. doi: 10.1073/pnas.0610282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott BW, Wojtowicz JM, Burnham WM. Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp Neurol. 2000;165:231–236. doi: 10.1006/exnr.2000.7458. [DOI] [PubMed] [Google Scholar]
- 14.Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry. 2000;47:1043–1049. doi: 10.1016/s0006-3223(00)00228-6. [DOI] [PubMed] [Google Scholar]
- 15.Segi-Nishida E, Warner-Schmidt JL, Duman RS. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc Natl Acad Sci USA. 2008;105:11352–11357. doi: 10.1073/pnas.0710858105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warner-Schmidt JL, Madsen TM, Duman RS. Electroconvulsive seizure restores neurogenesis and hippocampus-dependent fear memory after disruption by irradiation. Eur J Neurosci. 2008;27:1485–1493. doi: 10.1111/j.1460-9568.2008.06118.x. [DOI] [PubMed] [Google Scholar]
- 17.Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK. Enhancement of hippocampal neurogenesis by lithium. J Neurochem. 2000;75:1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x. [DOI] [PubMed] [Google Scholar]
- 18.Hashimoto R, Senatorov V, Kanai H, Leeds P, Chuang DM. Lithium stimulates progenitor proliferation in cultured brain neurons. Neuroscience. 2003;117:55–61. doi: 10.1016/s0306-4522(02)00577-8. [DOI] [PubMed] [Google Scholar]
- 19.Shimomura A, Nomura R, Senda T. Lithium inhibits apoptosis of mouse neural progenitor cells. Neuroreport. 2003;14:1779–1782. doi: 10.1097/00001756-200310060-00004. [DOI] [PubMed] [Google Scholar]
- 20.Dawirs RR, Hildebrandt K, Teuchert-Noodt G. Adult treatment with haloperidol increases dentate granule cell proliferation in the gerbil hippocampus. J Neural Transm. 1998;105:317–327. doi: 10.1007/s007020050061. [DOI] [PubMed] [Google Scholar]
- 21.Wakade CG, Mahadik SP, Waller JL, Chiu FC. Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res. 2002;69:72–79. doi: 10.1002/jnr.10281. [DOI] [PubMed] [Google Scholar]
- 22.Wang HD, Dunnavant FD, Jarman T, Deutch AY. Effects of antipsychotic drugs on neurogenesis in the forebrain of the adult rat. Neuropsychopharmacology. 2004;29:1230–1238. doi: 10.1038/sj.npp.1300449. [DOI] [PubMed] [Google Scholar]
- 23.Newton SS, Duman RS. Neurogenic actions of atypical antipsychotic drugs and therapeutic implications. CNS Drugs. 2007;21:715–725. doi: 10.2165/00023210-200721090-00002. [DOI] [PubMed] [Google Scholar]
- 24.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 25.Schänzer A, Wachs FP, Wilhelm D, Acker T, Cooper-Kuhn C, Beck H, Winkler J, Aigner L, Plate KH, Kuhn HG. Direct stimulation of adult neural stem cells in vitro and neurogenesis in vivo by vascular endothelial growth factor. Brain Pathol. 2004;14:237–248. doi: 10.1111/j.1750-3639.2004.tb00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Bras B, Barallobre MJ, Homman-Ludiye J, Ny A, Wyns S, Tammela T, Haiko P, Karkkainen MJ, Yuan L, Muriel MP, Chatzopoulou E, Bréant C, Zalc B, Carmeliet P, Alitalo K, Eichmann A, Thomas JL. VEGF-C is a trophic factor for neural progenitors in the vertebrate embryonic brain. Nat Neurosci. 2006;9:340–348. doi: 10.1038/nn1646. [DOI] [PubMed] [Google Scholar]
- 27.Muller S, Chakrapani BP, Schwegler H, Hofmann HD, Kirsch M. Neurogenesis in the dentate gyrus depends on CNTF and STAT3 signaling. Stem Cells in press. 2008 doi: 10.1634/stemcells.2008-0234. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF. TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron. 2008;59:399–412. doi: 10.1016/j.neuron.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol. 2005;192:348–356. doi: 10.1016/j.expneurol.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 30.Bartkowska K, Paquin A, Gauthier AS, Kaplan DR, Miller FD. Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development. 2007;134:4369–4380. doi: 10.1242/dev.008227. [DOI] [PubMed] [Google Scholar]
- 31.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001;50:260–265. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- 33.Balu DT, Hoshaw BA, Malberg JE, Rosenzweig-Lipson S, Schechter LE, Lucki I. Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res. 2008;23:37–43. doi: 10.1016/j.brainres.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Current Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 37.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Zhu W, Roh MS, Friedman AB, Rosborough KM, Jope RS. In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang UG, Seo MS, Roh MS, Kim Y, Yoon SC, Kim YS. The effects of clozapine on the GSK-3-mediated signaling pathway. FEBS Lett. 2004;560:115–119. doi: 10.1016/S0014-5793(04)00082-1. [DOI] [PubMed] [Google Scholar]
- 40.Alimohamad H, Rajakumar N, Seah YH, Rushlow W. Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry. 2005;57:533–542. doi: 10.1016/j.biopsych.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacology. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- 42.Roh MS, Seo MS, Kim Y, Kim SH, Jeon WJ, Ahn YM, Kang UG, Juhnn YS, Kim YS. Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp Mol Med. 2007;39:353–360. doi: 10.1038/emm.2007.39. [DOI] [PubMed] [Google Scholar]
- 43.Gould E. Serotonin and hippocampal neurogenesis. Neuropsychopharmacology. 1999;21 1:S46–51. doi: 10.1016/S0893-133X(99)00045-7. [DOI] [PubMed] [Google Scholar]
- 44.Eom TY, Roth KA, Jope RS. Neural precursor cells are protected from apoptosis induced by trophic factor withdrawal or genotoxic stress by inhibitors of glycogen synthase kinase 3. J Biol Chem. 2007;282:22856–22864. doi: 10.1074/jbc.M702973200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 46.Thomaidou D, Mione MC, Cavanagh JF, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuhn HG, Biebl M, Wilhelm D, Li M, Friedlander RM, Winkler J. Increased generation of granule cells in adult Bcl-2-overexpressing mice: a role for cell death during continued hippocampal neurogenesis. Eur J Neurosci. 2005;22:1907–1915. doi: 10.1111/j.1460-9568.2005.04377.x. [DOI] [PubMed] [Google Scholar]
- 48.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3 (GSK3) Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 49.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akhtar RS, Geng Y, Klocke BJ, Roth KA. Neural precursor cells possess multiple p53-dependent apoptotic pathways. Cell Death Differ. 2006;13:1727–1739. doi: 10.1038/sj.cdd.4401879. [DOI] [PubMed] [Google Scholar]
- 51.Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ. Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci USA. 2000;97:7579–7584. doi: 10.1073/pnas.120552597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 53.Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C, Zhang YJ, Nestler EJ, Duman RS. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci. 2002;22:3673–3682. doi: 10.1523/JNEUROSCI.22-09-03673.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kodama M, Fujioka T, Duman RS. Chronic olanzapine or fluoxetine administration increases cell proliferation in hippocampus and prefrontal cortex of adult rat. Biol Psychiatry. 2004;56:570–580. doi: 10.1016/j.biopsych.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 55.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol. 2007;18:391–418. doi: 10.1097/FBP.0b013e3282ee2aa8. [DOI] [PubMed] [Google Scholar]
- 57.Kempermann G, Krebs J, Fabel K. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr Opin Psychiatry. 2008;21:290–295. doi: 10.1097/YCO.0b013e3282fad375. [DOI] [PubMed] [Google Scholar]
- 58.Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3β and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- 59.Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci USA. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fabel K, Fabel K, Tam B, Kaufer D, Baiker A, Simmons N, Kuo CJ, Palmer TD. VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur J Neurosci. 2003;18:2803–2812. doi: 10.1111/j.1460-9568.2003.03041.x. [DOI] [PubMed] [Google Scholar]
- 61.Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, During MJ. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genetics. 2004;36:827–835. doi: 10.1038/ng1395. [DOI] [PubMed] [Google Scholar]
- 62.Miller BH, Schultz LE, Gulati A, Cameron MD, Pletcher MT. Genetic regulation of behavioral and neuronal responses to fluoxetine. Neuropharmacology. 2008;33:1312–1322. doi: 10.1038/sj.npp.1301497. [DOI] [PubMed] [Google Scholar]
- 63.Holick KA, Lee DC, Hen R, Dulawa SC. Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1A receptor. Neuropsychopharmacology. 2008;33:406–417. doi: 10.1038/sj.npp.1301399. [DOI] [PubMed] [Google Scholar]
- 64.Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell. 2009;136:1017–1031. doi: 10.1016/j.cell.2008.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.