

Abstract
Rationale
Sphingomyelin synthase 2 (SMS2) contributes to de novo SM biosynthesis and plasma membrane SM levels. SMS2 deficiency in macrophages diminishes NFκB and MAP kinase activation induced by inflammatory stimuli.
Objective
The effects of SMS2 deficiency on the development of atherosclerosis are investigated.
Methods and Results
We measured cholesterol efflux from macrophages of wild type (WT) and SMS2 knockout (KO) mice. We transplanted SMS2 KO mouse bone marrow into LDL receptor knockout mice (SMS2−/−→LDLr−/−), creating a mouse model of SMS2 deficiency in the macrophages. We found that SMS2 deficiency caused significant induction of cholesterol efflux in vitro and in vivo. Moreover, we found that SMS2 KO mice had less IL-6 and TNFα in the circulation before and after endotoxin stimulation, compared with controls. More importantly, after 3 months on a Western diet, SMS2−/−→LDLr−/− mice showed decreased atherosclerotic lesions in the aortic arch, root (57%, P<0.001), and the entire aorta (42%, P<0.01), compared with WT→LDLr−/− mice. Analysis of plaque morphology revealed that SMS2−/−→LDLr−/− mice had significantly less necrotic core area (71%, P<0.001), less macrophage content (37%, P<0.01), and more collagencontent (35%, P<0.05) in atherosclerotic lesions. We also found that SMS2−/−→LDLr−/− mice had significantly lower free cholesterol and cholesteryl ester levels in the brachiocephalic artery than WT→LDLr−/− ones (33 and 52%, P<0.01 and P<0.001, respectively).
Conclusions
SMS2 deficiency in the macrophages reduces atherosclerosis in mice. Macrophage SMS2 is thus a potential therapeutic target for treatment of this disease.
Keywords: Macrophage sphingomyelin synthase deficiency, Sphingomyelin biosynthesis, Cholesterol efflux, Inflammation, Sphingomyelin in plasma membranes, Atherosclerosis
Foam cell formation due to excessive accumulation of cholesterol in the macrophages is a pathological hallmark of atherosclerosis,1 which is also known to be an inflammatory disease.2 The accumulation of macrophage-derived foam cells in the vessel wall is always accompanied by the production of a wide range of chemokines and cytokines that regulate the turnover and differentiation of immigrating and resident cells, and subsequent plaque development.2 Thus, promoting cholesterol efflux from cholesterol-laden macrophages, as well as diminishing their inflammatory response, can both be significant antiatherogenic approaches.
The interaction between SM, cholesterol, and glycosphingolipiddrives the formation of plasma membrane rafts,3 and in some cells, caveolae.4, 5 SM is synthesized by sphingomyelin synthase (SMS), which transfers the phosphorylcholine moiety from phosphatidylcholine (PC) onto ceramide.6 Two SMS genes, SMS1 and SMS2, have been cloned, and their subcellular localization characterized.7, 8 SMS1 is found in the trans-golgi apparatus, while SMS2 is predominantly located in the plasma membranes.7, 9 Our lab and others have shown that SMS1 and SMS2 expression levels correlate positively with those of SM in the lipid rafts.10–12 Furthermore, SMS1 has been implicated in the regulation of lipid raft SM levels, as well as raft functions such as FAS receptor clustering,10 endocytosis, and apoptosis.11
SM-enriched lipid rafts may play important roles in cholesterol efflux. ATP-binding cassette transporters (ABC)A1, ABCG1, and scavenger receptor BI (SR-B1) are located in the plasma membranes, and exist either directly in rafts (SR-B1),13 or in association with the redistribution of lipids in plasma membranes (ABCA1 and ABCG1).14 It is therefore conceivable that changes in plasma membrane SM within the SMS2-null macrophages will influence the functions of these proteins and alter cholesterol efflux, thus influencing the development of atherosclerosis. Indeed, enhanced apoA-I-dependent cholesterol efflux by ABCA1 from SM-deficient Chinese hamster ovary (CHO) cells has been reported.15
Manipulation of plasma membrane SM levels can also alter the structure of lipid rafts and modify inflammatory responses. It has been reported that sphingomyelinase treatment causes the clustering of several receptors, including TNFα receptor,16 toll-like receptors (TLRs),17 and interleukin-1 receptor,18 thus influencing downstream signaling pathways. NFκB19 and MAP kinases20 are the key regulators of inflammation. We found that NFκB and MAP kinase activation is attenuated in macrophages from SMS2 KO mice in response to LPS stimulation.21 In line with these observations, we found that SMS2 deficiency substantially diminished the abundance of toll-like receptor 4 (TLR4)-MD2 complex levels on the surface of macrophages following LPS stimulation.21
For further evaluation of the relationship between macrophage SMS2 deficiency and atherosclerosis, we transplanted SMS2-deficient mouse bone marrow into LDL receptor-deficient mice (SMS2−/−→LDLr−/−), creating a model of SMS2 deficiency and LDL receptor expression exclusively in the macrophages. As a control, we also transplanted WT mouse bone marrow into LDL receptor-deficient mice (WT→LDLr−/−), creating a model of LDL receptor expression exclusively in the macrophages. We investigated the development of atherosclerosis in these animals.
Materials and Methods
Mice and Diets
LDL receptor-deficient (LDLr−/−) mice (8-week-old females)of a C57BL/6 background were purchased from Jackson Laboratory (Bar Harbor, ME). SMS2 KO mice,21 originally of a 129 background, were backcrossed with C57BL/6 mice for 4 generations. The animals (WT and KO) used in this study were littermates. All were fed a chow diet (Research Diets, Inc). Bone marrow transplantation was performed, and after 8 weeks all mice were switched to a Western diet (0.15% cholesterol, 20% saturated fat) for 3 months. Experiments involving animals were conducted with the approval of SUNY Downstate Medical Center IACUC.
Bone Marrow Transplantation to Replace Peripheral Macrophages
Bone marrow cells were harvested from the tibias of donor mice(SMS2−/− and WT), as previously described.22 A total of 20 LDLr−/− mice (age 8 weeks) were lethally irradiated with 1000 rads (10Gy). Ten of these animals were transplanted withSMS2−/− mouse bone marrow (5×106 cells), and the other 10 with WT bone marrow, via the femoral vein, all within3 hours of irradiation. We monitored the process of cell replacement by polymerase chain reaction (PCR), using genomic DNA from mouse white blood cells as a template. The primers used for SMS2 KO mouse screening were: Forward 5′-AGTGACAACGTCGAGCACAG-3′ and Reverse 5′-GGCCATTGAACAAGATGGAT-3′. The primers for WT mouse screening were: Forward 5′-GGCCATTGAACAAGATGGAT-3′ and Reverse 5′-GACGGTTGTCAGGATGAGGT-3′.
Lipid Analyses by LC/MS/MS
SM, PC, and ceramide levels were measured by LC/MS/MS, as previously described.21
mRNA Analyses
RNA was isolated from macrophages, using TriZol (Invitrogen). The primers used for mouse SMS2 RT-PCR were: Forward 5′-CAAAACTTGAAGGTCACTTGGA-3′ and Reverse 5′-GGTGGGGCTTGTGTAAGTGT-3′. The primers used for mouse SMS1 RT-PCR were: Forward 5′-GTGCTCAGACCGGAAGAAAG-3′ and Reverse 5′-ACTAGCTTCTCCGCGTGTTC-3′. 18S rRNA was used as an internal control. The forward and reverse primer sequences for 18S rRNA were: 5′-AGTCCCTGCCCTTTGTACACA-3′ and 5′-GATCCGAGGGCCTCACTAAAC-3′.
SMS Activity Assay
Macrophages were homogenized in a buffer containing 50 mM Tris-HCl, 1 mM EDTA, 5% sucrose, and a cocktail of protease inhibitors (Sigma). The homogenate was centrifuged at 5000 rpm for 10 minutes and the supernatant mixed in assay buffer containing 50 mM Tris-HCl (pH 7.4), 25 mM KCl, C6-NBD-ceramide (0.1 μg/μl), and phosphotidylcholine (0.01 μg/μl). The mixture was incubated at 37°C for 2 hours. Lipids were extracted in chloroform: methanol (2:1), dried under N2 gas, and separated by thin layer chromatography (TLC). Band intensity was quantified by Image–Pro Plus version 4.5 software (Media Cybernetics Inc.).
Lysenin Treatment and Cell Mortality Measurement
Macrophages were washed twice in PBS and incubated with lysenin, 50 ng/ml, for 1 hour. Cell viability was measured using WST-1 cell proliferation reagent according to the manufacturer’s instructions (Roche).
Cholesterol Efflux from Macrophages
Mouse peritoneal macrophages were labeled with [3H]cholesterol carried by acetylated LDL. After labeling, cells were washed with phosphate-buffered saline, equilibrated with DMEM, 0.2% bovine serum albumin for 1 hour, and incubated with 10 μg/ml purified human apoA-I or HDL in 0.5 ml of DMEM, 0.2% bovine serum albumin. The medium was collected at 8 hours and centrifuged at 6000 × g for10 minutes to remove cell debris and cholesterol crystals. Radioactivity in an aliquot of supernatant was determined by liquid scintillation counting. The cells were finally lysed in 0.5 ml of 0.1 M sodium hydroxide, 0.1% SDS, and the radioactivity in an aliquot was determined. Cholesterol efflux was expressed as the percentage of the radioactivity released from the cells into the medium relative to the total radioactivity in cells and medium.
Western Blot for Macrophage ABCA1, ABCG1, and SR-B1
Macrophages were lysed in 200 mM NaCl, 50 mM Tris (pH 7.5), 1 mM EDTA, and 1% (v/v) protease inhibitor cocktail (Sigma). Cell debris was cleared by centrifugation at 8,200 g for 10 minutes. Lysates were subjected to SDS/PAGE and then transferred to nitrocellulose membranes. The blots were probed with antibodies against ABCA1 (Abcom), ABCG1 (Abcom), and SR-B1 (Novus). β-Actin was used as a loading control. Blots were developed by a chemiluminescence detection system (SuperSignal West detection kit, Pierce). The maximum intensity of each band was measured by Image–Pro Plus version 4.5 software (Media Cybernetics Inc.).
In Vivo Macrophage Cholesterol Efflux Measurement
In vivo macrophage cholesterol efflux was measured by a method previously reported.23 Briefly, SMS2 KO or WT bone marrow-derived macrophages were loaded with cholesterol by incubation with acetylated LDL and [3H]cholesterol. The labeled macrophages were injected intraperitoneally into WT mice. Plasma was collected at 12, 24, and 48 hours. Feces were collected at 24 and 48 hours. All were analyzed for tracer counts.
Mouse Atherosclerotic Lesion Measurement
The aorta was dissected and the arch photographed, as previouslyreported.24 Aortic lesion en face assay was performed as previously described.24 For morphometric lesion analysis, sections were stained with Harris’ hematoxylin and eosin. Total intimal lesion area and acellular/anuclearareas (negative for hematoxylin-positive nuclei) per cross section were quantified by taking the average of 6 sections spaced 30 μm apart, beginning at the base of the aortic root. Histomorphological analysis of collagen was performed with Masson’s trichrome stain (Richard-Allan Scientific, Kalamazoo, MI). Images were viewed and captured with a Nikon Labophot 2 microscope equipped with a SPOT RT3 color video camera attached to a computerized imaging system with Image–Pro Plus version 4.5 software (Media Cybernetics Inc.).
Immunostaining of macrophage and smooth muscle cell (SMC) in the plaques
Sequential sections 10 μm thick was stained with macrophage-specific antibody (AIA31240, Accurate Chemical and Scientific Corporation) and anti-SMC actin antibody (1A4, Zymed). Primary antibodies were incubated for 1 hour at room temperature in3% serum matched to the species of the secondary antibodies. Biotinylated secondary antibodies were incubated for 30 minutes, followed by 45 minutes of horseradish peroxidase-conjugated streptavidinand visualization with diaminobenzidine. Nuclei were counterstained with hemalaune. The mean area of staining per section per animal from seven sections were determined for each animal. Staining areas were quantified with Image-Pro-Plus software.
Cell surface ABCA1 and SR-BI analysis by FACS
Acyltyl-LDL treated macrophages were scraped off the plates and re-suspended in PBS to make single-cell suspension. Cells were treated with Fc receptor block (mAb 2–4G2; BD Pharmingen) and stained with antibodies to SR-B1 (Rabbit pAb Novus Biologicals) or ABCA1 (Rabbit pAb Novus Biologicals) with 1:50 dilution. After wash, cells were stained with goat anti-rabbit second antibody conjugated with green fluorescence (Invitrogen) with 1:100 dilutions. After wash, cells were suspended in PBS containing 1ug/ml propidium iodide (PI). Cells were analyzed on a FACScan with CellQuest (Benton Dickinson). Dead cells were excluded from the analysis according to PI staining.
Brachiocephalic Artery (BCA) Total Cholesterol and Cholesteryl Ester Measurement
Total cholesterol, cholesteryl ester, SM, PC, and ceramide levels in BCA were measured according to a method previously reported by us.25, 12
Statistical Analysis
Each experiment was conducted at least 3 times. Data were typically expressed as mean±SD. Differences between groups were tested by the Mann–Whitney U test (nonparametric test). P values less than 0.05 were considered significant.
Results
SMS2-deficient Macrophages Produce More Cholesterol Efflux
Because plasma membrane SM levels regulate a spectrum of important genes involved in cholesterol efflux and signal transduction, we speculated that the SMS2, located in the plasma membranes, might be important in regulating macrophage cholesterol efflux. We therefore measured thisefflux from peritoneal macrophages of WT and SMS2-deficient mice. The deficiency of SMS2 caused a significant induction incholesterol efflux, compared with controls, using either apoA-I or HDL as the cholesterol acceptor (75 and 65%, P<0.01, respectively) (Figure 1A and 1B).
Figure 1.
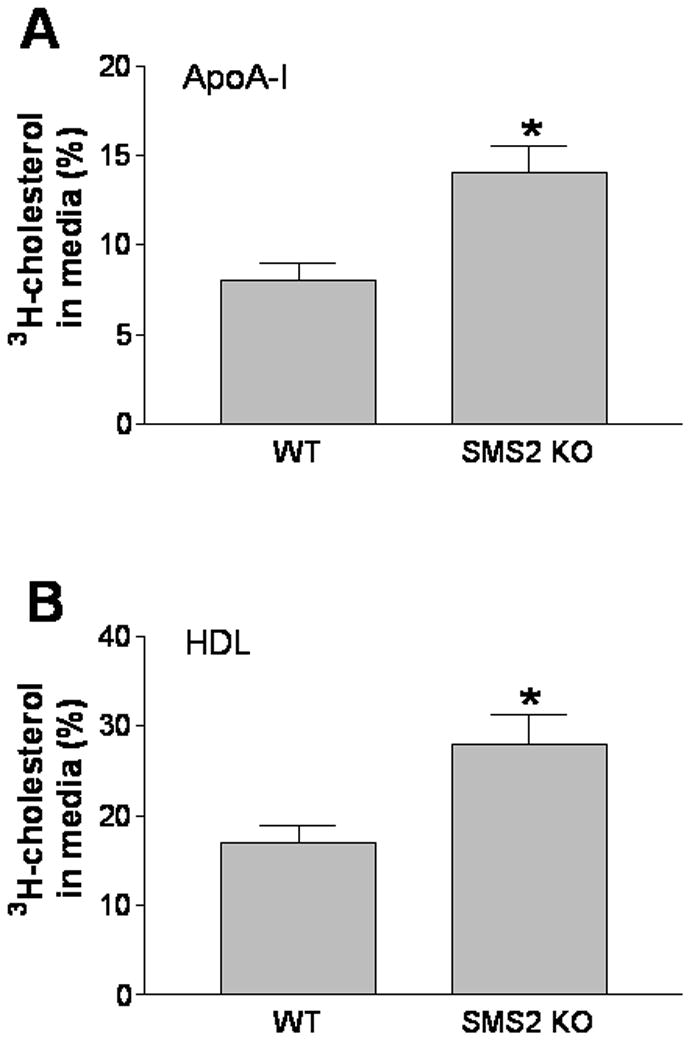
SMS2 deficiency significantly increased macrophage cholesterol efflux in vitro. Cholesterol efflux was measured as described in “Methods.” Panel A, cholesterol efflux toward apoA-I; Panel B, cholesterol efflux toward HDL. Values are mean ± SD, N=5. *P<0.01.
Promoting reverse cholesterol transport is considered to be an antiatherogenic process, one that might be altered through macrophage SMS2 deficiency. We utilized an approach reported by Rader’s group23 to investigate the effect of SMS2 deficiency on macrophage cholesterol efflux in vivo. Bone marrow-derived macrophages from both WT and SMS2 KO mice were loaded with [3H]cholesterol by incubation with acetylated LDL, and then injected intraperitoneally into WT animals. We found that SMS2 KO macrophages produce significantly more [3H]cholesterol efflux into the circulation at 24- and 48-hour time points (Figure 2A). Furthermore, feces from KO mice also accumulated more [3H]cholesterol 24 and 48 hours after injection (Figure 2B), compared with that of controls.
Figure 2.
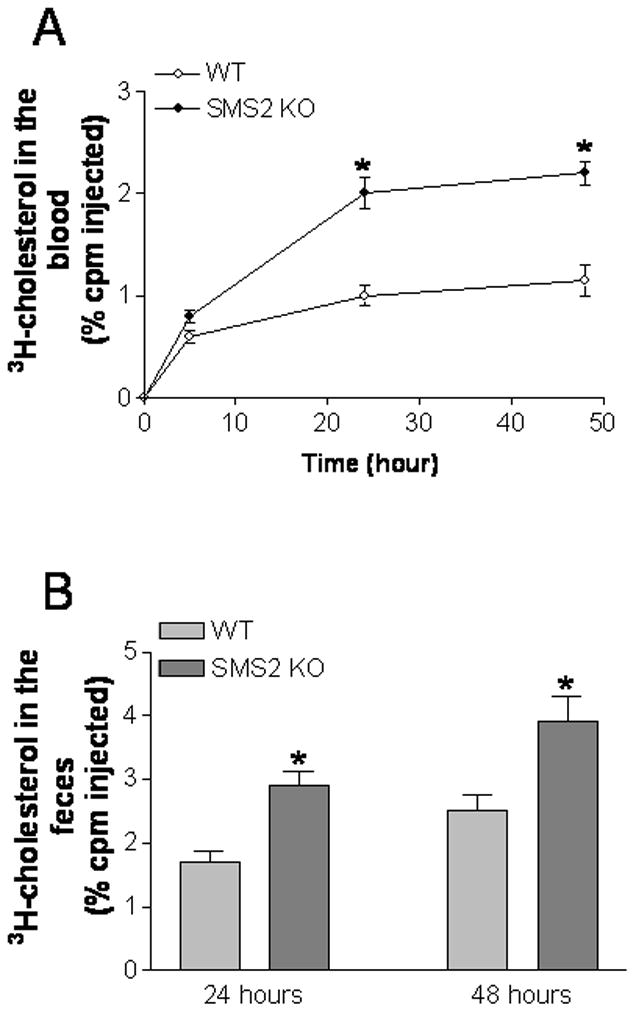
SMS2 deficiency significantly increased macrophage cholesterol efflux in vivo. SMS2 KO or WT bone marrow-derived macrophages were loaded with cholesterol by incubation with acetylated LDL and [3H]cholesterol. Labeled macrophages were injected intraperitoneally into WT mice. Plasma was collected at 12, 24, and 48 hours. Feces were collected at 24 and 48 hours. All were analyzed for tracer counts. Values are mean ± SD, N=5. *P<0.01.
To elucidate the possible mechanisms of cholesterol efflux induction in SMS2-deficient macrophages, we performed Western blot measurement of ABCA1, ABCG1, and SR-BI. SMS2 deficiency significantly increased ABCA1 (190±27%, P<0.01), ABCG1 (139±28%, P<0.01), and SR-BI (211±50%, P<0.01) in macrophages, compared with controls (Figure 3A and 3B). This suggests that all 3 molecules might contribute to higher cholesterol efflux from SMS2-null macrophages in vitro and in vivo. We also investigated ABCA1 and SR-BI levels on macrophage plasma membrane. FACS analysis showed that, after acyltyl-LDL stimulation, SMS2 KO macrophages have more ABCA1 and SR-BI on the cell surface than that on control macrophages (Fig. 3C and 3D), suggesting alteration of SM levels on plasma membrane influences both proteins, thus influencing cholesterol efflux.
Figure 3.
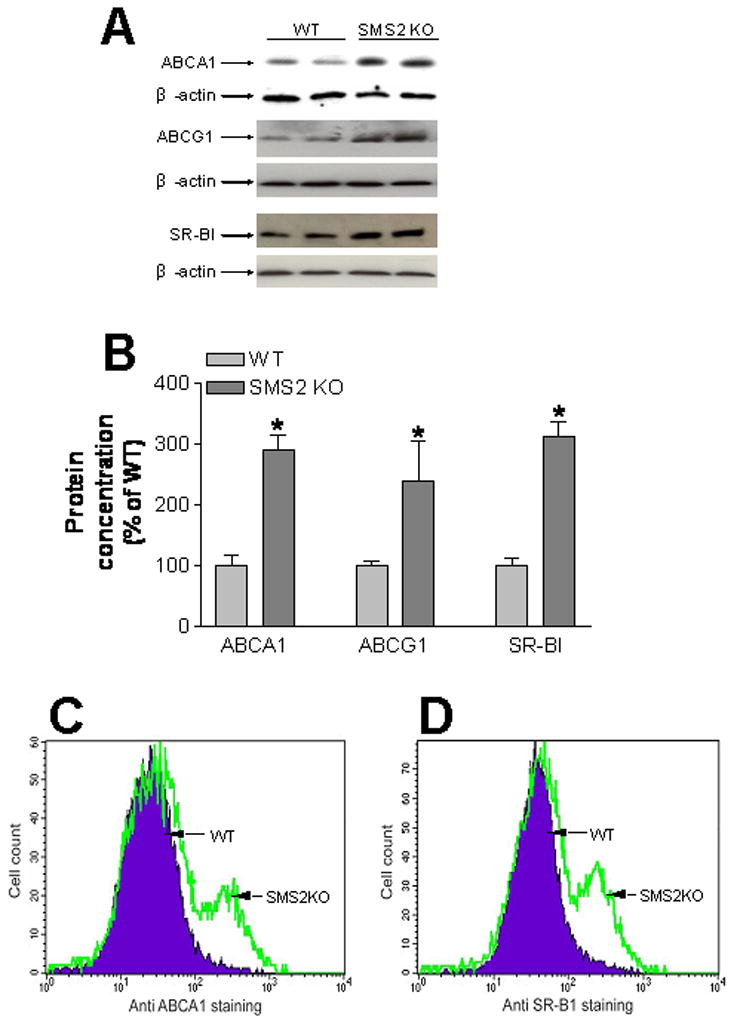
SMS2 deficiency significantly increased macrophage ABCA1, ABCG1, and SR-B1 protein levels. Macrophage lysates were subjected to SDS/PAGE and transferred to nitrocellulose membranes. Blots were probed with ABCA1, ABCG1, and SR-B1 antibodies. GAPDH was used as a loading control. Panel A, ABCA1, ABCG1, and SR-B1 fluorogram, which is representative of 3 independent experiments. Panel B, quantitative display of macrophage ABCA1, ABCG1, and SR-B1. Values are mean ± SD, N=4. *P<0.01.
SMS2 KO Mice Have Significantly Lower Inflammatory Cytokines in the Circulation
We have reported previously that SMS2 deficiency substantially diminished the abundance of toll-like receptor 4 (TLR4)-MD2 complex levels on the surface of macrophages after LPS stimulation. Likewise, NFκB and MAP kinase activation was attenuated in macrophages from SMS2 KO mice.21 In line with these findings, we have discovered in this study that plasma IL-6 and TNFα levels in SMS2 KO mice were significantly decreased before and after LPS stimulation (Table 1).
Table 1.
Plasma IL-6 and TNFα measurement in mice.
| Mice | IL-6 | TNFα |
|---|---|---|
| (pg/ml) | ||
| WT | 39±2 | 11±1 |
| SMS2KO | 15±5* | 6±3* |
| WT(LPS) | 1818±59 | 85±11 |
| SMS2KO(LPS) | 1376±99** | 48±5** |
| WT→LDLrKO | 41±3 | 9±2 |
| SMS2KO→LDLrKO | 16±17** | 5±1* |
Value: mean±SD; N=5.
P<0.05;
P<0.01.
Macrophage-specific SMS2 KO Mouse Preparation
Some 26 LDLr−/− mice were lethally irradiated. After3 hours, half the animals were transplanted with SMS2−/− mouse bone marrow cells (SMS2−/−→LDLr−/−), and the other half with WT ones (WT→LDLr−/−). We monitored the process of cell replacement by PCR, using genomic DNA from mouse white blood cells as a template, and measured them 8 weeks after transplantation. In the SMS2−/−→LDLr−/− group, the peripheral cells had been replaced by donor cells of an SMS2-deficient genotype, a 300 bp PCR product (Figure 4A). In the WT→LDLr−/− group, the replaced peripheral cells were of an SMS2 expression genotype, with a 500 bp PCR product (Figure 4A).
Figure 4.
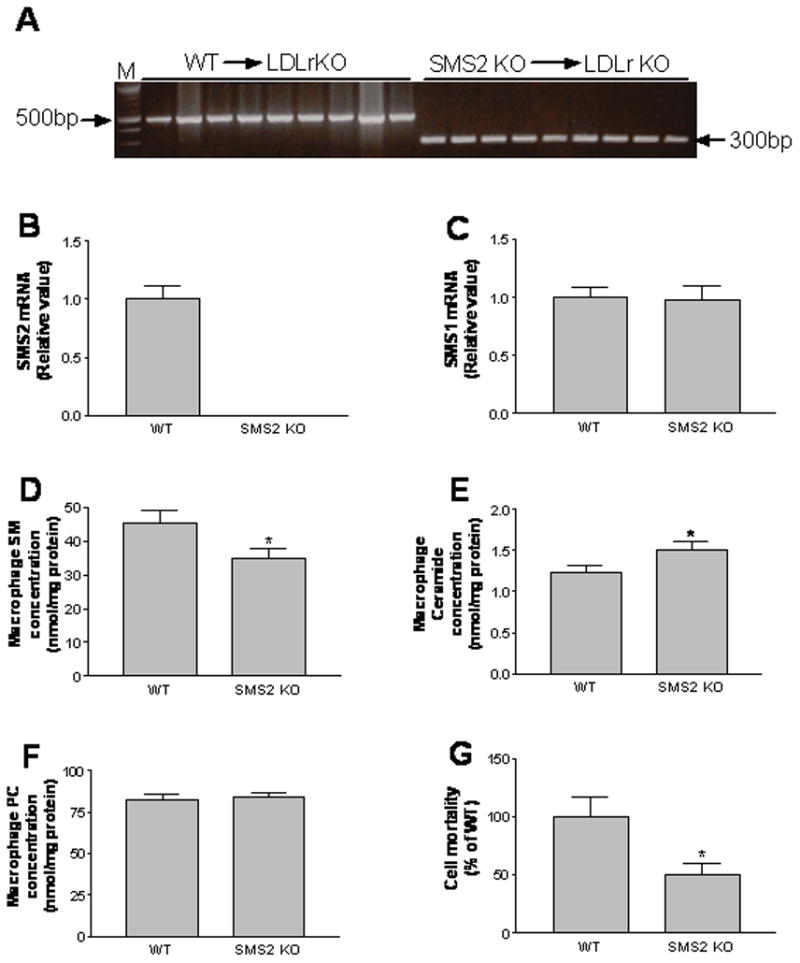
Preparation of LDL receptor KO mice with a macrophage-specific SMS2 deficiency. LDLr−/− animals were lethally irradiated. After3 hours, half of them were transplanted with SMS2−/− mouse bone marrow cells (SMS2−/−→LDLr−/−), and the other half with WT ones (WT→LDLr−/−). Panel A, genotype determination of mouse peripheral cells. Genomic DNA was extracted from white blood cells of the same mouse, before and after irradiation. The genotypes of SMS2 were determined with PCR, as described in “Methods.” WT mice had a 500 bp PCR product, and KO ones a 300 bp PCR product; Panel B, macrophage SMS2 mRNA levels measured by real time-PCR; Panel C, macrophage SMS1 mRNA levels measured by real time-PCR; Panels D–F, macrophage SM, ceramide, and PC measurement; Panel G, macrophage plasma membrane SM measurement (lysenin-mediated cell lysis assay21,26). Values in panel B-G are mean ± SD, N=5. *P<0.05.
As expected, bone marrow-derived SMS2−/− macrophages had no SMS2 expression (Figure 4B), but had normal SMS1 mRNA levels (Figure 4C). SMS2−/− or SMS2−/−→LDLr−/− peritoneal macrophages have significantly lower total SMS activity than that of controls (Supplement Fig. I, about 20%, P<0.05). Cellular SM levels were decreased (P<0.05) (Figure 4D, Supplement Table I), while ceramide levels were increased (P<0.05) (Figure 4E), mainly related to ceramide 16:0 increasing (Supplement Table II). There was no significant change in cellular PC levels (Figure 4F), but there was a significant decrease of diacylglycerol levels (Supplement Table IIIP<0.05). In order to measure SM on plasma membrane, we took the advantage of lysenin, a recently discovered SM-specific cytotoxin.26 Lysenin recognizes SM only when it forms aggregates or microdomains on the plasma membrane, and then lyse the cells.26 Lysenin-mediated cell mortality can indirectly reflect SM levels on the plasma membrane.12, 21 Based on lysenin-mediated cell lysis assay, we found that the SMS2−/−→LDLr−/− macrophages had significantly less SM in the plasma membranes, compared with controls (P<0.01) (Figure 4G). We also measured cellular cholesterol levels and did not find significant difference (Supplement Figure II). CE content was undetectable regardless of genotype, which is similar to a previous report.27
At this point, the rest of the animals were switched to a Western diet (0.15% cholesterol, 20% saturated fat) for 3 months. We found that SMS2−/−→LDLr−/− mice had significantly lower basal IL-6 and TNFα levels in the circulation (Table 1), compared with WT→LDLr−/− controls. We measured plasma lipid levels (cholesterol, SM, PC, and triglyceride) in these animals, finding no significant changes (Supplement Table IV).
Evaluation of Atherosclerosis in Macrophage-specific SMS2 KO Mice
To evaluate the impact of macrophage SMS2 deficiency on atherogenesis, we dissected mouse aortas and photographed them. We also measured proximal and whole aortic lesion areas. After 3 months on a Western diet, we found that all mice (18/18) had lesions in the aortic arch. However, the SMS2−/−→LDLr−/− animals had noticeably smaller ones than the WT→LDLr−/− mice (Figure 5A).
Figure 5.
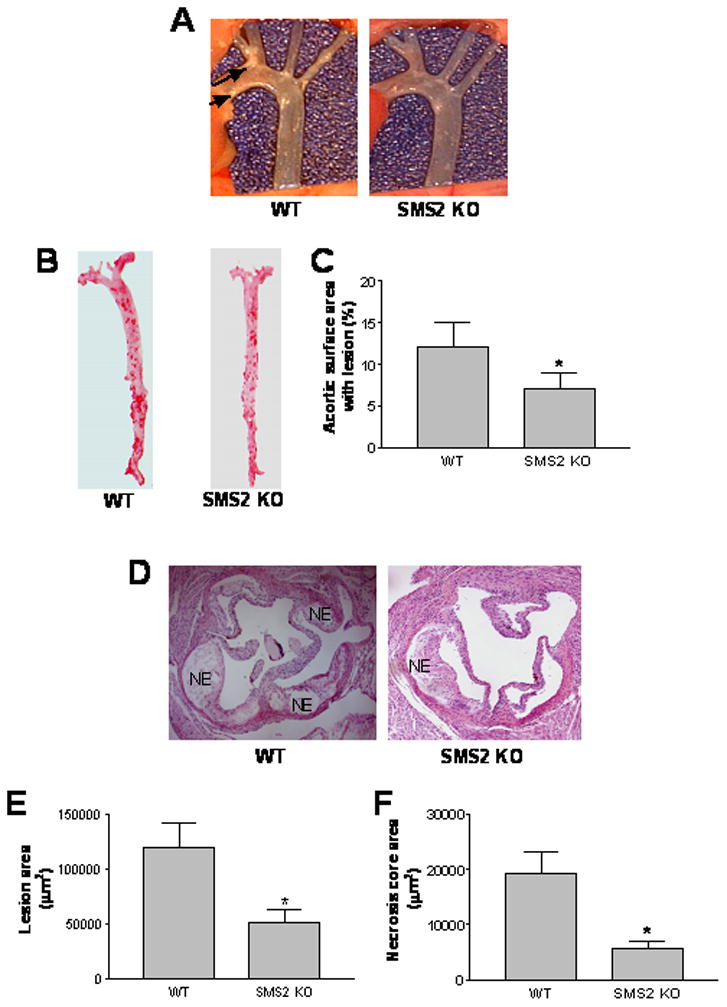
SMS2−/−→LDLr−/− mice demonstrated significantly decreased atherosclerotic lesion size and necrotic area. Panel A, mice were euthanized, and the aortas dissected and photographed. This set of pictures is representative of 9 sets. Atherosclerotic lesions are indicated by arrows; Panel B, Oil Red O staining for whole aorta (en face assay); Panel C, quantitative display of en face assay; Panel D, hematoxylin and eosin staining for proximal aorta (root assay) (NE: necrotic core area); Panel E, quantitative display of root assay; Panel F, quantitative display of necrotic core area. Values are mean ± SD, N=9. *P<0.01.
We also found that the SMS2−/−→LDLr−/− animals had 42% less lesion area in the whole aorta (Figure 5B and 5D), and 57% less in the proximal aorta (Figure 5D and 5E), compared with WT→LDLr−/− mice. These differences were highly significant (P<0.01 and P<0.001, respectively) (Figure 5C and 5E). Analysis of plaque morphology revealed substantial differences between the SMS2−/−→LDLr−/− and WT→LDLr−/− mice. As illustrated by the hematoxylin and eosin stained images, plaques from the SMS2−/−→LDLr−/− animals had a significant decrease in necrotic core areas that were anuclear, afibrotic, and eosin negative (71% P<0.01) (Figure 5F).
Furthermore, the SMS2−/−→LDLr−/− mice demonstrated substantially more collagen, as illustrated by the trichrome-stained images (Figure 6A, 35%, P<0.05). We did immunohistochemistry for macrophages and smooth muscle cells. We found that macrophage (Figure 6B) but not smooth muscle cell (Supplement Figure III) are significantly decreased in SMS2−/−→LDLr−/− mice, compared with controls. We determined the blood cell counts for both SMS2 KO and control mice. Since total SMS2 deficiency does not show the difference in blood cell counts (Supplement Table V), we do not expect that SMS2−/−→LDLr−/− mice have different blood cell counts compared with WT→LDLr−/− mice.
Figure 6.
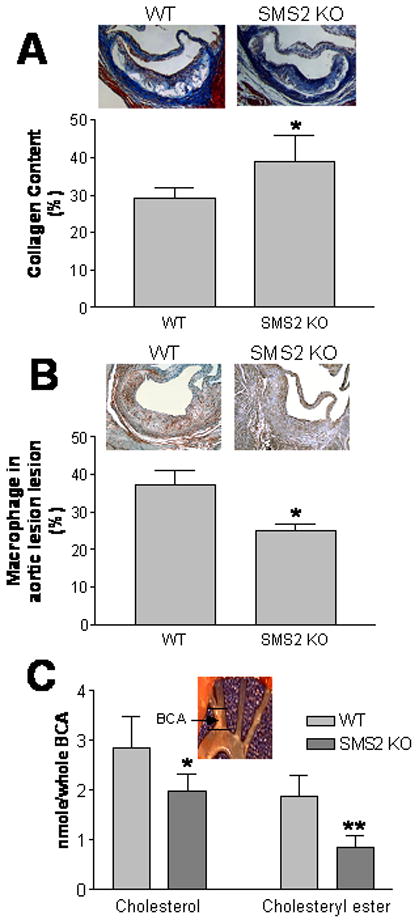
SMS2−/−→LDLr−/− mice demonstrated significantly increased collagen content in lesion areas, and decreased cholesterol and cholesteryl ester contents in the brachiocephalic artery (BCA). Panel A, collagen was stained with Masson’s trichrome and quantified by Image-Pro-Plus 4.5 software; Panel B, Macrophage immunostaining. The procedure was described in “Materials and Methods”; Panel C, total cholesterol and cholesteryl ester levels in BCA were measured according to our previous report.25 Values are mean ± SD, N=6. *P<0.05, **P<0.01.
We isolated the brachiocephalic arteries (BCA) from both groups and extracted lipid from them. Using LC/MS/MS to measure the free cholesterol and cholesteryl ester levels, we found that the SMS2−/−→LDLr−/− mice had significantly lower free cholesterol and cholesteryl ester levels in the BCA than that in the WT→LDLr−/− mice (33% and 52%, P<0.01 and P<0.001, respectively) (Figure 6B). We also measured SM, Ceramide, and PC in brachiocephalic artery (BCA). We did not find changes of PC (Table 2). We did find decrease of SM in SMS2−/−→LDLr−/− BCA, but such change did not reach to a statistical significance (P=0.063) (Table 2). Moreover, we found PC/SM ratio is significantly increase in SMS2−/−→LDLr−/− BCA compared with that in WT→LDLr−/− BCA (P<0.05) (Table 2). We also measure ceramide levels in BCA and we did not find changes (Table 2, Supplement Table VI).
Table 2.
Sphingomyelin, Ceramide, and phosphatidylcholine levels in mouse brachiocephalic artery
| Mice | SM | PC | Ceramide | PC/SM |
|---|---|---|---|---|
| (nmole/whole BCA) | ||||
| WT→LDLr−/− | 0.50±0.05 | 1.13±0.17 | 0.036±0.007 | 2.25±0.16 |
| SMS2−/−→LDLr−/− | 0.43±0.03 | 1.19±0.11 | 0.035±0.005 | 2.71±0.32* |
Value: mean±SD; N=6. Lipids were measured by LC/MS/MS.
P<0.05. SM, Sphingomyelin; PC, Phosphatidylcholine.
Discussion
As expected, SMS2 deficiency in bone marrow-derived cells led to a decrease in atherosclerosis in LDLr−/− mice fed a Western diet for 3 months. These findings appear to be explained by an increase in macrophage cholesterol efflux, in vitro and in vivo, and a decrease in inflammatory response.
We reported previously that plasma SM is an independent and positive risk factor for coronary heart disease and that SM levels may serve as a marker for atherogenic remnant lipoprotein accumulation.28,29 In this study, instead of study SM in the circulation, we focused on the macrophage plasma membrane SM levels. We believe that SM level reduction in the circulation could cause a global effect on atherosclerosis, while SM level reduction on cell membrane, such as macrophage membrane, could create a microenvironment, influencing cholesterol efflux and inflammation, thus causing anti-atherogenic consequences.
Macrophage plasma membrane SM levels are related to cholesterol efflux. Foam cell formation due to excessive accumulation of cholesterol by macrophages is a pathological hallmark of atherosclerosis.1 Macrophages cannot limit the uptake of cholesterol, and therefore depend on cholesterol efflux pathways to prevent their transformation into foam cells. We found that SMS2-deficient macrophages had significantly less SM in the plasma membranes (Figure 4G), and more cholesterol efflux than control cells in vitro (Figure 1) and in vivo (Figure 2). This observation could have important implications in terms of vessel wall homeostasis in response to atherogenic insult. It has been reported that lysosomal sphingomyelinase is involved in cholesterol transport from lysosomes to the plasma membranes.30 Sphingomyelinase hydrolyzes SM in late endosomes and lysosomes.31 Because SM avidly binds cholesterol,32 sphingomyelinase deficiency inhibits macrophage cholesterol efflux through promoting cholesterol sequestration by SM.30 It is conceivable that SMS2 deficiency may have the opposite effect as sphingomyelinase deficiency, in reference to macrophage cholesterol efflux, because SMS2 deficiency decreases de novo SM biosynthesis and decreases plasma membrane SM levels21 (Figure 4G).
Macrophage plasma membrane SM levels are related to the proteins associated with cholesterol efflux. In macrophages, ABCA1 exports cholesterol and phospholipid to lipid-free apolipoproteins, while ABCG1 exports cholesterol to phospholipid-containing acceptors.14 ABCA1-dependent cholesterol export involves an initial interaction of apoA-I with SM-rich lipid raft membrane domains.33 Such reorganization effectively expands the nonraft membrane fractions, and consequently preconditions cells for cholesterol efflux.34 ABCG1 exports cholesterol to HDL and other phospholipid-containing acceptors. These include particles generated during the lipidation of apoA-I by ABCA1, suggesting that the two transporters cooperate in cholesterol export.35 ABCG1 is mainly found intracellularly in the basal state, with little cell surface presentation. But on stimulation, for example by liver X receptor (LXR) agonist treatment, ABCG1 redistributes to the plasma membranes and increases cholesterol mass efflux to HDL.36 Scavenger receptor BI (SR-B1) also facilitates cholesterol efflux from macrophages.37 It is well-known that SR-B1 is located in SM-rich caveolae and lipid rafts.13 Inactivation of macrophage SR-B1 promotes atherosclerotic lesion development in apoE KO mice.38 In this study, we found that SMS2 deficient macrophages upregulate all these cholesterol-efflux-related transports and receptor (Fig. 3A and 3B). Moreover, SMS2 KO macrophages, after acyltyl-LDL stimulation, have more ABCA1 and SR-BI on the cell surface than that on control macrophages (Fig. 3C and 3D), suggesting that these proteins are located or associated with SM-rich microdomains (lipid rafts) on the macrophage plasma membrane and alteration of SM levels on these microdomains could influence both proteins, thus influencing cholesterol efflux.
SMS2 deficiency could alter the structure of lipid rafts and modify inflammatory responses. In our previous study, we found that SMS2 deficiency attenuates NFκB activation. We observed that upon stimulation by TNFα, the recruitment of TNFR1 receptor to lipid rafts following ligand stimulation was blocked in SMS2 knockdown cells,21 suggesting a mechanism for the modulation of NFκB activity by SMS2. This finding is in agreement with previous reports indicating that raft association of TNFR1 was found to be crucial for TNFα-mediated NFκB activation in human fibrosarcoma cells.39 We also found that LPS-induced plasma membrane recruitment of TLR4-MD-2 complex was diminished in SMS2 KO macrophages.21 SMS2 deficiency may also influence signal transduction pathways other than NFκB activation. The activation of MAP kinases was likewise attenuated in SMS2 KO macrophages.21 Taken together, these findings strongly suggest the critical role of SM, synthesized by SMS2, in the normal function of TNFR1 and TLR4 receptors in the plasma membranes following stimulation by their respective ligands. More importantly, SMS2 deficiency decreases IL-6 and TNFα levels in the circulation (Table 1).
It was noticed that after bone marrow transplantation the SMS2KO→LDLrKO macrophages express LDL receptor. Herijgers et al have reported that the presence or absence of the LDL receptors in the transplanted bone marrow does not influence atherogenesity in LDL receptor KO mice.40 Many researchers using this approach to study atherosclerotic lesion formation.40–42
The key findings of this study are the atherosclerotic plaque size and plaque morphology in SMS2−/−→LDLr−/− mice, compared with those in WT→LDLr−/− animals. Macrophage SMS2 deficiency decreased the plaque size and the necrotic core area (Figure 5), which is composed primarily of dead macrophages,43 and increased collagen content (Figure 6A), a sign of the integrity of the aorta. We measured cholesterol and cholesteryl ester contents in BCA and found that both are significantly decreased in the SMS2−/−→LDLr−/− mice (Figure 6B). We also found decrease of SM in SMS2−/−→LDLr−/− BCA, but such change did not reach to a statistical significance (P=0.063) (Table 2). Moreover, we found PC/SM ratio is significantly increase in SMS2−/−→LDLr−/− BCA compared with that in WT→LDLr−/− BCA (P<0.05) (Table 2). The significance of this finding deserves further investigation.
In conclusion, SMS2 physiologically contributed to de novo SM biosynthesis and plasma membrane SM levels. SMS2 deficiency in the macrophages caused blunted NFκB and MAP kinase responses to inflammatory/immunological stimuli, promoted cholesterol efflux, and reduced atherosclerosis in a mouse model. Thus, macrophage SMS2 is a potential therapeutic target for the treatment of atherosclerosis.
Supplementary Material
Acknowledgments
The authors are grateful to Thomas Beyer, Hai H. Bui, David A. Peake, Mike Kalbfleisch, and Robert Christie for their support in LC/MS/MS lipid analysis, to Ms. Yifan Fan for her assistance in aorta cutting and staining, to Ms. Ingar Hansen at Columbia University for her cellular cholesterol measurement (by gas-liquid chromatography) and to Mr. Calvin Yeang for his English editing.
Sources of Funding
This work was supported by American Heart Association Grant-in-Aid 0755922T (to Dr. Jiang), National Institutes of Health grants HL-64735 and HL-69817 (to Dr. Jiang).
Non-standard Abbreviations used
- KO
gene knockout
- SM
sphingomyelin
- SMS
sphingomyelin synthase
- ABCA1
ATP-binding cassette transporter A1
- ABCG1
ATP-binding cassette transporter G1
- SR-B1
scavenger receptor B1
Footnotes
Disclosures
None.
References
- 1.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–808. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 3.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 4.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 5.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 6.Merrill AH, Jones DD. An update of the enzymology and regulation sphingomyelin metabolism. Biochim Biophysi Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 7.Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004;23:33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamaoka S, Miyaji M, Kitano T, Umehara H, Okazaki T. Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growing in sphingomyelin synthase-defective lymphoid cells. J Biol Chem. 2004;279:18688–18693. doi: 10.1074/jbc.M401205200. [DOI] [PubMed] [Google Scholar]
- 9.Yeang C, Varshney S, Wang R, Zhang Y, Ye D, Jiang XC. The domain responsible for sphingomyelin synthase (SMS) activity. Biochim Biophys Acta. 2008;1781:610–617. doi: 10.1016/j.bbalip.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyaji M, Jin ZX, Yamaoka S, Amakawa R, Fukuhara S, Sato SB, Kobayashi T, Domae N, Mimori T, Bloom ET, Okazaki T, Umehara H. Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J Exp Med. 2005;202:249–259. doi: 10.1084/jem.20041685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van der Luit AH, Budde M, Zerp S, Caan W, Klarenbeek JB, Verheij M, Van Blitterswijk WJ. Resistance to alkyl-lysophospholipid-induced apoptosis due to downregulated sphingomyelin synthase 1 expression with consequent sphingomyelin- and cholesterol-deficiency in lipid rafts. Biochem J. 2007;401:541–549. doi: 10.1042/BJ20061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Z, Hailemariam TK, Zhou H, Li Y, Duckworth DC, Peake DA, Zhang Y, Kuo MS, Cao G, Jiang XC. Inhibition of sphingomyelin synthase (SMS) affects intracellular sphingomyelin accumulation and plasma membrane lipid organization. Biochim Biophysi Acta. 2007;1771:1186–1194. doi: 10.1016/j.bbalip.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matveev S, van der Westhuyzen DR, Smart EJ. Co-expression of scavenger receptor-BI and caveolin-1 is associated with enhanced selective cholesteryl ester uptake in THP-1 macrophages. J Lipid Res. 1999;40:1647–1654. [PubMed] [Google Scholar]
- 14.Jessup W, Gelissen IC, Gaus K, Kritharides L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr Opin Lipidol. 2006;17:247–257. doi: 10.1097/01.mol.0000226116.35555.eb. [DOI] [PubMed] [Google Scholar]
- 15.Nagao K, Takahashi K, Hanada K, Kioka N, Matsuo M, Ueda K. Enhanced apoA-I-dependent cholesterol efflux by ABCA1 from sphingomyelin-deficient Chinese hamster ovary cells. J Biol Chem. 2007;282:14868–14874. doi: 10.1074/jbc.M611230200. [DOI] [PubMed] [Google Scholar]
- 16.Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem. 2000;275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 17.Triantafilou M, Miyake K, Golenbock T, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–11. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- 18.Mathias S, Younes A, Kan CC, Orlow I, Joseph C, Kolesnick RN. Activation of the sphingomyelin signaling pathway in intact EL4 cells and in a cell-free system by IL-1 beta. Science. 1993;259:519–522. doi: 10.1126/science.8424175. [DOI] [PubMed] [Google Scholar]
- 19.Mayo MW, Baldwin AS. The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- 20.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endoctoxin and hyperosmolarity in mammalian cells. Science 1994. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 21.Hailemariam TK, Huan C, Liu J, Li Z, Roman C, Kalbfeisch M, Bui HH, Peake DA, Kuo MS, Cao G, Wadgaonkar R, Jiang XC. Sphingomyelin synthase 2 deficiency attenuates NFkappaB activation. Arterioscler Thromb Vasc Biol. 2008;28:1519–1526. doi: 10.1161/ATVBAHA.108.168682. [DOI] [PubMed] [Google Scholar]
- 22.Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267:1034–1037. doi: 10.1126/science.7863332. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 24.Hojjati MR, Li Z, Zhou H, Tang S, Huan C, Ooi E, Lu S, Jiang XC. Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem. 2005;280:10284–10289. doi: 10.1074/jbc.M412348200. [DOI] [PubMed] [Google Scholar]
- 25.Kuo MS, Kalbfleisch JM, Rutherford P, Gifford-Moore D, Huang XD, Christie R, Hui K, Gould K, Rekhter M. Chemical analysis of atherosclerotic plaque cholesterol combined with histology of the same tissue. J Lipid Res. 2008;49:1353–1363. doi: 10.1194/jlr.D700037-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Ishitsuka R, Yamaji-Hasegawa A, Makino A, Hirabayashi Y, Kobayashi T. A lipid-specific toxin reveals heterogeneity of sphingomyelin-containing membranes. Biophys J. 2004;86:296–307. doi: 10.1016/S0006-3495(04)74105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang XC, Paultre F, Pearson TA, Reed RG, Francis CK, Lin M, Berglund L, Tall AR. Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol. 2000;20:2614–2618. doi: 10.1161/01.atv.20.12.2614. [DOI] [PubMed] [Google Scholar]
- 29.Schlitt A, Hojjati MR, von Gizycki H, Lackner KJ, Blankenberg S, Schwaab B, Meyer J, Rupprecht HJ, Jiang XC. Serum sphingomyelin levels are related to the clearance of postprandial remnant-like particles. J Lipid Res. 2005;46:196–200. doi: 10.1194/jlr.C400011-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Leventhal AR, Chen W, Tall AR, Tabas I. Acid sphingomyelinase-deficient macrophages have defective cholesterol trafficking and efflux. J Biol Chem. 2001;276:44976–44983. doi: 10.1074/jbc.M106455200. [DOI] [PubMed] [Google Scholar]
- 31.Schuchman EH, Desnick RJ. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill; New York: 1995. pp. 2601–2624. [Google Scholar]
- 32.Slotte JP. Sphingomyelin-cholesterol interactions in biological and model membranes. Chem Phys Lipids. 1999;102:13–27. doi: 10.1016/s0009-3084(99)00071-7. [DOI] [PubMed] [Google Scholar]
- 33.Gaus K, Kritharides L, Schmitz G, Boettcher A, Drobnik W, Langmann T, Quinn CM, Death A, Dean RT, Jessup W. Apolipoprotein A-1 interaction with plasma membrane lipid rafts controls cholesterol export from macrophages. FASEB J. 2004;18:574–576. doi: 10.1096/fj.03-0486fje. [DOI] [PubMed] [Google Scholar]
- 34.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ABCA1 expression disrupts raft membrane microdomains through its ATPase-related functions. J Biol Chem. 2006;281:36091–36101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- 35.Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol. 2006;26:534–540. doi: 10.1161/01.ATV.0000200082.58536.e1. [DOI] [PubMed] [Google Scholar]
- 36.Wang N, Ranalletta M, Matsuura F, Peng F, Tall AR. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler Thromb Vasc Biol. 2006;26:1310–1316. doi: 10.1161/01.ATV.0000218998.75963.02. [DOI] [PubMed] [Google Scholar]
- 37.Huang ZH, Gu D, Lange Y, Mazzone T. Expression of scavenger receptor BI facilitates sterol movement between the plasma membrane and the endoplasmic reticulum in macrophages. Biochemistry. 2003;42:3949–3955. doi: 10.1021/bi0269207. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S, Linton MF. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2003;108:2258–2263. doi: 10.1161/01.CIR.0000093189.97429.9D. [DOI] [PubMed] [Google Scholar]
- 39.Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity. 2003;18:655–664. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 40.Herijgers N, de Winther MP, Van Eck M, Havekes LM, Hofker MH, Hoogerbrugge PM, Van Berkel TJ. Effect of human scavenger receptor class A overexpression in bone marrow-derived cells on lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knockout mice. J Lipid Res. 2000;41:1402–1409. [PubMed] [Google Scholar]
- 41.Ranalletta M, Wang N, Han S, Yvan-Charvet L, Welch C, Tall AR. Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1−/− bone marrow. Arterioscler Thromb Vasc Biol. 2006;26:2308–2315. doi: 10.1161/01.ATV.0000242275.92915.43. [DOI] [PubMed] [Google Scholar]
- 42.Baldán A, Pei L, Lee R, Tarr P, Tangirala RK, Weinstein MM, Frank J, Li AC, Tontonoz P, Edwards PA. Impaired development of atherosclerosis in hyperlipidemic Ldlr−/− and ApoE−/− mice transplanted with Abcg1−/− bone marrow. Arterioscler Thromb Vasc Biol. 2006;26:2301–2307. doi: 10.1161/01.ATV.0000240051.22944.dc. [DOI] [PubMed] [Google Scholar]
- 43.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.