

Abstract
The accumulation of misfolded proteins in intracellular amyloid inclusions, typical of many neurodegenerative disorders including Huntington's and prion disease, is thought to occur after failure of the cellular protein quality control mechanisms. Here we examine the formation of misfolded protein inclusions in the eukaryotic cytosol of yeast and mammalian cell culture models. We identify two intracellular compartments for the sequestration of misfolded cytosolic proteins. Partition of quality control substrates to either compartment seems to depend on their ubiquitination status and aggregation state. Soluble ubiquitinated misfolded proteins accumulate in a juxtanuclear compartment where proteasomes are concentrated. In contrast, terminally aggregated proteins are sequestered in a perivacuolar inclusion. Notably, disease-associated Huntingtin and prion proteins are preferentially directed to the perivacuolar compartment. Enhancing ubiquitination of a prion protein suffices to promote its delivery to the juxtanuclear inclusion. Our findings provide a framework for understanding the preferential accumulation of amyloidogenic proteins in inclusions linked to human disease.
The strong correlation between the accumulation of aggregated proteins in amyloid inclusions and the onset of several neurodegenerative diseases calls for a better understanding of the mechanisms and functions of inclusion formation. Research indicating that soluble aggregation intermediates have a toxic ‘gain of function’ activity suggests that regulated formation of protein inclusions serves cytoprotective functions, such as sequestering misfolded species1–7, and it may also facilitate their clearance8–12. It is unknown whether inclusions contain only terminally aggregated proteins or whether they also sequester soluble misfolded conformations13. Intriguingly, although all proteins can form amyloid-like inclusions after misfolding1,14, only a handful of proteins cause amyloidosis and disease2. In principle, these amyloidogenic disease-related proteins may interact differently with the cellular quality control machinery. Thus, characterization of the pathways leading to inclusion formation is critical for understanding the basis of protein conformation disorders.
Cellular inclusions form in an organized process that seems to be conserved from yeast to mammalian cells2,8,15. Distinct inclusions with specific characteristics have been observed13,16–19, including insoluble perinuclear inclusions (called aggresomes) that co-localize with the microtubule organizing centre20, perinuclear inclusions containing soluble endoplasmic reticulum associated protein degradation (ERAD) substrates18,21, and inclusions co-localizing with autophagic markers9,10. It is unclear whether all these observations pertain to the same compartment or what underlies the distinct solubility and long-term fates observed for different quality control substrates in these inclusions.
Unlike amyloidogenic proteins, little is known about the fate of ‘normal’ misfolded cytosolic globular proteins22. Protein misfolding can arise as a consequence of stress-induced denaturation, destabilizing missense mutations or lack of oligomeric assembly partners. To examine how cytosolic quality control proceeds in these different scenarios, we chose a panel of model substrates corresponding to each case (Fig. 1) and compared their fate to that of model amyloidogenic proteins (Fig. 2). Our findings show that the quality control machinery partitions misfolded proteins, on the basis of their ubiquitination state and solubility, among two distinct quality control compartments. Interestingly, amyloidogenic proteins are preferentially sorted to only one of these compartments. These distinct quality control compartments may represent two cellular strategies for the sequestration of aggregation prone, potentially toxic polypeptides.
Figure 1. A panel of quality control substrates defines two distinct compartments for the sequestration of misfolded cytosolic proteins.
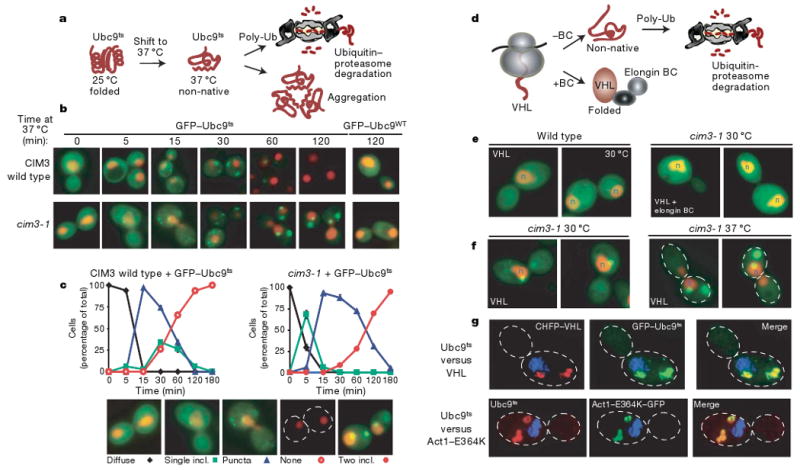
a, The temperature-sensitive mutant of Ubc9(Y68L) (Ubc9ts) is folded and long-lived at 25 °C. After temperature shift to 37 °C, the Ubc9ts protein misfolds and is degraded by the ubiquitin–proteasome pathway. b, Time-dependent changes in localization of folded and misfolded GFP–Ubc9 in wild-type (WT) and cim3-1 cells. Nuclei were visualized by co-expressing NLS–tdTomato (NLS–TFP). Ubc9 expression was shut off by addition of 2% glucose before temperature shift in all experiments. c, Quantification of Ubc9ts localization after misfolding in wild-type and cim3-1 cells. Graphs represent three separate experiments conducted as in b. The phenotypes (see panels) of 100 cells were scored at each time point. d, Quality control of the VHL tumour suppressor. VHL folds after elongin BC binding to form the VBC complex. In the absence of elongin BC, VHL is degraded by the ubiquitin–proteasome pathway25. e, VHL localization in wild-type and cim3-1 cells, and at 30 °C and 37 °C in cim3-1 cells (f). Two panels are shown for each experiment. g, Misfolded VHL, Ubc9 and actin co-localize in the same two inclusions. VHL tagged with mCherry (CHFP–VHL, red) with GFP–Ubc9ts (green, upper panel) or with Act1–E364K–GFP (green, lower panel) in cim3-1 yeast, after 2 h at 37 °C. Images collected as a Z-series and deconvoluted are shown as a two-dimensional projection.
Figure 2. Amyloidogenic proteins are preferentially directed to a single inclusion.
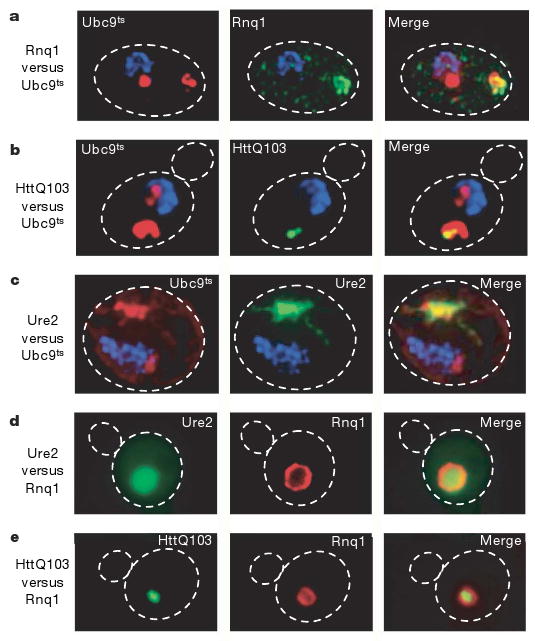
a, Co-localization of inclusions of the yeast prion Rnq1 (green, tagged with GFP) and misfolded Ubc9ts (red, tagged with CHFP), and HttQ103–GFP with CHFP–Ubc9ts(b). Ure2–GFP with CHFP–Ubc9ts (c), in cim3-1 yeast after 2 h at 37 °C. Images were collected as a Z-series and de-convoluted. d, Co-localization of the yeast prions Ure2–GFP and Rnq1–CHFP in the peripheral inclusion. A direct fluorescence image is shown for e. e, Co-localization of HttQ103–GFP with Rnq1–CHFP in the peripheral inclusion.
Two compartments for misfolded cytosolic proteins
To determine the fate of cytosolic misfolded substrates, we initially followed a destabilized Ubc9 variant that misfolds above 30 °C (refs 23, 24; Fig. 1a). Ubc9ts, fused to green fluorescent protein (GFP) to facilitate detection (GFP–Ubc9ts), was expressed under the control of a galactose-regulated promoter. Glucose addition repressed expression, allowing us to follow the fate of GFP–Ubc9ts from the earliest stages of protein misfolding after shift to 37 °C (Fig. 1a). At permissive temperatures, GFP–Ubc9ts was native and diffuse, similar to wild-type GFP–Ubc9 (Fig. 1b, 0 min, compare with wild type panel 120 min). GFP–Ubc9ts misfolding led to degradation by the ubiquitin–proteasome pathway, as reported for untagged Ubc9ts (Fig. 1b, compare 5 min and 60 min; and Fig. 1d, left panel)23,24. During degradation we observed transient accumulation of Ubc9ts in distinct cytosolic puncta and inclusions that were eventually cleared (for example, Fig. 1b, 30 min and Fig. 1c). Most cells contained a juxtanuclear inclusion as well as smaller puncta throughout the cytosol, whereas some cells contained only the juxtanuclear inclusion (Fig. 1b, c). Impairment of proteasome-mediated degradation either in cim3-1 cells or by treatment with the proteasome inhibitor MG132 stabilized GFP–Ubc9ts and led to its reproducible accumulation in two distinct inclusions in virtually every cell (Fig. 1b, 60 min and 120 min and Supplementary Fig. 1a). At early time points after misfolding in proteasome-defective cells, GFP–Ubc9ts accumulated in structures resembling those observed during degradation in control cells (Fig 1b, compare 15 min and 30 min). Quantification indicated that the juxtanuclear inclusion formed first, closely followed by cytosolic puncta (Fig. 1c). However, at later incubation times at 37 °C the juxtanuclear inclusion remained, but the puncta were no longer observed. Instead, a second large perivacuolar inclusion was now formed at the periphery of the cell (Fig. 1b, c). Once formed, both inclusions persisted well beyond the time course shown in Fig. 1. Notably, formation of both inclusions was an active process, as it was reversibly inhibited by the microtubule-depolymerizing drug benomyl (Supplementary Fig. 2). These two inclusions may represent distinct compartments for the sequestration of misfolded proteins.
We next examined other types of cytosolic quality control substrates. We initially followed the unassembled von Hippel-Lindau (VHL) tumour suppressor25,26. VHL only folds after binding to its cofactor elongin BC27 (Fig. 1d). Tumour-causing mutations impairing elongin BC binding, or expression in cells lacking elongin BC, lead to misfolded VHL ubiquitination and degradation25 (Fig. 1d), resulting in reduced levels of diffuse fluorescence (Fig. 1e, compare left panel, misfolded without elongin BC, with right panel, folded VHL with elongin BC). Inhibition of the proteasome in cim3-1 cells (Fig. 1f), or with MG132 (Supplementary Fig. 1c), led to formation of a single juxtanuclear GFP–VHL inclusion. Importantly, proteasome impairment did not produce GFP–VHL inclusions under conditions leading to productive VHL folding (Fig. 1e, plus elongin BC, right panel).
It was puzzling that at 30 °C VHL consistently formed a single juxtanuclear inclusion whereas Ubc9ts formed two distinct inclusions. Ubc9ts destabilization requires thermal stress, hence formation of two inclusions might result from the increased load of denatured quality control substrates at 37 °C. Indeed, when unassembled VHL was expressed at 37 °C it also accumulated in two inclusions as observed for Ubc9ts (Fig. 1f, c). Three-dimensional fluorescence deconvolution microscopy demonstrated that the inclusions formed by VHL and Ubc9ts overlap spatially in the same compartments (Fig. 1g and Supplementary Movie 1).
A missense mutation of actin, actin(E364K), also degraded via the ubiquitin–proteasome pathway25, similarly accumulated in the same inclusions as Ubc9ts (Fig. 1g). As clearance of misfolded Ubc9, VHL and actin requires ubiquitination, we considered whether proteasome impairment or stress cause widespread aggregation of ubiquitinated proteins (Supplementary Fig. 1b). This is not the case, as native substrates of the ubiquitin–proteasome pathway28, such as Arg–GFP (R–GFP), Ub–G76A–GFP (Ub–GFP) and Deg1–GFP (Supplementary Fig. 1b and data not shown), remained soluble and diffuse after proteasome impairment, even under conditions of stress (Supplementary Figs 1b and 3c). We conclude that different classes of misfolded cytosolic proteins are sequestered in two defined cellular inclusions, one juxtanuclear and one at the periphery of the cell. The juxtanuclear inclusion seems to form first and is more prevalent under normal cellular conditions. However, stress conditions lead to protein accumulation in the second peripheral inclusion. In principle, the differential partitioning of non-native quality control substrates between these two compartments may be determined by a change in their intrinsic properties, such as aggregation state, or by their interaction with saturatable quality control components, or both.
We explored the relationship between inclusions formed by disease-related amyloidogenic proteins and those characterized here for misfolded cytosolic proteins (Fig. 2a–c). The relative spatial localization of the aggregates formed by glutamine-rich yeast prion proteins Rnq1 and Ure2, as well as polyQ expanded Huntingtin (HttQ103) relative to the Ubc9ts inclusions was determined by deconvolution microscopy. All the amyloidogenic proteins tested formed an inclusion that consistently co-localized with the perivacuolar peripheral inclusion of Ubc9ts (Fig. 2a–c; Supplementary Movie 2). We did not observe any cases of co-localization of either the prion proteins or Htt with the juxtanuclear inclusion.
Unlike normal quality control substrates, amyloidogenic proteins (including Huntingtin (Htt) and prions) form large insoluble inclusions even in the absence of proteasome inhibition10,13,17,29. Thus, amyloidogenic proteins were also analysed in the absence of proteasome inhibition and under normal growth temperatures (Fig. 2d, e). Rnq1, Ure2 and HttQ103 also accumulated under these normal conditions in aggregates localized exclusively in the peripheral compartment (Fig. 2d, e). Additionally, Rnq1 was also found in small puncta throughout the cell (Fig. 2a). The accumulation of amyloidogenic proteins in the peripheral inclusion in the absence of either stress or proteasome impairment (Fig. 2d, e) indicates that this compartment can also form under normal conditions. Notably, Rnq1 always surrounded the Ure2 and HttQ103 deposits (red fluorescence in Fig. 2d, e), suggesting that Rnq1 is targeted to this perivacuolar compartment with slower kinetics than the other amyloidogenic proteins. These observations suggest that some unique feature of amyloidogenic proteins earmarks them for exclusive delivery to the peripheral inclusion.
Distinct quality control compartments in Mammalian cells
We next determined whether differential sequestration of misfolded and amyloidogenic proteins in distinct quality control compartments is conserved in mammalian cells (Fig. 3, Supplementary Fig. 1d, e). Misfolded Ubc9ts and VHL showed diffuse fluorescence when expressed in untreated HeLa cells (Fig. 3a, upper panel), with VHL more prominent around the endoplasmic reticulum and nucleus. After proteasome inhibition both proteins co-localized in the perinuclear region, as observed in yeast cells (Fig. 3b, lower panel). Importantly, wild-type folded Ubc9 did not co-aggregate with VHL under these conditions (Supplementary Fig. 1d). We then compared the distribution of HttQ103 and misfolded VHL. As observed in yeast cells, HttQ103 and misfolded VHL were generally sequestered in two different inclusions after proteasome inhibition in mammalian cells (Fig. 3b, lower panel; Supplementary Fig. 1e). In the absence of proteasome impairment, VHL was degraded whereas the HttQ103 inclusion was still observed. It thus appears that the differential sequestration of misfolded proteins in two quality control compartments is conserved from yeast to mammals.
Figure 3. Mammalian cells differentially sequester misfolded proteins in two distinct compartments.
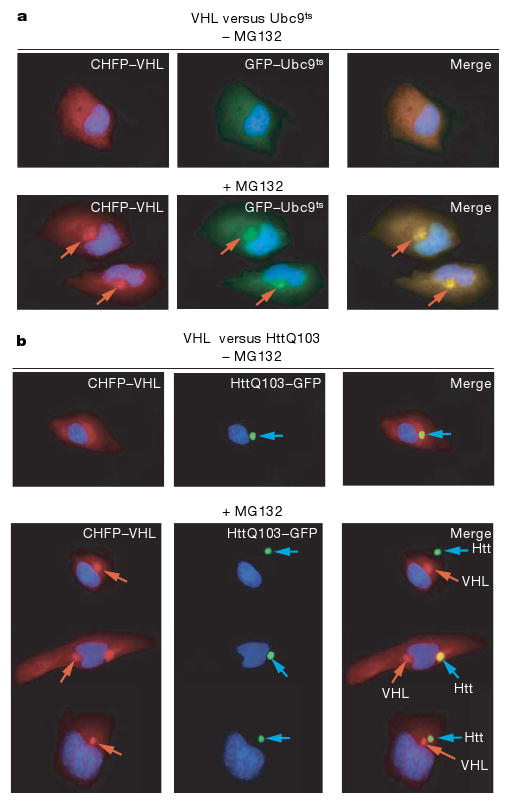
a, CHFP–VHL and GFP–Ubc9ts show diffuse localization in the absence of proteasome inhibition (upper panel), and form co-localizing perinuclear puncta and inclusions next to the endoplasmic reticulum after proteasome inhibition (+MG132, lower panel).
b, HttQ103–GFP forms one hyper-fluorescent inclusion (upper panel, note inclusion not always perinuclear). After proteasome inhibition (+MG132), CHFP–VHL forms an inclusion that is distinct from that of HttQ103–GFP (lower panel).
Quality control compartments show distinct diffusion properties
We next examined the solubility state of misfolded proteins in either inclusion by determining their diffusion properties using Fluorescence Loss in Photobleaching (FLIP)30. In brief, a laser pulse was used to photobleach GFP–Ubc9ts from a small section of cytosol outside of the two GFP–Ubc9ts inclusions (Fig. 4a, square). The ensuing changes in fluorescence intensity of the different cellular compartments, assessed as a function of time, provide a measure of their relative exchange rate with the bleached cytoplasmic portion (Fig. 4a). Bleaching caused a rapid loss of the diffuse cytosolic fluorescence corresponding to soluble GFP–Ubc9ts (Fig. 4a, black trace). A rapid fluorescence loss was also observed for the juxtanuclear inclusion (Fig. 4a, red trace), indicating that a substantial fraction of GFP–Ubc9ts in this compartment is soluble and can exchange with the cytosolic pool. We therefore refer to this inclusion as the ‘juxtanuclear quality control’ compartment, or JUNQ. In contrast, following a small initial reduction in fluorescence, the peripheral perivacuolar compartment retained most (>70%) of its fluorescent signal (Fig. 4a, blue trace). This suggests that this inclusion contains a large fraction of non-diffusing, possibly insoluble GFP–Ubc9ts. Accordingly, we named this inclusion the ‘insoluble protein deposit’, or IPOD. As the conservation of fluorescent signal within the IPOD could result from a barrier to exchange with the cytosolic pool, for example, by a membrane, we examined the internal mobility of the protein within the IPOD using FRAP30. When a small sector within the IPOD was directly bleached, we did not observe any redistribution of the fluorescent signal within the IPOD from the non-bleached part of the inclusion. This indicates that the protein in this structure is immobile, consistent with this compartment containing aggregated species (Fig. 4b).
Figure 4. Differential solubility of misfolded substrates in the distinct quality control compartments.
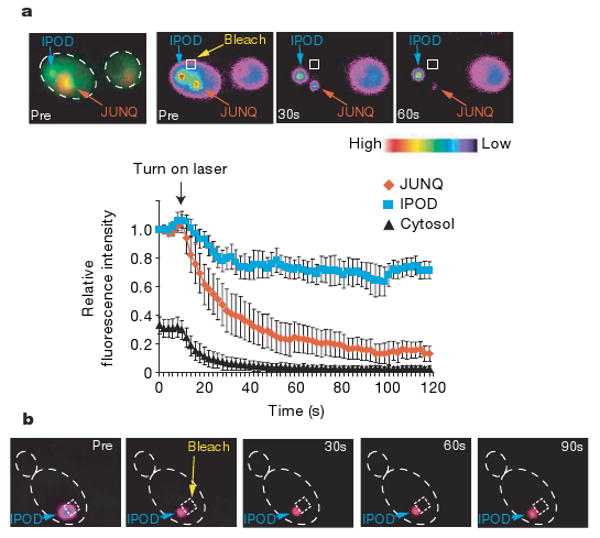
a, Qualitative FLIP analysis indicates that misfolded protein in the JUNQ and the IPOD exhibit different relative exchange rates with the cytosolic pool. Pre- and post-bleach images of a representative FLIP experiment with GFP–Ubc9ts are shown. The fluorescence intensity scale is pseudocoloured as shown. A square designates the location of the photobleaching laser spot. GFP–Ubc9ts was expressed in cim3-1 yeast. Relative fluorescence of JUNQ (orange), IPOD (blue) and cytosol (black) from ten FLIP experiments is shown over time. b, Protein in the IPOD inclusion is immobile. Pre- and post-bleach images of a representative FRAP experiment and subsequent recovery of GFP–Ubc9ts are shown. GFP–Ubc9ts was expressed in Δubc4/5 cells and shifted to 37 °C to form the IPOD.
Biochemical analyses supported the conclusion that the JUNQ and IPOD accumulate proteins in distinct solubility states (Supplementary Fig. 3). VHL localized only to the JUNQ was in a Triton-soluble state (Supplementary Fig. 3a), whereas accumulation in the IPOD correlated with a shift to the insoluble fraction (Supplementary Fig. 3a; see also Fig. 6b and Supplementary Fig. 6g for Ubc9 and Htt). We conclude that one compartment, the JUNQ, contains a large fraction of soluble misfolded protein, whereas the IPOD compartment contains non-diffusing, insoluble species. The observation that amyloidogenic proteins appear to be targeted exclusively to the IPOD suggests that this compartment is the preferred cellular destination for protein aggregates.
Figure 6. Partitioning between JUNQ and IPOD is regulated by ubiquitination.
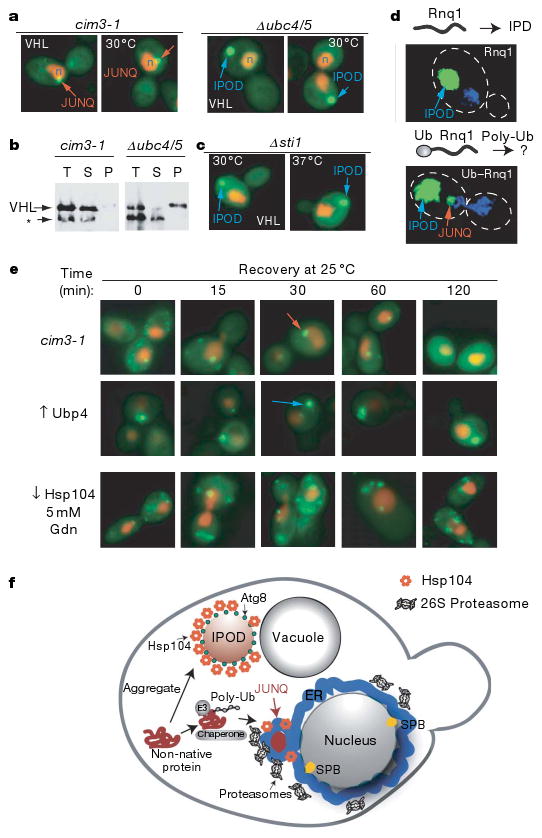
a, Blocking ubiquitination of misfolded VHL prevents its localization to JUNQ, and redirects these proteins to the IPOD. b, VHL in the IPOD accumulates in a Triton-insoluble fraction. Asterisk denotes cross-reacting band unrelated to VHL. c, Deletion of Sti1, required for VHL degradation25, reroutes misfolded VHL to the IPOD. d, Ubiquitination suffices to promote prion delivery to the JUNQ. Rnq1–GFP localizes exclusively to the IPOD. A ubiquitination signal (Ub–G76A) engineered in the yeast prion Rnq1–GFP causes Ub–G76A–Rnq1–GFP localization to both JUNQ and IPOD. e, Recovery of diffuse cytosolic fluorescence by thermally denatured GFP–Ubc9ts accumulated in the JUNQ (top) but not in the IPOD (middle) after return to the permissive temperature. The recovery of GFP–Ubc9ts requires Hsp104 disaggregase activity (bottom). f, Model for sorting of cytosolic misfolded proteins to distinct quality control compartments.
JUNQ and IPOD are defined subcellular compartments
Because different quality control substrates reproducibly accumulated in the same two compartments, we examined the relationship of both JUNQ and IPOD with known cellular structures and components (Fig. 5 and Supplementary Fig. 5). Deconvolution microscopy indicated that the JUNQ is formed in an indentation of the nucleus (Fig. 5a; see also Supplementary Movies 1 and 2). In dividing cells both JUNQ and IPOD were invariably retained in the mother cell, raising the possibility that these compartments provide a mechanism to retain misfolded proteins in the mother cells during cell division (Supplementary Fig. 4a and data not shown). This could explain previous observations indicating that oxidatively damaged proteins are prevented from entering the daughter cell31,32. Notably, neither the JUNQ nor the IPOD were localized to the spindle pole body (Fig. 5b) unlike the aggresome which co-localizes with the microtubule organizing centre20.
Figure 5. The JUNQ and IPOD are defined subcellular compartments.
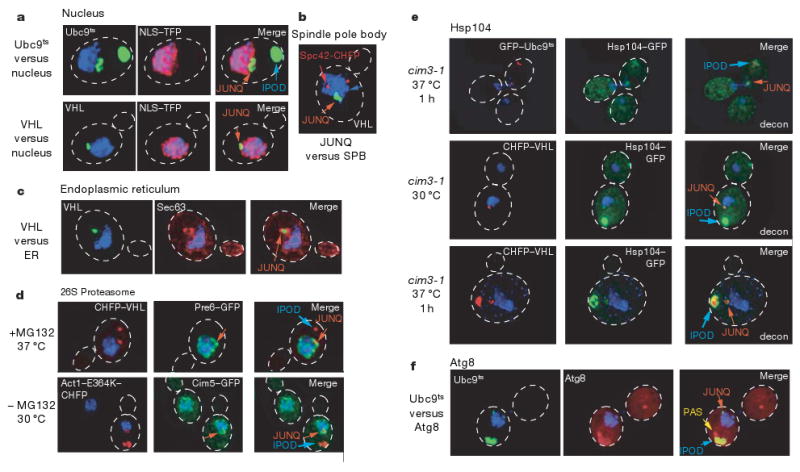
a, JUNQ compartment tightly co-localizes with nuclear membrane. DNA is visualized with DAPI (blue), nucleoplasm with NLS–TFP (red). GFP–Ubc9ts (top) at 37 °C and GFP–VHL (bottom) in cim3-1 cells. Two-dimensional projections of de-convoluted Z-series are shown. b, The JUNQ compartment, shown here for GFP–VHL, does not localize to the spindle pole body (marked by Spc42–CHFP), and is in close proximity to the endoplasmic reticulum visualized with Sec63–CHFP (c). d, The JUNQ compartment (upper panel, CHFP–VHL, red), but not the IPOD (Act1–E364K–CHFP, lower panel), concentrates 26S proteasomes (visualized with Cim5–GFP for regulatory particle and Pre6–GFP for core particle, green). e, Hsp104 localizes to both compartments. JUNQ and IPOD were formed by expressing CHFP–Ubc9ts (upper panel) or CHFP–VHL (lower panel). Note Hsp104 also accumulates in an IPOD structure (blue arrow) independently of ectopically-expressed aggregating protein when CHFP–VHL is expressed in cim3-1 cells at 30 °C (middle panel). f, The IPOD, shown here for GFP–Ubc9ts, co-localizes with CHFP–Atg8. Some CHFP–Atg8 can also be seen in the pre-autophagosomal structure (PAS)12.
A similar analysis using the endoplasmic reticulum marker Sec63 (ref. 19) indicated that the JUNQ is in close proximity to the endoplasmic reticulum. Sec63 redistributed around the JUNQ relative to the remaining nuclear envelope (Fig. 5c; Supplementary Fig. 4). Sec63 usually marks the ERAC structures that accumulate ERAD substrates19, suggesting that the JUNQ forms at a defined cellular location in close proximity to the region that participates in ERAD. Perhaps the localization of cytosolic and endoplasmic reticulum misfolded proteins to one cellular location serves to concentrate cellular quality control components with their substrates to both enhance the efficiency of misfolded protein clearance and sequester them from the cellular milieu.
Given the centrality of proteasomal degradation in protein turnover, we examined the cellular distribution of 26S proteasomes using previously characterized GFP-tagged proteasomes (Fig. 5d and Supplementary Fig. 4c–e)33. Deconvolution microscopy revealed that most proteasomes in the cells localize to the endoplasmic reticulum surface, even though there is a fraction of diffuse proteasomes. For all misfolded proteins examined, we observed a re-distribution of proteasomes to the site of JUNQ protein accumulation both at 30 °C and 37 °C (Fig. 5d; Supplementary Fig. 4c–e). Mutant actin occasionally accumulated in the quality control compartments even without proteasome inhibition (Fig. 5d, bottom). This also led to proteasome re-distribution to the JUNQ, suggesting that misfolded protein accumulation can recruit proteasomes to the JUNQ. In contrast, proteasomes did not co-localize with the IPOD, indicating that soluble misfolded proteins, rather than insoluble amyloid aggregates, cause a re-distribution of cellular proteasomes. Our data suggest that the perinuclear JUNQ compartment acts as a major site of proteasome concentration and misfolded protein degradation.
The chaperone Hsp104, another quality control component that interacts with misfolded and aggregated proteins, was examined using a functional GFP-tagged Hsp104 (ref. 34). Hsp104 co-localized with both JUNQ and IPOD (Fig. 5e; Supplementary Fig. 4e–g). Most Hsp104 accumulated around the IPOD compartment, often in an arrangement around the protein inclusion (Fig. 5e). Hsp104 was often found in IPOD- or JUNQ- like inclusions in the absence of ectopically expressed misfolded proteins (Fig. 5e, middle panel, and data not shown), indicating that these quality control compartments are normally present in cells. The co-localization of Hsp104 with the IPOD resonates with its role in disaggregating or fragmenting aggregates of prion proteins4, which also localize to this compartment, and with its role in preventing inheritance of oxidatively damaged proteins31,32. At the JUNQ, Hsp104 may serve to keep proteins soluble for either refolding or degradation (Fig. 6e).
Autophagy has been implicated in the clearance of protein aggregates35 and possibly also ubiquitinated misfolded proteins36–38. The IPOD, but not the JUNQ, co-localized with the autophagic marker Atg8 (ref. 12) and the pre-autophagosomal structure (PAS; Fig. 5f; see also Supplementary Fig. 5 for electron microscopy). The association of the IPOD with an autophagic marker (Supplementary Fig. 5b for electron microscopy) provides an intriguing link between aggregated proteins in the IPOD and the autophagy pathway. Neither Atg8 nor Atg7, both essential components of the autophagic pathway, were required for IPOD formation (Supplementary Fig. 5c). Thus, it is also possible that that Atg8 co-localization has an autophagy-independent function in the IPOD, as recently proposed for its mammalian homologue LC3 (refs 36–38).
Immunoelectron microscopy analysis confirmed that the JUNQ is closely associated with the nucleus, and may be flanked by proliferations of the nuclear membrane (Supplementary Fig. 5a, b). The IPOD (Supplementary Fig. 5a) was made up of electron-dense material, consistent with our FRAP and biochemical characterization. Occasionally, both electron microscopy and fluorescence analyses of prion IPODs showed labelling in a circular hollow pattern around a densely packed core (Supplementary Fig. 5a, Ure2–GFP). We hypothesize that the ectopically expressed prions are occasionally layered over aggregates in pre-existing IPODs containing endogenous proteins (for example, Hsp104 in Fig. 5e).
Role of ubiquitination in substrate partitioning
We considered whether ubiquitination of misfolded proteins15 plays a role in their partitioning to either the JUNQ or the IPOD (Fig. 6 and Supplementary Fig. 6a). Degradation of misfolded VHL, Ubc9 and actin requires ubiquitination by the E2 pair Ubc4/5 (refs 23, 25 and 26). Ubiquitination was impaired by expressing these proteins in Δubc4/5 cells (Fig. 6a, VHL; Supplementary Fig. 6,Ubc9ts; actin, data not shown) or by overexpression of Ubp4 (ref. 39; Supplementary Fig. 6e). Similar results were obtained using either strategy for all misfolded proteins (Fig. 6a; Supplementary Fig. 6b, c). Impairing misfolded protein ubiquitination blocked their accumulation in the JUNQ and instead resulted in exclusive accumulation in the IPOD, even at 30 °C and in the absence of proteasome inhibition (Fig. 6, Supplementary Fig. 6). The IPODs formed under these conditions exhibited the same morphology as observed previously, and were also Atg8 and Hsp104-positive (Supplementary Figs 6d and 7e).
Consistent with its relocalization to the IPOD, blocking ubiquitination reduced the solubility of misfolded VHL (Fig. 6b) and Ubc9 (Supplementary Fig. 6g). Misfolded VHL was Triton-soluble under conditions where it only forms the JUNQ (Fig. 6c, left panel), but was almost entirely insoluble when targeted to the IPOD once ubiquitination was blocked (Fig. 6b, right panel). Similar results were obtained with Ubc9ts; blocking its ubiquitination rendered it as insoluble as aggregated HttQ53 (Supplementary Fig. 6g). Alternatively, blocking the ubiquitination of a native degradation substrate, Ub–GFP, in the same Δubc4/5 cells did not impair its solubility (Supplementary Fig. 3c). Thus, ubiquitination is an important determinant for maintaining solubility of misfolded proteins and sorting them to the JUNQ, whereas non-ubiquitinated species are directed to the IPOD.
We next exploited the observation that the chaperone Sti1 is required for VHL degradation, but not for VHL folding25. Sti1 deletion also directed VHL to the IPOD, as observed in Δubc4/5 cells (Supplementary Fig. 7d, e), but had no effect on the localization of Ubc9ts (Supplementary Fig. 7c). This result indicates that ubiquitination and partitioning of misfolded proteins between JUNQ and IPOD is modulated by specific interactions with the cellular chaperone network. The finding that amyloidogenic proteins are primarily targeted to the IPOD may thus reflect their inefficient interaction with quality control chaperone and ubiquitination components. This property could distinguish prions and other amyloidogenic proteins from the bulk of misfolded quality control substrates that normally do not accumulate in amyloids.
Unexpectedly, blocking the ubiquitination of misfolded proteins caused them to behave like amyloidogenic proteins. We considered whether enhancing ubiquitination of the prion Rnq1 by engineering a synthetic ubiquitination signal suffices to promote partitioning to the JUNQ (Fig. 6d, Supplementary Fig. 8). Rnq1 normally only accumulates in IPOD inclusions (Figs 2b–f and 6d, left panel), but enhancing its ubiquitination with an amino-terminal ubiquitin-fusion degradation (UFD) signal (Supplementary Fig. 8a) directed a fraction of Ub–Rnq1 to the JUNQ (Fig. 6d, right panel). Biochemical analysis confirmed this observation (Supplementary Fig. 8b). Rnq1 is predominantly in the insoluble fraction (Supplementary Fig. 8b, left panel) whereas Ub–Rnq1 is distributed between soluble and insoluble fractions (Supplementary Fig. 8b, right panel). Notably, higher molecular weight poly-ubiquitinated Ub–Rnq1 was found only in the soluble fraction. As non-ubiquitinated Ub–Rnq1 nevertheless has one N-terminal ubiquitin, it seems that poly-ubiquitination is required for Rnq1 delivery to the JUNQ. These experiments indicate that poly-ubiquitination is a key determinant for partitioning misfolded proteins between the two quality control compartments, as it is necessary for sorting misfolded proteins to the JUNQ and sufficient to redirect a prion protein from the IPOD to the JUNQ.
Consequences of protein accumulation in the JUNQ or IPOD
We next examined the functional consequences of accumulation in the JUNQ or the IPOD (Fig. 6e) by exploiting the observation that the thermal denaturation of Ubc9ts is reversible (data not shown)24. Thermally denatured GFP–Ubc9ts was directed to either the JUNQ or the IPOD by changing its ubiquitination state; the fate of Ubc9ts in either compartment was then examined after return to the permissive temperature (Fig. 6e). Cells containing Ubc9ts in puncta and the JUNQ gradually recovered diffuse GFP–Ubc9ts fluorescence (Fig. 6e, top), In contrast, cells with GFP–Ubc9ts in the IPOD did not recover diffuse fluorescence (Fig. 6e, data not shown for Δubc4/5). Ubc9ts refolding from the JUNQ required Hsp104 and was inhibited by 5 mM guanidine, suggesting that Hsp104 facilitates refolding of proteins in the JUNQ. Thus, misfolded Ubc9ts that is ubiquitinated and sorted into the JUNQ can be refolded by the cellular chaperone machinery, whereas Ubc9ts sorted to the IPOD is terminally sequestered from the cytoplasm. These experiments suggest that poly-ubiquitination not only targets proteins for degradation, but may also contribute to their re-folding competence.
Discussion
Cellular quality control networks have a key role in maintaining protein homeostasis40. We find two cytosolic quality control compartments, the JUNQ and the IPOD, carry out general but distinct functions managing misfolded and aggregated proteins and are conserved from yeast to mammalian cells (Fig. 6f). After misfolding, most proteins will be recognized and ubiquitinated by the quality control machinery, which directs them to the JUNQ, a region that concentrates disaggregating chaperones and 26S proteasomes and is in close proximity to the perinuclear endoplasmic reticulum region involved in ERAD (Fig. 6f). Accumulation of misfolded proteins and quality control components at the endoplasmic reticulum membrane may facilitate both degradation and refolding by increasing their local concentrations and enhancing encounter rates by restricting diffusion. Cellular increase of misfolded protein loads, for example, by stress or during ageing, may saturate the quality control machinery needed for sorting to the JUNQ, resulting in accumulation of aggregated and potentially toxic species. These misfolded proteins are directed to the IPOD, which seems to terminally sequester protein aggregates. The spatial sequestration of these aggregates from the site where most proteasomal degradation takes place may serve a protective function. Spatial sequestration may also facilitate aggregate clearance, either through the autophagic pathway or by dilution through retention in the dividing mother cell.
The identification of two distinct quality control compartments resonates with a number of previous observations. Studies in Caenorhabditis elegans suggest two hierarchical pathways for degradation of amyloidogenic Aβ species41,42. Alternative degradation pathways were reported for ERAD substrates21 and α-synuclein43, with soluble species of the same protein degraded via the proteasome and insoluble aggregates by autophagy43,44. For mammalian cells, there have been disparate reports on the solubility and structural properties of protein inclusions13,16,18,36. Thus, polyQ inclusions in mammalian cells are immobile17, similar to the IPOD in yeast. The distinct localization and solubility of misfolded proteins in the JUNQ also resembles observations that mammalian SOD1 inclusions contain soluble protein and are spatially distinct from polyQ aggregates13,16. Soluble misfolded VHL is also reported to accumulate in an ubiquitination-dependent manner on the cytosolic surface of the mammalian endoplasmic reticulum, similar to our findings in yeast13,16. Importantly, our findings provide a framework for integrating these various observations into a conserved cellular pathway of quality control. Further, we identify solubility and the ubiquitination state of a quality control substrate as key determinants of its delivery to either the JUNQ or the IPOD. Future studies should determine whether poly-ubiquitination exerts these effects through interactions with cellular components or by directly enhancing protein solubility.
The preferential targeting of amyloidogenic proteins to the IPOD may arise from their reduced affinity for quality control components. In turn, this may lead to higher levels of toxic misfolded conformations, consistent with findings that the toxicity of amyloidogenic proteins resides in small soluble species45. Accordingly, overexpression of chaperones and ubiquitination components, which alleviate toxicity, could compensate for their reduced affinity for the amyloidogenic species4,22,46,47. The observation that amyloidogenic and globular misfolded proteins are differentially engaged by cellular quality control pathways provides new perspectives on the molecular basis of protein conformation diseases, and may have useful implications for the understanding of neurodegeneration, ageing and stress.
Methods Summary
Yeast growth, manipulation and protein expression were performed as described25,48,49. Misfolded and amyloidogenic proteins used in this study were tagged with EGFP, mCherry or tdTomato50. Conventional epifluorescence micrographs were obtained from live yeast cells on a Zeiss Axiovert microscope with a ×100 oil lens (NA1.4). Deconvoluted images were acquired using an Olympus microscope. Digital images (12 bit) were digitally deconvoluted by using DELTAVISION software (Applied Precision). Live-cell imaging was performed using the Marianas system from Intelligent Imaging Innovations equipped with the MicroPoint FRAP laser system (Photonic Instruments).
Supplementary Material
Acknowledgments
We thank R. Tsien, S. Michaelis, J. Glover, C. Enenkel and V. Albanese for reagents; J. Mulholland for electron microscopy and deconvolution microscopy help; S. Yamada and W. J. Nelson for help with the live cell microscopy. We thank R. Andino, W. Burkholder, J. England, R. Geller, M. Kaganovich, E. Bennett, J. Nelson and members of the Frydman laboratory for discussions and comments on the manuscript. This work was supported by grants from NIH to R.K. and J.F.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions J.F. directed the project; D.K. and J.F. designed and interpreted all experiments; R.K. participated in the initial fluorescence microscopy experiments; D.K. performed all experiments. D.K. and J.F. wrote the paper.
References
- 1.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 4.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nature Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 5.Outeiro TF, Lindquist S. Yeast cells provide insight into α-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaffar G, et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell. 2004;15:95–105. doi: 10.1016/j.molcel.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 7.Lesne S, et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 8.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 9.Iwata A, et al. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc Natl Acad Sci USA. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor JP, et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 11.Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 12.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsumoto G, Kim S, Morimoto RI. Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J Biol Chem. 2006;281:4477–4485. doi: 10.1074/jbc.M509201200. [DOI] [PubMed] [Google Scholar]
- 14.Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 15.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto G, Stojanovic A, Holmberg CI, Kim S, Morimoto RI. Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J Cell Biol. 2005;171:75–85. doi: 10.1083/jcb.200504050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci USA. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamhi-Nesher S, et al. A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell. 2001;12:1711–1723. doi: 10.1091/mbc.12.6.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huyer G, et al. A striking quality control subcompartment in Saccharomyces cerevisiae: the endoplasmic reticulum-associated compartment. Mol Biol Cell. 2004;15:908–921. doi: 10.1091/mbc.E03-07-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 21.Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human α-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–212. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nature Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- 23.Betting J, Seufert W. A yeast Ubc9 mutant protein with temperature-sensitive in vivo function is subject to conditional proteolysis by a ubiquitin- and proteasome-dependent pathway. J Biol Chem. 1996;271:25790–25796. doi: 10.1074/jbc.271.42.25790. [DOI] [PubMed] [Google Scholar]
- 24.Tongaonkar P, Beck K, Shinde UP, Madura K. Characterization of a temperature-sensitive mutant of a ubiquitin-conjugating enzyme and its use as a heat-inducible degradation signal. Anal Biochem. 1999;272:263–269. doi: 10.1006/abio.1999.4190. [DOI] [PubMed] [Google Scholar]
- 25.McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 26.Vang S, et al. Actin mutations in hypertrophic and dilated cardiomyopathy cause inefficient protein folding and perturbed filament formation. FEBS J. 2005;272:2037–2049. doi: 10.1111/j.1742-4658.2005.04630.x. [DOI] [PubMed] [Google Scholar]
- 27.Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J. Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell. 1999;4:1051–1061. doi: 10.1016/s1097-2765(00)80233-6. [DOI] [PubMed] [Google Scholar]
- 28.Mateus C, Avery SV. Destabilized green fluorescent protein for monitoring dynamic changes in yeast gene expression with flow cytometry. Yeast. 2000;16:1313–1323. doi: 10.1002/1097-0061(200010)16:14<1313::AID-YEA626>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 29.Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. Proc Natl Acad Sci USA. 2006;103:11051–11056. doi: 10.1073/pnas.0604548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lippincott-Schwartz J, Patterson GH. Development and use of fluorescent protein markers in living cells. Science. 2003;300:87–91. doi: 10.1126/science.1082520. [DOI] [PubMed] [Google Scholar]
- 31.Rujano MA, et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 2006;4:e417. doi: 10.1371/journal.pbio.0040417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erjavec N, Nystrom T. Sir2p-dependent protein segregation gives rise to a superior reactive oxygen species management in the progeny of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2007;104:10877–10881. doi: 10.1073/pnas.0701634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enenkel C, Lehmann A, Kloetzel PM. Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelope-ER network in yeast. EMBO J. 1998;17:6144–6154. doi: 10.1093/emboj/17.21.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tkach JM, Glover JR. Amino acid substitutions in the C-terminal AAA1 module of Hsp104 prevent substrate recognition by disrupting oligomerization and cause high temperature inactivation. J Biol Chem. 2004;279:35692–35701. doi: 10.1074/jbc.M400782200. [DOI] [PubMed] [Google Scholar]
- 35.Sarkar S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nature Chem Biol. 2007;3:331–338. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szeto J, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2:189–199. doi: 10.4161/auto.2731. [DOI] [PubMed] [Google Scholar]
- 37.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 38.Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 39.Swaminathan S, Amerik AY, Hochstrasser M. The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol Biol Cell. 1999;10:2583–2594. doi: 10.1091/mbc.10.8.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 41.Siegel SJ, Bieschke J, Powers ET, Kelly JW. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry. 2007;46:1503–1510. doi: 10.1021/bi061853s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 43.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. α-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 44.Kruse KB, Brodsky JL, McCracken AA. Autophagy: an ER protein quality control process. Autophagy. 2006;2:135–137. doi: 10.4161/auto.2.2.2388. [DOI] [PubMed] [Google Scholar]
- 45.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 46.Barral JM, Broadley SA, Schaffar G, Hartl FU. Roles of molecular chaperones in protein misfolding diseases. Semin Cell Dev Biol. 2004;15:17–29. doi: 10.1016/j.semcdb.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 47.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nature Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Melville MW, McClellan AJ, Meyer AS, Darveau A, Frydman J. The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Mol Cell Biol. 2003;23:3141–3151. doi: 10.1128/MCB.23.9.3141-3151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams A, Gottschling D, Kaiser C, Stearns T. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press; 1997. [Google Scholar]
- 50.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp red fluorescent protein. Nature Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.