

Abstract
AIM: To investigate the molecular mechanism and functional consequences of heme oxygenase-1 (HO-1) activation by lansoprazole in endothelial cells and macrophages.
METHODS: Expression of HO-1 mRNA was analyzed by Northern blotting. Western blotting was used to determine the HO-1 and ferritin protein levels. NADPH-dependent reactive oxygen species (ROS) formation was measured with lucigenin-enhanced chemiluminescence. HO-1 promoter activity in mouse fibroblasts, stably transfected with a 15-kb HO-1 gene that drives expression of the reporter gene luciferase, was assessed using in vivo bioluminescence imaging.
RESULTS: Lansoprazole increased HO-1 mRNA levels in endothelial cells and HO-1 protein levels in macrophages. In addition, lansoprazole-induced ferritin protein levels in both cell systems. Moreover, induction of the antioxidant proteins HO-1 and ferritin by lansoprazole was followed by a decrease in NADPH-mediated ROS formation. The radical scavenging properties of lansoprazole were diminished in the presence of the HO inhibitor, chromium mesoporphyrin IX. Induction of HO-1 gene expression by lansoprazole was not related to oxidative stress or to the activation of the mitogen-activated protein kinase pathway. However, the phosphatidylinositol 3-kinase inhibitor LY294002 showed a concentration-dependent inhibition of HO-1 mRNA and promoter activity.
CONCLUSION: Activation of HO-1 and ferritin may account for the gastric protection of lansoprazole and is dependent on a pathway blocked by LY294002.
Keywords: Antioxidants, Ferritin, Heme oxygenase-1, Lansoprazole, Reactive oxygen species
INTRODUCTION
Peptic ulcer disease remains common worldwide. It is caused most frequently by stress, alcohol, Helicobacter pylori (H pylori) infection, or the use of non-steroidal anti-inflammatory drugs (NSAIDs). It is believed that the integrity of the gastric mucosa depends on a delicate balance between aggressive and defensive mechanisms. Although the cellular and molecular bases of gastric mucosal defense are well understood, the mechanisms by which mucosal damage is mediated by aggressive factors remain largely unclear. The increased production of reactive oxygen species (ROS), termed oxidative stress, is considered to be a major causative factor for mucosal lesions induced by stress[1], NSAIDs[2], and H pylori[3].
Proton pump inhibitors (PPIs) are the drugs of choice for the therapeutic control of gastroesophageal reflux disease, peptic ulcer disease, and eradication of H pylori, and as mucosal protective agents when using NSAIDs. They have been shown to be more effective than other anti-secretory drugs (i.e. H2 receptor antagonists) in the management of acid-related diseases[4]. The anti-secretory action is mediated by their irreversible inhibition of H+/K+-ATPase, the terminal proton pump of parietal cells[5]. However, emerging evidence suggests that the benefit of PPIs is not only mediated by their potent blockade of the gastric H+/K+-ATPase, but also by their ability to provide anti-inflammatory, anti-apoptotic, and antioxidative effects[6,7]. By scavenging ROS and thus protecting the mucosa, these pleiotropic effects of PPIs may be responsible for maintaining the anatomical and functional integrity of the gastric mucosa. In addition to a variety of endogenous factors such as prostaglandins, nitric oxide, and sulfhydryl compounds, which have been proposed to account for the gastroprotective effects of PPIs[7,8], we previously have shown that PPIs also induce the antioxidant protein heme oxygenase-1 (HO-1) in gastric epithelial and endothelial cells[9].
The inducible stress protein HO-1 is the rate-limiting enzyme involved in heme breakdown to generate equimolar amounts of biliverdin, free iron, and CO[10]. Biliverdin is subsequently converted by biliverdin reductase to bilirubin, which is a potent free radical scavenger and acts as a strong endogenous antioxidant[11]. CO has been recognized as a protective agent in hemorrhagic shock and as a modulator of vascular tone. Moreover, it shows anti-apoptotic and anti-inflammatory activity[12]. The HO-1-dependent release of free iron leads to the expression of a second antioxidant protein, ferritin[13]. Ferritin rapidly sequesters free cytosolic iron, thus limiting the formation of oxygen-centered radicals via the Fenton reaction. Thereby, ferritin has emerged as a critical and fast-acting endogenous cytoprotectant that plays an important role in cellular antioxidant defense mechanisms[13].
The activation of the HO-1 gene is regulated primarily at the level of transcription involving various signaling pathways. In particular, phosphorylation-dependent signaling cascades that bind to the transcription factors regulating the HO-1 gene seem to play a key role in HO-1 gene stimulation. In an inducer- and cell-specific fashion, signaling pathways that are implicated in regulating HO-1 gene expression are those important for proliferation and cell survival. Many studies have focused on the activation of the mitogen-activated protein kinases (MAPKs). Recently, other investigators have demonstrated a link between the phosphatidylinositol 3-kinase (PI3K) cell survival pathway and regulation of the HO-1 gene. Some reports suggest a role of cAMP-dependent protein kinase A, protein kinase C, cGMP-dependent PKG, or tyrosine kinases in HO-1 transcriptional regulation[14].
The aim of this study was to elucidate the mechanism of gastric protection by PPIs beyond their effective acid reduction properties, using lansoprazole as a model compound. We focused on the activation of the antioxidant proteins, HO-1 and ferritin, by lansoprazole in cell systems that lack the actual PPI target, the H+/K+-ATPase pump. We then further assessed the underlying mechanism of the upregulation of HO-1 by lansoprazole.
MATERIALS AND METHODS
Materials
Fetal bovine serum (FBS), cell culture media, and penicillin and streptomycin were obtained from GIBCO (Eggenstein, Germany). Chemiluminescence Western Blotting Kit and D-luciferin were purchased from GE Healthcare (Freiburg, Germany) and BioSynth (Naperville, IL, USA), respectively. Wortmannin and primary HO-1 antibody were obtained from Axxora (Grünberg, Germany). PeqGOLD TriFast was purchased from Peqlab (Erlangen, Germany). Chromium mesoporphyrin IX (CrMP) was purchased from Frontier Scientific (Carnforth, UK). For HO-1 probes, the template was an EcoRI restriction fragment of the human HO-1 cDNA (clone 2/10), which was kindly provided by Dr. Rex Tyrrell (University of Bath, UK)[15]. The polyclonal antibody against human ferritin and all other chemicals were obtained from Sigma (Taufkirchen, Germany). The stock solutions of lansoprazole (300 mmol/L), ranitidine (300 mmol/L), MAPK inhibitors [SB203580 (22 mmol/L), SB202190 (20 mmol/L), SP 600125 (45 mmol/L), PD 098059 (22.5 mmol/L)], and PI3K inhibitors [LY294002 (20 mmol/L), wortmannin (100 μmol/L)] were all prepared in dimethyl sulfoxide and stored at -20°C in the dark. The stock solution of CrMP (10 mmol/L) was prepared by dissolving 6.5 mg in 50 μL 2 mol/L NaOH, adjusted to 1.0 mL with sterile water, and stored at -20°C in the dark until use or for up to 12 mo.
Cell culture
Human endothelial-like ECV304 cells were obtained from the European Collection of Cell Cultures (Salisbury, UK). ECV304 cells have been used as a convenient model for the study of vascular endothelial cells[16,17], because they show endothelium-like properties such as producing endothelium specific Weibel-Palade bodies, endothelium-related antigens and angiotensin-converting enzyme, in addition to having human-umbilical-vein-endothelial-cell-like morphology[18,19]. Cells were grown in M199 medium that contained 10% FBS, streptomycin (100 μg/mL), and penicillin (100 U/mL). Murine bone-marrow-derived macrophages (J774), obtained from the American Type Culture Collection (Manassas, VA, USA), were maintained in Dulbecco’s Minimal Essential Medium (DMEM) that contained 10% FBS, streptomycin (100 μg/mL), and penicillin (100 U/mL). Embryonic mouse fibroblast cells (NIH3T3-HO-1-luc), stably transfected with a transgene that contained the full-length (15 kb) mouse HO-1 promoter that drives expression of the reporter gene luciferase, were grown in DMEM that contained 10% FBS, streptomycin (100 μg/mL), and penicillin (100 U/mL). All cells were maintained in a humidified incubator at 37°C and 5% CO2.
HO-1 mRNA analysis
Sub-confluent endothelial cells in 150-mm culture dishes were incubated for 8 h in the presence of control medium, lansoprazole, or ranitidine. Superoxide dismutase (SOD), as well as PI3K and MAPK inhibitors, was added 20 min before lansoprazole treatment. Total RNA was extracted using Trizol reagent according to manufacturer’s instructions (Peqlab). For Northern blotting, samples that contained equal amounts of RNA (20-30 μg) were separated on a 1% denaturing formaldehyde gel and then transferred onto a positively charged nylon membrane by vacuum (500 mbar). The transferred RNA was fixed by baking at 80°C for 30 min. After incubation, membranes were hybridized with a randomly primed 32P-labeled human HO-1 cDNA probe[15] overnight at 65°C. Equal loading of RNA was assessed by staining 18S and 28S rRNAs with ethidium bromide and by a second hybridization using a 32P-labeled β-actin cDNA probe.
Formation of ROS
NADPH-dependent ROS formation was measured by monitoring lucigenin-derived chemiluminescence at 37°C using a Berthold LB96V luminometer according to previously published protocols[20]. Cells were first cultured in six-well plates. After pretreatment with lansoprazole or ranitidine for 12 h, cells were washed and suspended in PBS and then lucigenin (5 μmol/L) and NADPH (10 μmol/L) were added. CrMP (3 μmol/L) was added 20 min before lansoprazole treatment. Chemiluminescence was measured in relative light units (RLU) every 5 min over a period of 20 min. Data were expressed as the mean of peak values in the 20-min measurement normalized to the maximal light emission (RLUmax%) of time-matched NADPH-treated control cells.
HO-1 and ferritin protein analysis
ECV304 cells and macrophages were cultured in 100-mm dishes as described above. After 4-24 h of incubation with control medium or lansoprazole, cells were washed and extracted as described previously[21]. One hundred micrograms of HO-1 protein or 20 μg ferritin protein were separated by SDS-PAGE. Proteins were then transferred to a nitrocellulose membrane and incubated with a polyclonal antibody to HO-1 or ferritin. Antigen/antibody complexes were visualized using a horseradish peroxidase chemiluminescence system according to the manufacturer’s instructions. The densitometric quantitation was performed using Quantity One Basic software (Bio-Rad, USA).
In vivo HO-1 promoter activity
NIH3T3-HO-1-luc cells, stably transfected with a 15-kb HO-1 gene upstream of the transcription initiation site that drives expression of the reporter gene luciferase, were treated with control medium or lansoprazole. PI3K and MAPK inhibitors were added 20 min before lansoprazole. After 24 h incubation, luciferin (300 μg/mL) was added to the cells. Light emission, used as an index of HO-1 promoter activity in living cells, was collected using the In Vivo Imaging System (IVIS™, Caliper Life Sciences, Alameda, CA, USA), quantitated using LivingImage software (Caliper Life Sciences), and expressed as photons emitted/5 min, as previously described[22].
Statistical analysis
Results are expressed as mean ± SD. Data were analyzed using ANOVA and by Bonferroni’s correction for multiple comparisons. All statistical calculations were performed using GraphPad Prism 3.02. Differences were considered significant at P < 0.05. Analyses were based on three to six independent experiments using different cell passages on different days.
RESULTS
Effect of lansoprazole and ranitidine on HO-1 mRNA levels
In endothelial cells, the effects of ranitidine (an H2 receptor antagonist) and lansoprazole on HO-1 mRNA levels were compared following 8 h incubation with each compound. Compared to untreated control cells, ranitidine treatment did not affect HO-1 mRNA levels at any concentration (Figure 1). In contrast, lansoprazole treatment elevated HO-1 mRNA levels in a concentration-dependent manner with nearly a sixfold induction at 100 μmol/L. This increase in HO-1 mRNA levels was comparable to the induction observed following treatment with the positive control CdCl2 (10 μmol/L).
Figure 1.

Effect of ranitidine and lansoprazole on HO-1 mRNA induction in endothelial cells after 8 h of incubation. Fold induction from control levels is shown as mean ± SD of three separate experiments. aP < 0.05 vs control. Treatment with CdCl2 was used as a positive control. Representative Northern blotting analysis is shown in the upper panel.
Effect of lansoprazole on HO-1 protein expression in macrophages
When we investigated the effect of lansoprazole (5-100 μmol/L) on HO-1 protein expression in J774 macrophages, a cell system contributing positively to the mucosal defense, a significant 1.75-2.2-fold induction in HO-1 protein levels was found following 12 h of incubation with lansoprazole at concentrations of 30, 50 and 100 μmol/L (Figure 2).
Figure 2.

Effect of lansoprazole on HO-1 protein expression in macrophages (J774 cells) after 12 h of incubation. Fold induction from control levels is shown as mean ± SD of three to six separate experiments. aP < 0.05 vs control. Representative Western blotting analysis is shown in the upper panel.
Effect of ranitidine and lansoprazole on NADPH-mediated ROS formation
Activated phagocytes represent a major source of ROS production and are therefore used to investigate oxidative stress in cell systems. To analyze the free-radical scavenging effects of the anti-secretory drugs ranitidine and lansoprazole, macrophages were preincubated with these compounds for 12 h. Ranitidine (30 μmol/L) did not affect NADPH-mediated ROS formation (Figure 3). In contrast, lansoprazole (30 μmol/L) significantly decreased NADPH-mediated ROS production (20%). To examine whether HO-1 accounted for the cell protection mediated by lansoprazole, the selective HO inhibitor CrMP was used. CrMP (3 μmol/L) eliminated the antioxidant action of lansoprazole against NADPH-mediated free-radical formation. No change in NADPH-mediated ROS formation was observed in cells treated with CrMP alone.
Figure 3.

Lansoprazole, but not ranitidine, decreased NADPH-dependent ROS formation in macrophages. This effect was reversed in the presence of CrMP. Measurements of lucigenin-enhanced chemiluminescence were performed. aP < 0.05 vs control; cP < 0.05 vs lansoprazole alone. Data are shown as mean ± SD of three to six separate experiments.
Effect of lansoprazole on ferritin protein expression in endothelial cells and macrophages
Increased ferritin protein levels were found in tandem with increased HO-1 protein in macrophages in a concentration-dependent manner after 12 h of incubation with lansoprazole (Figure 4A). Ferritin protein expression in endothelial cells also increased in a time-dependent manner following exposure to lansoprazole (Figure 4B). The induction of ferritin protein expression occurred at a lansoprazole concentration of 100 μmol/L, which significantly induced HO-1 mRNA and protein levels.
Figure 4.

Lansoprazole increased ferritin protein expression in a concentration- and time-dependent manner in macrophages (A) and endothelial cells (B). Data are shown as mean ± SD of three to six separate experiments. aP < 0.05 vs control. A representative Western blotting analysis is shown in the upper panel.
Regulation of the lansoprazole-induced HO-1 gene expression: effect of oxidative stress
Since HO-1 is highly induced by oxidative stress, we evaluated the role of superoxide anion in the PPI-mediated upregulation of HO-1 mRNA. Endothelial cells were treated with lansoprazole (30-100 μmol/L) in the presence of SOD for 8 h (Figure 5). Preincubation with SOD (15 U/mL) did not change the basal or lansoprazole-mediated increases in HO-1 mRNA levels. These results indicate that superoxide anion is not involved in the regulation of HO-1 expression by lansoprazole.
Figure 5.

Effect of SOD on lansoprazole-induced HO-1 mRNA expression. After pretreatment with SOD for 20 min, endothelial cells were incubated with lansoprazole for 8 h. Fold induction compared to control is shown as mean ± SD of three separate experiments. aP < 0.05 vs control. A representative Northern blotting analysis is shown in the upper panel.
Effect of MAPK inhibitors on the lansoprazole-induced HO-1 gene activation
Induction of HO-1 mRNA in endothelial cells after treatment with lansoprazole for 8 h was not influenced by pretreatment with the ERK inhibitor PD098059 (10 μmol/L) or the JNK inhibitor SP600125 (10 μmol/L) (Figure 6A and B). At a concentration of 10 μmol/L, the p38 inhibitor SB203580 had no effect on HO-1 mRNA induction by lansoprazole (50 μmol/L). Higher concentrations of the inhibitor decreased HO-1 mRNA levels in comparison to that following treatment with 50 μmol/L lansoprazole alone (Figure 6C). Thus, to further explore the role of p38 MAPK on PPI-mediated HO-1 gene activation, we treated endothelial cells with a second p38 MAPK inhibitor (α and β subunit), SB202190 (0.1-10 μmol/L). SB202190 did not affect the induction of HO-1 mRNA by lansoprazole (Figure 6C). Moreover, when we investigated the effect of both p38 MAPK inhibitors on HO-1 promoter activity using NIH3T3-HO-1-luc cells, incubation with 30 μmol/L lansoprazole alone for 24 h significantly increased HO-1 promoter activity by 1.7-fold compared with untreated control levels (data not shown). Neither p38 inhibitor diminished HO-1 promoter activation by lansoprazole (Table 1). All MAPK inhibitors alone did not affect HO-1 mRNA levels or HO-1 promoter activity.
Figure 6.
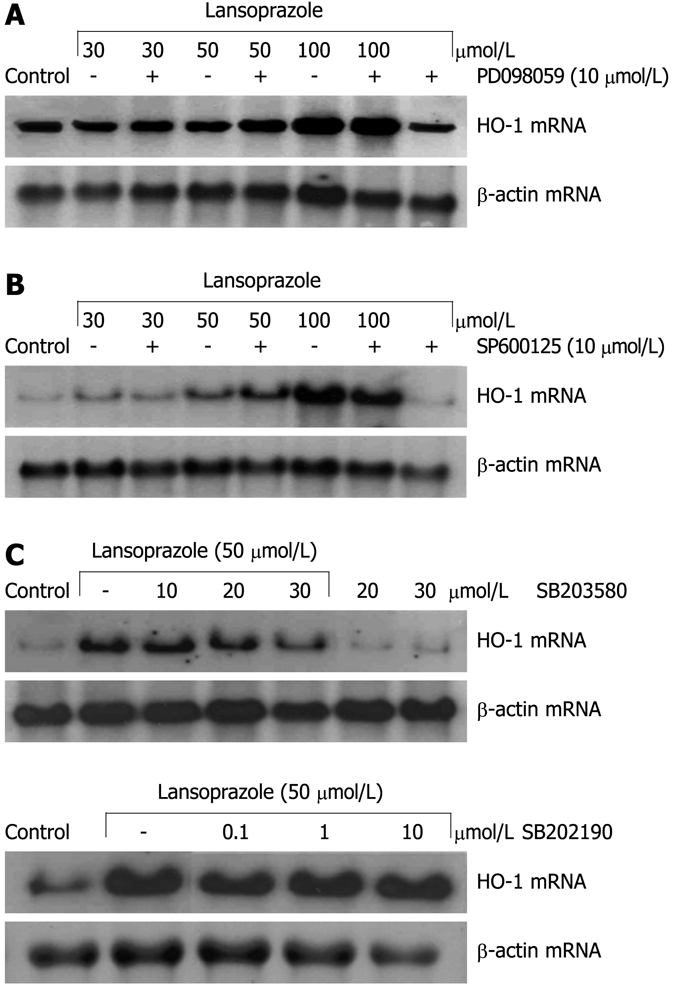
Effect of different MAPK inhibitors on lansoprazole-induced HO-1 mRNA expression. After pretreatment with ERK inhibitor PD098059 (A), JNK inhibitor SP600125 (B), or p38 inhibitors SB203580 and SB202190 (C) for 20 min, endothelial cells were incubated with lansoprazole for 8 h. The Northern blotting analysis shown is representative of three to six independent experiments.
Table 1.
Effect of p38 inhibitors SB203580 and SB202190 on lansoprazole-induced HO-1 promoter activity in NIH3T3-HO-1-luc cells (mean ± SD, %)
| Treatment | HO-1 promoter activity |
| Lansoprazole (30 μmol/L) | 100.0 ± 8.4 |
| Lansoprazole (30 μmol/L) + SB203580 (10 μmol/L) | 109.2 ± 9.0 |
| Lansoprazole (30 μmol/L) + SB203580 (20 μmol/L) | 108.8 ± 12.6 |
| Lansoprazole (30 μmol/L) + SB203580 (30 μmol/L) | 101.8 ± 14.4 |
| Lansoprazole (30 μmol/L) + SB202190 (0.1 μmol/L) | 91.0 ± 3.7 |
| Lansoprazole (30 μmol/L) + SB202190 (1.0 μmol/L) | 112.4 ± 15.1 |
Change from HO-1 promoter activity measured in cells treated with 30 μmol/L lansoprazole.
Involvement of the PI3K pathway in the regulation of HO-1 induction by lansoprazole
Preincubation of endothelial cells with the PI3K inhibitor LY294002 (5-50 μmol/L) diminished the lansoprazole-induced (50 μmol/L) increase in HO-1 mRNA expression in a concentration-dependent manner (Figure 7A and C). Similar effects were observed in NIH3T3-HO-1-luc cells (Figure 8A and C). The lansoprazole-mediated (30 μmol/L) promoter activation was abolished by up to 50% in the presence of LY294002 (5-25 μmol/L) (Figure 8C). Preincubation with the fungal metabolite wortmannin (0.01 and 0.1 μmol/L) did not affect the lansoprazole-mediated increase in HO-1 mRNA and HO-1 promoter activity levels (Figure 7B and C, Figure 8B and C). Both PI3K inhibitors alone did not affect HO-1 promoter activity or transcriptional levels under identical experimental conditions.
Figure 7.
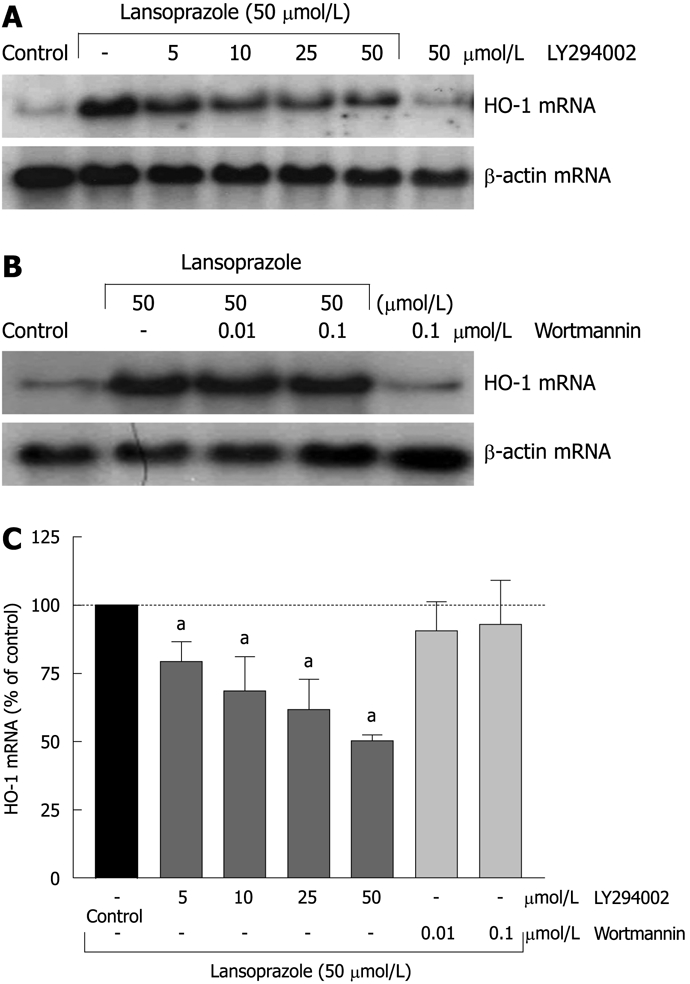
Effect of PI3K inhibitors LY294002 and wortmannin on lansoprazole-induced HO-1 mRNA expression. After pretreatment with PI3K inhibitors for 20 min, endothelial cells were incubated with lansoprazole for 8 h. Representative Northern blotting analysis is shown (A and B). Results are expressed as mean ± SD of three to six separate experiments compared to lansoprazole (control = gene expression of cells stimulated with 50 μmol/L lansoprazole = 100%) (C). aP < 0.05 vs lansoprazole alone.
Figure 8.
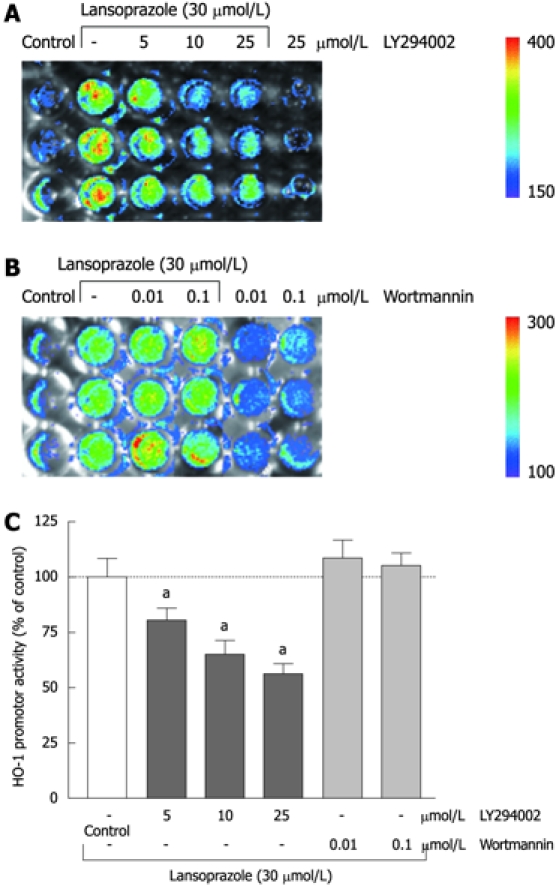
Effect of PI3K-inhibitors LY294002 and wortmannin on lansoprazole-induced HO-1 promoter activity in stable transfected cell lines. Representative images of three to six separate experiments are shown (A and B). Light emission (shown by the rainbow bars: blue, least intense; red, most intense) was measured in NIH3T3-HO-1-luc cells with the IVIS. Results are expressed as mean ± SD of three to six separate experiments compared to lansoprazole (control = HO-1 promoter activity of cells stimulated with 30 μmol/L lansoprazole = 100%) (C). aP < 0.05 vs lansoprazole alone.
DISCUSSION
Since ROS and inflammation are important causative factors for the development of mucosal damage, the pleiotropic effects of PPIs are of particular clinical interest for the control of gastroduodenal ulcers. Thus, investigation of the molecular mechanism of PPI-mediated gastric protection is required.
In this study, we used endothelial cells and macrophages, which play important roles in mucosal defense, to evaluate the effects of PPIs on the HO-1/ferritin system. The blood flow through the endothelial cells that innervate the mucosa is crucial for supplying nutrients, limiting damage, and facilitating repair. Macrophages play key roles in the mucosal immune system, sensing and in response to foreign materials[23]. Neither cell type expresses H+/K+-ATPase, therefore, the observed induction of HO-1 and ferritin by PPIs is assumed to be independent of their antisecretory properties. We have shown previously that PPIs induce HO-1 mRNA, protein and enzymatic activity in gastric epithelial cells and endothelial cells[9]. In this study, we provided further evidence that activation of the HO-1 gene by PPIs might account for their beneficial effects in the treatment of peptic ulcer disease. We demonstrated that the H2 receptor antagonist ranitidine, in contrast to lansoprazole, failed to increase HO-1 mRNA levels. Moreover, lansoprazole also significantly upregulated HO-1 protein levels as a consequence of elevated HO-1 mRNA levels in J774 cells (data not shown). Macrophage-derived ROS production promotes the killing of microorganisms on one hand, but on the other hand, contributes to oxidative stress in inflammatory sites and alters basic cell functions such as adhesion and proliferation[24,25]. In acute inflammatory illnesses, HO-1 mRNA levels are significantly elevated, which suggests that monocytes exert potent anti-inflammatory effects via HO-1 activation, and thereby regulate the production of pro-inflammatory cytokines to protect organs and cells from irreversible damage[26]. Thus, upregulation of HO-1 expression by lansoprazole in macrophages could be beneficial in the management of mucosal inflammation.
In addition, we demonstrated that lansoprazole performed free-radical scavenging in macrophages. The protection against NADPH-mediated ROS production by lansoprazole occurred after 12 h of incubation and after washout of lansoprazole. Moreover, the inhibition of the antioxidant effect of lansoprazole in the presence of the HO inhibitor CrMP[27] demonstrates that HO-1 and its enzymatic products are indeed of functional relevance for the antioxidant effects of lansoprazole.
All experiments in this study have been performed with lansoprazole, but studies using omeprazole have shown similar effects on HO-1 mRNA and HO-1 and ferritin protein expression, as well as on the reduction of ROS (data not shown). Taken together, these data indicate that the induction of HO-1 might be responsible, at least in part, for the antioxidative action of PPIs and their advantage over H2-antagonists in the therapy of ulcer disease and NSAID-related mucosal damage.
Our data support other work that has shown that HO-1 might be a mediator of gastroprotective pathways. Immunohistochemical studies have demonstrated that HO-1 is expressed constitutively in normal gastric and colonic mucosa and its upregulation occurs in these tissues during inflammation or the healing phase of gastric ulcers[28,29].
In the present study, significant HO-1 induction following treatment with lansoprazole in endothelial cells and macrophages, as well as antioxidant protection occurred at a concentration of 30 μmol/L. Plasma concentrations after oral administration of lansoprazole (30 mg/d) ranged from 2.2 to 5.6 μmol/L[30]. Depending on the dose (30-90 mg/d) and the route of administration, higher plasma concentration levels (e.g. 15-21 μmol/L) may be attainable[31,32]. These concentrations approximate the concentrations found to be effective at increasing the level of HO-1 expression in the present study. Furthermore, Blandizzi and co-workers have suggested that the ED50 of lansoprazole needed to provide gastroprotection is 2-4 times higher compared to that needed for inhibition of acid secretion[7].
The anti-inflammatory and antiproliferative actions of the HO-1 product CO, as well as the potent antioxidant effect of bilirubin, are thought to contribute to the overall protective effect of HO-1[12]. CO has also been shown to provide vasodilatory activities through the activation of soluble guanylyl cyclase[33]. Omeprazole and lansoprazole induce relaxation of isolated human arteries[34] at the same concentration range (30-300 μmol/L lansoprazole) that we have found to be effective in the induction of HO-1. Although Naseri and co-workers have suggested the regulation of intracellular Ca2+ as an underlying mechanism, it might also be possible that the generation of CO by HO-1 activity contributes to the vasodilatory effect of PPIs.
The third direct product of heme metabolism, free Fe2+, leads to the induction of a multimeric iron-chelating protein, ferritin[35]. In the present study, the ferritin protein induction by lansoprazole occurred in a concentration- and time-dependent manner in macrophages and endothelial cells. The activation of ferritin protein synthesis was obtained after long incubations (12-24 h) and at concentrations that have also been shown to result in HO-1 mRNA and protein increase, which suggests ferritin induction as a functional consequence of elevated HO-1 activity.
Besides induction through its physiological substrate heme, HO-1 gene expression can also be stimulated by a variety of stress inducers including, but not limited to, heavy metals, ultraviolet irradiation, endotoxin, and oxidants such as hydrogen peroxide[14]. In contrast, antioxidants such as α-tocopherol and allopurinol prevent the upregulation of HO-1[36]. Thus, we investigated the role of ROS in PPI-mediated HO-1 induction. Pretreatment with the antioxidant enzyme SOD did not affect lansoprazole-mediated HO-1 induction, precluding a mediator role of superoxide.
Recently, it has been shown by Yeo and co-workers that PPIs influence MAPK-dependent pathways[37]. However, in the present study, the ERK inhibitor PD098059 and the JNK inhibitor SP600125 had no inhibitory effect on lansoprazole-induced HO-1 mRNA. All inhibitors have been used in concentrations previously shown to be effective in the inhibition of the final kinase of the respective MAPK cascade[38,39]. The p38 kinase inhibitor (α- and β-subunit) SB203580, at a concentration of 30 μmol/L, impaired the lansoprazole-mediated HO-1 mRNA induction. SB 203580 has been shown to inhibit its established targets, the α- and β-subunit of p38 with IC50 values of 50 and 500 nmol/L, respectively. Yet, other kinases like lymphocyte-specific protein tyrosine kinase, glycogen synthase kinase-3β (GSK3β) and Akt1 (protein kinase Bα) were also inhibited by SB 203580, with IC50 values that were 100 ± 500-fold higher than that for p38[40]. As SB203580 interacts with other kinases at higher concentrations, we determined the effect of a second p38 inhibitor, SB202190, on HO-1 mRNA induction by lansoprazole. SB202190 was without effect on the lansoprazole-mediated increase in HO-1 mRNA. In agreement with this, both p38 inhibitors have been without effect on lansoprazole-induced HO-1 promoter activity in NIH3T3-HO-1-luc-cells, which indicates that MAPK activation is not involved in PPI-mediated HO-1 induction.
The PI3K inhibitor LY294002 diminished the increase of HO-1 mRNA level and HO-1 promoter activity induced by lansoprazole in a concentration-dependent manner. Surprisingly, a second PI3K inhibitor, wortmannin, did not affect HO-1 expression. Although both PI3K inhibitors have been shown to block the phosphorylation of PI3K completely at the concentrations used in our study[41,42], the potency to interact with other kinases is different. An inhibitory effect by LY294002 but not by wortmannin on HO-1 induction has already been described by other groups, which suggests that PI3K is not involved in HO-1 gene activation in these particular cases[43,44]. LY294002 is also known to block casein kinase 2 (CK2) and GSK3β by up to 50% at a concentration of 50 μmol/L[40]. Moreover, the phosphorylation of PI3K downstream kinase p70s6k is inhibited almost completely at a concentration of 10-25 μmol/L LY294002, whereas the inhibition of Akt phosphorylation requires LY294002 doses of up to 150-200 μmol/L[45]. Therefore, a different influence of special PI3K downstream kinases or an involvement of CK2 or GSK3β on the PPI-mediated HO-1 gene activation is conceivable. Indeed there is evidence that CK2 might exert a significant influence on HO-1 gene activation by the phorbol 12-myristate 13-acetate[46]. Further studies are needed to clarify the influence of GSK3β, CK2 and PI3K downstream kinases on HO-1 induction by PPIs.
In summary, we demonstrated that lansoprazole is a potent inducer of the antioxidant proteins HO-1 and ferritin. The induction of HO-1 by lansoprazole is independent of oxidative stress, and involves a signaling pathway that is blocked by LY294002. The activation of the HO-1/ferritin pathway occurs in endothelial cells and macrophages; cells relevant to the mucosal microcirculation and the mucosal immune system. Neither cells express H+/K+-ATPase, therefore, the observed induction of HO-1 and ferritin by lansoprazole can be assumed to occur independently of the antisecretory effect. It is plausible that HO-1 and its enzymatic products are responsible for, or contribute to, the gastric protection seen with lansoprazole and other PPIs.
COMMENTS
Background
Proton pump inhibitors (PPIs) irreversibly inhibit the gastric proton pump in parietal cells and are therefore potent antisecretory drugs. However, comparisons with other acid-reducing agents suggest that the gastroprotective effect of PPIs cannot be explained solely by their antisecretory action. Recent studies have revealed anti-apoptotic, anti-inflammatory, and free-radical scavenging properties of PPIs. The inducible stress protein, heme oxygenase-1 (HO-1), catalyzes the degradation of heme. As a result of the antioxidative, anti-inflammatory, and vasodilatory effects of its products, bilirubin and CO, HO-1 is believed to be a mediator of cytoprotective pathways.
Research frontiers
The pathogenesis of gastric ulcer disease is generally characterized by a decrease in mucosal defense and an increase in aggressive mechanisms. Although the cellular and molecular basis of the gastric mucosal defense are well understood, the mechanisms of mucosal damage mediated by aggressive factors remain largely unclear. The increased production of reactive oxygen species (ROS), termed oxidative stress, is considered to play a crucial role in the development of mucosal damage.
Innovations and breakthroughs
The induction of the antioxidant proteins HO-1 and ferritin by PPIs occurs independently of their antisecretory effects. It is plausible that HO-1 and its enzymatic products are responsible for, or at least contribute to, the gastric protection observed with PPI treatment. There is evidence that PPIs activate the HO-1 gene through a phosphatidylinositol 3-kinase (PI3K)-dependent pathway.
Applications
The present results suggest that the cytoprotective effects of PPIs can be ascribed to a reduction of oxidative stress via induction of the HO-1/ferritin system in endothelial cells and macrophages. PPIs might be beneficial for cardiac patients and patients on daily pain medication by enhancing gastric mucosal protection and reducing the risk of gastrointestinal bleeding caused by chronic use of aspirin and other NSAIDs. Moreover, PPI-dependent HO-1 induction in endothelial cells of the systemic circulation might exert vascular-protective actions.
Terminology
HO-1 is also know as heat shock protein 32. The enzyme catalyzes the degradation of heme and leads to the accumulation of free iron, CO, and bilirubin. HO-1 is the inducible of two isoforms of the enzyme. PI3K catalyzes the production of the plasma membrane lipid phosphatidylinositol-3,4,5-trisphosphate. Signaling pathways downstream of PI3K affect cell growth, survival and movement.
Peer review
The manuscript investigated molecular mechanism(s) for lansoprazole-induced gastric protection. The authors used endothelial cells and macrophages as models for cytoprotection and showed that lansoprazole induced HO-1 and ferritin and thereby decreased ROS formation in those cell systems. This HO-1 gene activation involved the PI3K pathway. The authors’ hypothesis that lansoprazole upregulates HO-1 and thereby protects gastric mucosa is provocative.
Acknowledgments
We thank Hendrik J Vreman, PhD (Department of Pediatrics, Stanford University School of Medicine, Stanford, CA, USA) for valuable discussions and for critically reviewing the manuscript, and Petra Schwartz for her excellent technical assistance.
Footnotes
Supported by The German Academic Exchange Program (DAAD, S.S.G.)
Peer reviewer: Shingo Tsuji, MD, PhD, AGAF, Professor, Department of Internal Medicine and Therapeutics, Osaka University Graduate School of Medicine(A8), 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan
S- Editor Tian L L- Editor Kerr C E- Editor Zheng XM
References
- 1.Das D, Bandyopadhyay D, Bhattacharjee M, Banerjee RK. Hydroxyl radical is the major causative factor in stress-induced gastric ulceration. Free Radic Biol Med. 1997;23:8–18. doi: 10.1016/s0891-5849(96)00547-3. [DOI] [PubMed] [Google Scholar]
- 2.Langman MJ, Brooks P, Hawkey CJ, Silverstein F, Yeomans N. Non-steroid anti-inflammatory drug associated ulcer: epidemiology, causation and treatment. J Gastroenterol Hepatol. 1991;6:442–449. doi: 10.1111/j.1440-1746.1991.tb00885.x. [DOI] [PubMed] [Google Scholar]
- 3.Konturek PC, Bielański W, Konturek SJ, Hahn EG. Helicobacter pylori associated gastric pathology. J Physiol Pharmacol. 1999;50:695–710. [PubMed] [Google Scholar]
- 4.Gisbert JP, González L, Calvet X, Roqué M, Gabriel R, Pajares JM. Proton pump inhibitors versus H2-antagonists: a meta-analysis of their efficacy in treating bleeding peptic ulcer. Aliment Pharmacol Ther. 2001;15:917–926. doi: 10.1046/j.1365-2036.2001.01012.x. [DOI] [PubMed] [Google Scholar]
- 5.Sachs G, Shin JM, Vagin O, Lambrecht N, Yakubov I, Munson K. The gastric H,K ATPase as a drug target: past, present, and future. J Clin Gastroenterol. 2007;41 Suppl 2:S226–S242. doi: 10.1097/MCG.0b013e31803233b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biswas K, Bandyopadhyay U, Chattopadhyay I, Varadaraj A, Ali E, Banerjee RK. A novel antioxidant and antiapoptotic role of omeprazole to block gastric ulcer through scavenging of hydroxyl radical. J Biol Chem. 2003;278:10993–11001. doi: 10.1074/jbc.M210328200. [DOI] [PubMed] [Google Scholar]
- 7.Blandizzi C, Natale G, Gherardi G, Lazzeri G, Marveggio C, Colucci R, Carignani D, Del Tacca M. Acid-independent gastroprotective effects of lansoprazole in experimental mucosal injury. Dig Dis Sci. 1999;44:2039–2050. doi: 10.1023/a:1026626519534. [DOI] [PubMed] [Google Scholar]
- 8.Tsuji S, Sun WH, Tsujii M, Kawai N, Kimura A, Kakiuchi Y, Yasumaru S, Komori M, Murata H, Sasaki Y, et al. Lansoprazole induces mucosal protection through gastrin receptor-dependent up-regulation of cyclooxygenase-2 in rats. J Pharmacol Exp Ther. 2002;303:1301–1308. doi: 10.1124/jpet.102.035204. [DOI] [PubMed] [Google Scholar]
- 9.Becker JC, Grosser N, Waltke C, Schulz S, Erdmann K, Domschke W, Schröder H, Pohle T. Beyond gastric acid reduction: proton pump inhibitors induce heme oxygenase-1 in gastric and endothelial cells. Biochem Biophys Res Commun. 2006;345:1014–1021. doi: 10.1016/j.bbrc.2006.04.170. [DOI] [PubMed] [Google Scholar]
- 10.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 12.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 13.Eisenstein RS, Garcia-Mayol D, Pettingell W, Munro HN. Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc Natl Acad Sci USA. 1991;88:688–692. doi: 10.1073/pnas.88.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 15.Keyse SM, Tyrrell RM. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA. 1989;86:99–103. doi: 10.1073/pnas.86.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes SE. Functional characterization of the spontaneously transformed human umbilical vein endothelial cell line ECV304: use in an in vitro model of angiogenesis. Exp Cell Res. 1996;225:171–185. doi: 10.1006/excr.1996.0168. [DOI] [PubMed] [Google Scholar]
- 17.Yang Q, Zhu P, Wang Z, Jiang J. Lipopolysaccharide upregulates the expression of Toll-like receptor 4 in human vascular endothelial cells. Chin Med J (Engl) 2002;115:286–289. [PubMed] [Google Scholar]
- 18.Meghji P, Burnstock G. Inhibition of extracellular ATP degradation in endothelial cells. Life Sci. 1995;57:763–771. doi: 10.1016/0024-3205(95)02004-3. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi K, Sawasaki Y. Human endothelial cell line, ECV304, produces pro-urokinase. In Vitro Cell Dev Biol. 1991;27A:766–768. doi: 10.1007/BF02631240. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Zhu H, Kuppusamy P, Roubaud V, Zweier JL, Trush MA. Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem. 1998;273:2015–2023. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- 21.Grosser N, Abate A, Oberle S, Vreman HJ, Dennery PA, Becker JC, Pohle T, Seidman DS, Schröder H. Heme oxygenase-1 induction may explain the antioxidant profile of aspirin. Biochem Biophys Res Commun. 2003;308:956–960. doi: 10.1016/s0006-291x(03)01504-3. [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, Feng JQ, Harris SE, Contag PR, Stevenson DK, Contag CH. Rapid in vivo functional analysis of transgenes in mice using whole body imaging of luciferase expression. Transgenic Res. 2001;10:423–434. doi: 10.1023/a:1012042506002. [DOI] [PubMed] [Google Scholar]
- 23.Wallace JL, Ma L. Inflammatory mediators in gastrointestinal defense and injury. Exp Biol Med (Maywood) 2001;226:1003–1015. doi: 10.1177/153537020122601107. [DOI] [PubMed] [Google Scholar]
- 24.Cathcart MK. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:23–28. doi: 10.1161/01.ATV.0000097769.47306.12. [DOI] [PubMed] [Google Scholar]
- 25.Daniel DS, Dai G, Singh CR, Lindsey DR, Smith AK, Dhandayuthapani S, Hunter RL Jr, Jagannath C. The reduced bactericidal function of complement C5-deficient murine macrophages is associated with defects in the synthesis and delivery of reactive oxygen radicals to mycobacterial phagosomes. J Immunol. 2006;177:4688–4698. doi: 10.4049/jimmunol.177.7.4688. [DOI] [PubMed] [Google Scholar]
- 26.Yachie A, Toma T, Mizuno K, Okamoto H, Shimura S, Ohta K, Kasahara Y, Koizumi S. Heme oxygenase-1 production by peripheral blood monocytes during acute inflammatory illnesses of children. Exp Biol Med (Maywood) 2003;228:550–556. doi: 10.1177/15353702-0322805-26. [DOI] [PubMed] [Google Scholar]
- 27.Appleton SD, Chretien ML, McLaughlin BE, Vreman HJ, Stevenson DK, Brien JF, Nakatsu K, Maurice DH, Marks GS. Selective inhibition of heme oxygenase, without inhibition of nitric oxide synthase or soluble guanylyl cyclase, by metalloporphyrins at low concentrations. Drug Metab Dispos. 1999;27:1214–1219. [PubMed] [Google Scholar]
- 28.Barton SG, Rampton DS, Winrow VR, Domizio P, Feakins RM. Expression of heat shock protein 32 (hemoxygenase-1) in the normal and inflamed human stomach and colon: an immunohistochemical study. Cell Stress Chaperones. 2003;8:329–334. doi: 10.1379/1466-1268(2003)008<0329:eohsph>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo JS, Cho CH, Wang WP, Shen XZ, Cheng CL, Koo MW. Expression and activities of three inducible enzymes in the healing of gastric ulcers in rats. World J Gastroenterol. 2003;9:1767–1771. doi: 10.3748/wjg.v9.i8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson T, Holmberg J, Röhss K, Walan A. Pharmacokinetics and effect on caffeine metabolism of the proton pump inhibitors, omeprazole, lansoprazole, and pantoprazole. Br J Clin Pharmacol. 1998;45:369–375. doi: 10.1046/j.1365-2125.1998.t01-1-00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howden CW, Metz DC, Hunt B, Vakily M, Kukulka M, Amer F, Samra N. Dose-response evaluation of the antisecretory effect of continuous infusion intravenous lansoprazole regimens over 48 h. Aliment Pharmacol Ther. 2006;23:975–984. doi: 10.1111/j.1365-2036.2006.02849.x. [DOI] [PubMed] [Google Scholar]
- 32.Yacyshyn BR, Thomson AB. The clinical importance of proton pump inhibitor pharmacokinetics. Digestion. 2002;66:67–78. doi: 10.1159/000065588. [DOI] [PubMed] [Google Scholar]
- 33.Cardell LO, Lou YP, Takeyama K, Ueki IF, Lausier J, Nadel JA. Carbon monoxide, a cyclic GMP-related messenger, involved in hypoxic bronchodilation in vivo. Pulm Pharmacol Ther. 1998;11:309–315. doi: 10.1006/pupt.1998.0152. [DOI] [PubMed] [Google Scholar]
- 34.Naseri E, Yenisehirli A. Proton pump inhibitors omeprazole and lansoprazole induce relaxation of isolated human arteries. Eur J Pharmacol. 2006;531:226–231. doi: 10.1016/j.ejphar.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 35.Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 36.Tomaro ML, Frydman J, Frydman RB. Heme oxygenase induction by CoCl2, Co-protoporphyrin IX, phenylhydrazine, and diamide: evidence for oxidative stress involvement. Arch Biochem Biophys. 1991;286:610–617. doi: 10.1016/0003-9861(91)90088-z. [DOI] [PubMed] [Google Scholar]
- 37.Yeo M, Kim DK, Han SU, Lee JE, Kim YB, Cho YK, Kim JH, Cho SW, Hahm KB. Novel action of gastric proton pump inhibitor on suppression of Helicobacter pylori induced angiogenesis. Gut. 2006;55:26–33. doi: 10.1136/gut.2005.067454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296(Pt 2):297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 43.Abate A, Yang G, Wong RJ, Schroder H, Stevenson DK, Dennery PA. Apigenin decreases hemin-mediated heme oxygenase-1 induction. Free Radic Biol Med. 2005;39:711–718. doi: 10.1016/j.freeradbiomed.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 44.Andreadi CK, Howells LM, Atherfold PA, Manson MM. Involvement of Nrf2, p38, B-Raf, and nuclear factor-kappaB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol. 2006;69:1033–1040. doi: 10.1124/mol.105.018374. [DOI] [PubMed] [Google Scholar]
- 45.Adi S, Wu NY, Rosenthal SM. Growth factor-stimulated phosphorylation of Akt and p70(S6K) is differentially inhibited by LY294002 and Wortmannin. Endocrinology. 2001;142:498–501. doi: 10.1210/endo.142.1.8051. [DOI] [PubMed] [Google Scholar]
- 46.Naidu S, Wijayanti N, Santoso S, Kietzmann T, Immenschuh S. An atypical NF-kappa B-regulated pathway mediates phorbol ester-dependent heme oxygenase-1 gene activation in monocytes. J Immunol. 2008;181:4113–4123. doi: 10.4049/jimmunol.181.6.4113. [DOI] [PubMed] [Google Scholar]