

Abstract
The prevalence of obesity among children, adolescents and adults has been dramatically increasing worldwide during the last several decades. The obesity epidemic has been recognized as one of the major global health problems, because its health hazard is linked to a number of common diseases including breast and prostate cancers. Obesity is caused by combination of genetic and environmental factors. While genetic contribution to obesity has been known to be significant, the genetic factors remain relatively unchanged. Recent studies have highlighted the involvement of environmental “obesogens”, i.e. the xenobiotic chemicals that can disrupt the normal development and homeostatic control over adipogenesis and energy balance. Several lines of evidence suggest that increasing exposure to chemicals with endocrine-disrupting activities (endocrine disrupting chemicals, EDCs) contributes to the increased obesity. The cellular and molecular mechanisms underlying obesogen-associated obesity are just now being appreciated. In this paper, we comprehensively reviewed current knowledge about the role of estrogen receptors alpha and beta (ERα and ERβ) in regulation of energy metabolism pathways, including glucose transport, glycolysis, tricarboxylic acid (TCA) cycle, mitochondrial respiratory chain (MRC adenosine nucleotide translocator (ANT) and fatty acid β-oxidation and synthesis, by estrogens and then examined the disturbance of E2/ER-mediated energy metabolism pathways by environmental obesogens; and finally, we discussed the potential implications of disturbance of energy metabolism pathways by obesogens in obesity and pointed out several key aspects of this area that need to be further explored. A better understanding of the cellular and molecular mechanisms underlying obesogen-associated obesity will lead to new approaches for slow down and/or prevention of the increased trend of obesity associated with exposure to obesogens.
Keywords: Adenosine nucleotide translocase (ANT), Estrogens, Estrogen receptor alpha and beta, Endocrine disrupting chemicals, Glucose transport, Glycolysis, Mitochondrial respiratory chain, Obesity, Tricarboxylic acid cycle
I. Introduction
The prevalence of obesity (defined as excessive body fat with more than 25% of body fat in men and more than 30% of body fat in women) among children, adolescents and adults has been dramatically increasing during the last several decades [1-3]. Worldwide, more than one billion adults are overweight and over 300 million are obese [4]. The majority of developed countries particularly The United States [5] and England [6] and the rapidly developing countries such as China [7] and India [8] have been experiencing dramatic increases in obesity. The obesity epidemic has been recognized as one of the major global health problems, because its health hazard is linked to several types of common diseases including type 2 diabetes, heart disease, high blood pressure, stroke, pulmonary problem, liver disease, psychological/social problems, and reproductive problems [4, 9], as well as with breast [9, 10] and prostate [11] cancers. Thus, great efforts must be made to prevent and/or slow down this trend.
Obesity is caused by combination of genetic and environmental factors. While genetic contribution to obesity has been known to be significant, the genetic factors remain relatively unchanged [12]. Thus, the current epidemic of obesity and associated metabolic diseases could be attributed to the dramatic changes in the environmental and nutritional factors over the last several decades [3, 14, 15]. The increased caloric intake and decreased physical activities are thought to be one of the major causes of this dramatic increase. However, there are several lines of evidence that highlight the potential involvement of environmental “obesogens”, i.e. the xenobiotic chemicals that can disrupt the normal development and homeostatic control over adipogenesis and energy balance [16, 17]. Increasing exposure to the industrial and agricultural chemicals with endocrine-disrupting activities (endocrine disrupting chemicals, EDCs) released into the environment could be another factor contributing to the increased obesity [3, 13, 14]. In order to prevent the increased trend of obesity related to exposure to obesogens (e.g. EDCs), it is important to understand the cellular and molecular mechanisms underlying obesogen-associated obesity. In this review, we firstly gleaned several lines of evidence indicating the role of estrogen receptors alpha and beta (ERα and ERβ) in regulation of energy metabolism pathways by estrogens; Secondly, we examined the disturbance of E2/ER-mediated energy metabolism pathways by environmental obesogens; Finally, we discussed the potential implications of disturbance of energy metabolism pathways by obesogens in relation to obesity that may be causally related to exposure to environmental obesogens.
II. Role of Estrogens, Estrogenic Chemicals and ERs in Obesity
2.1. Involvement of Estrogens, Estrogenic Chemicals in Regulation of body weight and obesity
There is accumulating evidence indicating that endogenous estrogens, e.g. 17β-estradiol (E2), and environmental estrogenic EDCs are involved in the regulation of body weight and obesity. For instance, it has been observed that treatment of Syrian hamsters with estrogens caused significant decreases in body weight and fat content without affecting food intake[15]. Deficiency or loss of estrogens in adult animals, e.g. in follicle-stimulating hormone receptor knock out mice [20], ovariectatomied (Ovx) mice and Syrian hamster [21-24], and aromatase knock out (ARKO) mice [16-18], is associated with bodyweight gain and the development of obesity. Furthermore, more interestingly, Gao et al. [19] reported that estrogens regulated the body weight and energy metabolism in the brain in a way similar to that of the leptin, the hormone that regulates energy metabolism in the brain. During menopause women tend to gain body fat. The increase in adiposity is likely due to the decline in endogenous estrogens [20]. On the other hand, estrogen therapy has positive effects on carbohydrate and lipid metabolism in overweight-obese younger postmenopausal women [21, 22].
2.2. Role of ERα and ERβ in E2 Regulation of Body Weight and Obesity
The biological effects of estrogens are mainly mediated via estrogen ERs, i.e. ERα and ERβ, by regulating the expression of estrogen responsive genes via nuclear ERs [23-27] and by eliciting several rapid, non-nuclear genomic signaling pathways via the plasma membrane-associated ERs [28-32]. Mitochondria are also important targets for the action of estrogens and ERs [33, 34]. Several lines of evidence have pointed to the involvement of ERα and ERβ in mediating estrogen’s effects on body weight. First, tamoxifen (TAM), an anti-estrogen, significantly attenuated the effects of E2 on reducing body weight when given concurrently with E2 [15]. Second, ICI 182,780, another pure estrogen antagonist, has been shown to antagonize the effects of E2 on estrous behavior and energy balance in Syrian hamsters [35]. Third, the role of ERα in E2 regulation of body weight and obesity is supported by following observations: a) both male and female mice that have been genetically altered to reduce the ability to produce estrogen by knocking out aromatase (ARKO), an enzyme that catalyzes the conversion of androgen to estrogen, became obese when fed the same amounts as normal mice [36, 37]; b) Increased white adipose tissue (WAT) and body fat were seen in both sexual mature male and female ERα knocked out (ERαKO) mice [38, 39]. ERα absence caused adipocyte hyperplasia/hypertrophy, insulin resistance, and glucose intolerance and reduced energy expenditure in both sexes, clearly indicating that E2/ERα signaling is critical in female and male WAT and that obesity in ERαKO mice involves a mechanism of reduced energy expenditure [39, 40].
The role of ERβ in E2 regulation of body weight and obesity is less clear and somewhat controversial. Unlike ERαKO mice, ERβKO mice did not show a clear increase in total body fat and enhanced serum leptin levels [38, 40]. Because ERαKO mice undergo a 10-fold increase in E2, a persistent or even increased E2 signaling through ERβ could be a factor in obesity of ERαKO mice. A study by Naaz et al. [41] has revealed a role of ERβ in adipose tissue. These investigators ovariectomized (Ovx) or sham-ovariectomized (sham-Ovx) adult female ERαKO mice and then treated them with vehicle or E2, and measured bodyweights and food consumption at 28 days after treatment. They observed that sham-Ovx ERαKO mice had increase in bodyweight whereas Ovx ERαKO mice showed a 6 % decrease, and E2 replacement restored body weight to sham levels. Fat pads of Ovx ERαKO mice showed 45% and 16% decreases in bodyweight and adipocyte circumference, respectively, compared to sham-Ovx or E2-replaced Ovx ERαKO mice. Ovx ERαKO mice showed a trend towards decreased feed consumption that did not reach significance. Blood glucose levels were lower both before and after glucose injection in Ovx mice compared to sham ERαKO mice, and E2 treatment reversed this. Insulin levels following glucose challenge were lower in Ovx mice compared to sham-Ovx ERαKO mice, indicating that ovariectomy ameliorated the glucose intolerance and insulin resistance in ERαKO mice. Immunohistochemical analysis revealed strong staining for ERβ in adipose tissue. These observations indicate that removing E2/ERβ signaling in ERαKO mice by ovariectomy decreases body and fat-pad weights and adipocyte size, while improving insulin and glucose metabolism. Thus, ERβ mediated effects on adipose tissue are opposite those of ERα, although E2 effects on adipose tissue are predominately through ERα. Another study by Foryst-Ludwig et al. [42] revealed that ERβ inhibited ligand-mediated transcriptional activity induced by proxisomal proliferator activating receptor-gamma (PPARγ), a master regulator of insulin signaling/glucose metabolism, resulting in a blockade of PPARγ-induced adipocytic gene expression and decreased adipogenesis. Several types of mutations in human ERβ gene have been identified in some obese adolescents and women with bulimic disease [43, 44]. Liang et al. [45] observed that Ovx in 8-week old female Wistar rats induced hyperphagia in association with increased bodyweight and abdominal fat accumulation. These effects were fully reversed by subcutaneous E2 replacement. These investigators examined the effects of intracerebroventricular infusion of E2, alone or in combination with anti-sense oligonucleotides for either ERα or ERβ in Ovx rats. The E2-treated group showed a 10-20% lower daily food intake after the 2-week infusion period, and a 14% reduction in body weight with similar reduction in abdominal fat compared to the control group. The inhibitory effects of E2 on food intake and bodyweight were blocked by co-administration of anti-sense oligonucleotides of ERβ but not of those of ERα. These observations suggest that ERβ is involved in the anorectic action of estrogen and in the modulation of bodyweight. It has been shown that the ratio of ERα to ERβ in adipose tissue was associated with obesity as well as the serum level and production of leptin in omental adipose tissue [46]. Together, these observations indicate that regulation of bodyweight and obesity by estrogens and estrogenic chemicals are differentially mediated via ERα and ERβ.
As mentioned above, both ERα and ERβ are localized in nucleus, plasma membrane and mitochondria. While the relative contributions of these different pools of ERs to estrogen regulation of body weight and obesity have yet clearly addressed, a recent study by Pedram et al. [47] indicated that expression of ERα functional domain in plasma membrane in ERαKO mice did not rescue these mice from developing obesity. These investigators generated a transgenic mouse expressing only a functional E domain of ERα at the plasma membrane (MOER). These MOER female mice exhibited comparable activation of ERK and phosphatidylinositol 3-kinase, which was not seen in cells from ERαKO mice, indicating that several membrane ERα-mediated signaling pathways were retained in these mice. However, ovaries of MOER, like those in homozygous Strasbourg ERαKO mice, showed multiple hemorrhagic cysts and no corpus luteum. Their mammary gland development was also rudimentary. More importantly, MOER mice showed plentiful abdominal visceral and other depots of fat and increased body weight compared to wild type mice despite comparable food consumption, indicating that the membrane pool of ERαis likely not involved in E2 regulation of body weight and obesity. As described below, the nuclear and mitochondrial ERs are involved in the regulation of proteins/enzymes involved in several energy metabolism pathways, it is likely that the nuclear and mitochondrial pools of ERα may be responsible for E2 regulation of body weight and obesity.
III. Imbalance of Energy Metabolism/Utilization and Obesity
Obesity is associated with the accumulation of excessive body fat resulting from chronic imbalance of energy whereby the intake of energy exceeds expenditure. A balanced body energy budget controlled by limitation of calorie uptake and/or increment of energy expenditure is the most effective way against obesity. An elevation of energy expenditure and/or a suppression of energy intake are the ways to reduce obesity.
The energy metabolism pathways in the cells include: a) glucose transport, b) glycolysis; c) tricarboxylic acid (TCA) cycle, d) mitochondrial respiratory chain (MRC) mediated electron transfer/oxidative phosphorylation; and e) ATP translocation and utilization. The biochemical pathways involved in energy storage include fatty acid (FA) biosynthesis/oxidation and triglyceride biosynthesis/disposition. A well balance of energy flow requires the coordination of these biochemical pathways involved in the entire energy metabolism pathways. Overabundance in some pathways coupled with the deficiency in other pathways will cause energy imbalance. Metabolic energy expenditure flow (glucose transport → glycolysis → TCA cycle → MRC→ ATP utilization) negatively regulates energy balance, whereas energy storage processes (FA biosynthesis→FA disposition→cholesterol and triglycerides synthesis/storage) and storage of low-density lipoproteins could opposite the energy balance. Catabolic pathways split carbohydrate molecules into small molecules and generate reduced coenzymes (NADH & FADH) and ATP. For example, it has been shown [48] that enhancing hepatic glycolysis by over-expressing the key enzymes involved in glycolysis, e.g. glucokinase and 6-phospho fructose-2-kinase/fructose-2,6-bisphosphatase, reduced bodyweight and adiposity in obese mice. On the other hand, inhibition and/or deficiency in other links of the energy expenditure flow chain may lead to abnormal energy flow. Chronic physical, biochemical and cellular activities that disturb the energy balance could either reduce or enhance obesity.
IV. Regulation of Energy Metabolism Pathways and Energy Utilization by Estrogens and ERs and Their Disturbance of by Endocrine Disrupting Chemicals
It has been observed that during pregnancy or after experimental manipulations of ovarian hormones (e.g. Ovx and replacement therapy with E2 or with E2 + progesterone), Syrian hamsters exhibited changes in energy balance and body fat content without modifying their food intake, suggesting that estrogens play an important role in the regulation of energy balance [49]. In fact, estrogens and ERs are involved in the regulation of energy metabolism and utilization from glucose transport→glycolysis/glucoegenesis→TCA cycle →MRC-mediated electron transport/oxidative phosphorylation→ATP translocation (ANT) and utilization by directly or indirectly regulating the activity and expression of the key enzymes/proteins involved in these processes (Fig. 1).
Figure 1. Proposed Models for the Regulation of Energy Metabolism Pathways and Utilization by Estrogens and ERs.
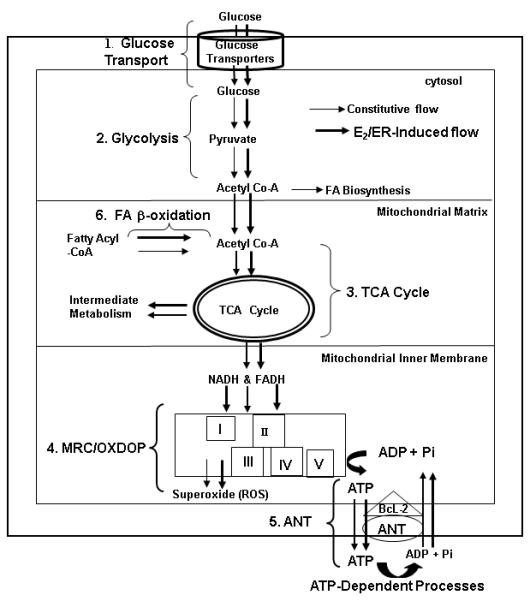
There could be two energy flows: constitutive energy flow, indicated by the black arrows; and E2/ER-indcued energy flow, indicated by the bold red arrows
A large number of exogenous chemicals are capable of altering endocrine functions and causing adverse effects at the levels of organism, its progeny and/or subpopulation of organisms. Some of which posses estrogenic and/or anti-estrogenic activities and are capable of disrupting endocrine systems and thus, are defined as endocrine disrupting chemicals (EDCs) [50]. It has been reported [51] that exposure of animals to several EDCs, including phytoestrogens, synthetic estrogens (e.g. diethylstilbestrol, DES) and environmental EDCs caused changes in bodyweight and development of obesity: i) developmental exposure to low dose of DES is associated with decreased body weight gain, in association with significantly elevated metabolic rate [52, 53]; ii) Neonatal exposure to soybean products enriched in phytoestrogens altered body weight, adiposity and adipokines in adult female mice, indicating that brief exposure to phytoestrogens early in life affect mouse white adipose mass and serum adenosines [54, 55]; and iii) A significant decrease in maternal body weight gain and food consumption was seen in pregnant rats treated with butyl benzyl phthalate (BBP) by gastric incubation at 1000 mg/kg on days 15 to 17 of pregnancy. This treatment also caused significant decreases in the weighs of their male and female fetuses [56]. It has been reported [57] that the offspring of Sprague-Dawley female rats exposed prerinatally to low doses (0.1 mg/kg body weight/day) of bisphenol A(BPA), a monomer used in the manufacture of polycarbonate plastics and resins, from day 6 of pregnancy through the period of lactation, exhibited an increased body weight that was apparent soon after birth and continued into adulthood. The female offspring from female rats exposed perinatally to high dose (1.2 mg /kg body weigh/day) to BPA exhibited altered patterns of estrous cycle and decreasing levels of plasma luteinizing hormone in adulthood. Decreased maternal body weight, body weight gain and reduced food consumption in pregnant rats and their fetuses was also observed in pregnant female rats exposed perinatally to BPA [58, 59]. There is increasing concern for the adverse health outcomes after developmental exposure to these environmental EDCs capable of perturbing the endocrine systems. The known adverse effects caused by exposure to these EDCs include the low reproductive activity in female offspring and high increase of reproductive tract dysfunction in both male and female offsprings. Another important consequence of exposure of animals to these EDCs is the altered body weight (either gain or loss). However, the cellular and molecular mechanisms by which E2 and EDCs are involved in the regulation of body weight and obesity are poorly understood, pointing to the urgent need of a comprehensive investigation into this area. As described below, EDCs are capable of disturbing several key aspects of E2/ER-mediated energy metabolism pathways.
4.1. Regulation of Glucose Transport by E2/ERs and Potential Disturbance by EDCs
4.1.1. Regulation of Glucose Transport by E2/ERs
Hexoses including glucose and fructose play an essential role in energy metabolism and other cellular metabolism. Glucose enters eukaryotic cells via so-called “glucose transporters”, the membrane-associated carrier proteins. There are two different types of glucose transporters: the Na+-coupled glucose transporters (SGLT) and glucose transporter facilitators (GLUT). SGLT family consists of three members: SGLT1, SGLT2 and SGLT3. SGLT1 and SGLT2 function as sugar transporters whereas SGLT-3 serves as a glucose sensor. Human GLUT family consists of 14 members (designated GLUT-1 to GLUT-14), among which eleven are involved in sugar transport. The individual isoforms of glucose transporters exhibit different substrate specificity, kinetic characteristics, and expression profiles, thereby allowing a tissue-specific adaptation of glucose uptake through regulation of their gene expression. In addition, some transporters (e. g. GLUT-4 and GLUT-8) are regulated by their subcellular distribution. In addition to mediating glucose entry into cells, some isoforms (e.g, GLUT-2) appear to be responsible for glucose sensing of pancreatic β-cells, neuronal, or other cells, thereby playing a major role in the hormonal and neural control (for review see [60]). More importantly, several congenital defects of sugar metabolism are associated with aberrant transporter genes. For instance, aberrant SGLT1 is related to the glucose-galactose malabsorption syndrome whereas deficiency in GLUT-1 is related to the glucose transporter 1 deficiency syndrome. Fanconi-Bickel syndrome is caused by defects in GLUT-2. A malfunction of glucose transporter expression or regulation of GLUT-4 appears to be related the insulin resistance syndrome [60]. Increased uptake of glucose compared to cells in normal tissue is a defining characteristic feature of malignant cells and high expression of the glucose transporters were specifically identified in human breast cancer cells [61-66]. Over-expression of GLUT-1 was found to be a characteristic feature in breast cancer biopsies, with a positive correlation between its expression and tumor grade, extent of proliferation and invasiveness [67-72] whereas decreased expression of GLUT-4 was seen early in the development of feline obesity[73].
Estrogens are known to influence glucose homeostasis with dominant effects in the liver. While the mechanisms of these effects are unknown, estrogens have been known to stimulate glucose transport in various target organs by altering the expression of genes encoding proteins and enzymes involved in glucose metabolism [63, 67, 68]. Kumagai et al. [69] examined the effects of E2 and progesterone (P) on insulin sensitivity in Ovx rats by the euglycaemic hyperinsulinaemic clamp technique and measurements of insulin-stimulated 2-deoxy-D-glucose (2-DOG) transport and glycogen synthesis in white and red parts of the gastrocnemius, the extensor digitorum longus and soleus muscles and in the liver (only glycogen synthesis). They observed that Ovx was followed by insulin resistance and paralleled by a decreased insulin-stimulated content of 2-DOG in muscles, an index of glucose transport. Glycogen synthesis in muscle was also decreased, although to less extent. E2, alone or in combination with P, restored this to values of intact controls. Ovx led to decrease in liver glycogen synthesis, which was restored by the treatment with E2 plus P. Thus, E2 plays an important role in the maintenance of normal insulin sensitivity while P alone seems to be followed by insulin resistance. E2 plus P may be of importance for maintenance of normal glycogen synthesis in the liver. In investigating the clinical significance of GLUT-1 expression in human breast carcinoma, Kang et al. [77] observed a significant correlation between GLUT-1 expression and ER and PR status. E2 stimulated GLUT-1 expression and this effect was inhibited by tamoxifen [65, 70, 71]. Cheng et al. [70] observed that E2 treatment induced two- to four fold increases in Glut3 and Glut4 mRNA levels and lesser but significant increases in Glut3 and 4 protein levels in primate cerebral cortex. Estrogens induced approximately 3-to 4-fold increases in mRNA and protein of GLUT-1 in the immature rat uterus [71]. E2 and epidermal growth factor also increased protein levels of GLUT12, a novel glucose transporter protein located intracellularly and at the cell surface, in cultured breast cancer cells [72].
The expression of Glut-2 is also regulated by peroxisome proliferator activator receptor gamma (PPAR-γ in the liver. It has been reported that rosiglitazone, a PPAR-γ agonist, enhanced the GLUT-2 mRNA level in the primary cultured hepatocytes and Alexander cells, when they were transfected with PPAR-γ/RXR-α. A peroxisome proliferator response element was identified in the mouse GLUT-2 promoter. PPAR-γ bound to the -197-184 region of the promoter. A steroid hormone responsive element was also identified in the promoter of this gene [73, 74]. These results suggest that liver GLUT-2 may be a direct target of PPAR-γ ligand contributing to glucose transport into liver in a condition when PAPR-γ expression is increased as in type-2 diabetes or in severe obesity. There is signal cross-talk between ERs and PPAR-γ in the regulation of estrogen-responsive genes and peroxisome proliferator-responsive genes [75, 76]. It is likely that E2 and ERs are involved in the regulation of GLUT-2 and, perhaps, other glucose transporter genes as well.
Barros et al. [85] investigated the influence ERα and ERβ on regulation of GLUT-4 and caveolin-1, a structural protein associated with GLUT-4, in mouse gastrocnemius muscle. Immunohistochemical analysis revealed that both ERs were co-expressed in the nuclei of most muscle cells, and that their levels were not affected by the absence of E2 in ArKO mice. GLUT-4 expression on the muscle cell membrane was not affected by loss of ERβ but was greatly reduced in ERαKO mice and elevated in ArKO mice, accompanied by a parallel reduction in Glut-4 mRNA levels in ERαKO mice. Upon treatment of ArKO mice with the ERα agonist, 2,3-bis (4-hydroxyphenyl) propionitrile (PPT), GLUT-4 expression was reduced. Caveolin-1 expression was higher in ArKO mice and lower in ERαKO and ERβKO mice than in WT littermates. GLUT-4 and caveolin-1 were co-localized in WT and ArKO mice but not in ERαKO and ERβKO mice. These results reveal that ERα is a positive regulator of GLUT-4 expression, whereas ERβ has a suppressive role. Both ERα and ERβ are necessary for optimal caveolin-1 expression. These results indicate that co-localization of caveolin-1 and GLUT-4 is not an absolute requirement for muscle glucose metabolism but reduction in GLUT-4 may contribute to the insulin resistance observed in ERαKO mice.
4.1.2. Potential Disturbance of Glucose Transport by EDCs
Glucose transporters could be the potential targets of estrogenic and anti-estrogenic EDCs. Sakurai et al. reported [77] that treatment of mouse 3T3-F442A adipocytes with BPA caused increased GLUT-4 protein levels and enhanced basal and insulin-stimulated glucose uptake. Bogacka et al. [78] observed decreased expression of GLUT-2 mRNA and increased expression of glucokinase in the hindbrain of obese rats. By changing the expression of glucose transporter genes, e.g. GLUT-1, GLUT-2 and GLUT-4, estrogens and estrogenic EDCs are able to alter glucose transport and glucose metabolism, leading to abnormal glucose metabolism which could be associated with the development of obesity [79].
4. 2. Regulation of Glycolysis by E2/ ERs and Potential Disturbance by EDCs
4.2.1. Regulation of Glycolysis by E2/ ERs
Glycolysis is the biochemical pathway catalyzed by a number of enzymes, leading to the breaking down of the imported glucose and fructose into three-carbon molecules, pyruvate, with generation of 2 molecules of NADH and ATP (Fig. 2). The enzymes participating in glycolysis include hexokinase (HK)(e.g. glucose kinase, GK), phosphoglucoisomerase(PGI), phosphofructokinase(PFK), aldolase(AD), glyceraldehyde-3-phosphate dehydrogenase(GAPD), phosphoglycerate kinase (PGK), phosphoglycerate mutase(PGM), enolase(EN) and pyruvate kinase(PK). 6-phospho fructose-2-kinase, fructose-2,6-bisphosphatase(PFKFB) and glucose-6-phosphatase are also involved. PFKFB is a bifunctional enzyme responsible for maintaining the cellular level of fructose-2, 6-bisphosphate, a powerful allosteric activator of glycolysis. Minchenko et al. [80] observed over-expression of PFKFB-isozyme-4 (PFKFB-4) in the human breast and colon tumors as compared to corresponding non-malignant tissues. They have shown that breast cancer MCF-7 cells constitutively express PFKFB-4 mRNA and that the expression of this gene was highly induced by hypoxia. Over-expression of PFKFB-4 transcript levels in breast and colon tumors correlated with the enhanced expression of PFKFB-3, hypoxia-inducible factor (HIF)-1, HIF-1 dependent-Glut-1 and vascular endothelial growth factor (VEGF). These data clearly demonstrates over-expression of PFKFB-4 in the breast and colon malignant tumors.
Figure 2. Regulation of Glycolytic Enzymes by E2/ERs.

Biochemical reactions of glycolysis and the enzymes that catalyze these reactions are shown. The stimulating effects of E2 on expression and/or activity of the enzymes are indicated by arrows
Several studies have suggested that estrogens and ERs play a role in the regulation of glycolysis. For instance, Furman et al [63] observed that estrogens induced glycolysis in MCF-7 cells, which were inhibited by temoxifen (TAM). Kostanyan and Nazaryan [81] examined the effect of E2 on the activities of the following glycolytic enzymes in female rat brain: HK, PFK, AD, GAPD, PGK, PGM and PK. The activities of HK (both soluble and membrane-bound forms), PFK and PK were increased 4h after E2 treatment, while the others remained unchanged. The changes in activity of these enzymes were not seen in the presence of actinomycin D. The significant rise in the activities of the key glycolytic enzymes was also observed in the cell culture of mouse neuroblastoma C1300 treated with E2. Only three of the studied isozymes, namely, HKII, B4 and K4, for HK, PFK and PK, respectively were found to be E2-sensitive. The results suggest that rat brain glycolysis is regulated by E2 and is carried out in neurons due to an induction of definite isozymes. Reiss [82] examined the effects of the ontogeny and estrogen responsiveness of PGK, PGM, En, and PK in rat brain and uterus. In 30-day-old rats, their brain and uterus express the fetal isoforms CK-B, PGK-A, PGM-B, En-β and PKM-2, and the differentiated isoforms En-α and PKM-1. The activity of glycolytic enzymes in uterus of two-day-old rats is as in brain, while CK activity was 3 times higher in brain. The activities of the glycolytic enzymes in brain began to increase (3-4-fold) 10 days after birth, in a coordinated manner. In contrast to brain, the levels of glycolytic enzymes in uterus were highest at birth, suggesting the action of a tissue-specific mechanism for regulation of the constitutive levels of glycolytic isozymes. All enzymes but PGM showed an increase in total activity in uterus in response to E2. Bogacka et al. observed [78] increased expression of GK in the hindbrain of obese rats, suggesting that altered expression of this protein could be related to obesity More recently, Pastorelli et al. [83] identified, using proteomic analysis, the in vivo E2-regulated proteins in female mice and found that EN-β and PKM-2 were E2-regulated. Yan and Vatner [84] performed proteomic analysis to study changes in proteins in hearts of four groups of monkeys: young males (YMs); old males (OMs) and young females (YFs) and old females (OFs). They found decreased expression of key enzymes in glycolysis (e.g. PK, EN-α, and triosephosphate isomerase) in Oms. These changes in glycolytic enzymes were not observed in Yms, YFs and OFs groups. Thus, E2 enhances the expression and activities of several key enzymes involved in glycolysis.
4.2.2. Potential Disturbance of Glycolysis by EDCs
Estrogenic and anti-estrogenic EDCs have been shown to have substantial effects on glycolysis. Kim et al. [93] tested effects of phenolic compounds such as BPA, 4-nonylphenol (NP), 4-octylphenol (OP) and 4-propylphenol (PP) on glucose-6-phosphate dehydrogenase (G6PD) in MCF-7 cells and female immature Sprague-Dawley (SD) rats. They observed that at the concentration higher than 1 × 10-8 M, BPA significantly increased the G6PD activity in a dose-dependent manner. NP (at concentration above 1 × 10-9 M) also enhanced the G6PD activity by about 1.8 times over the control. OP produced weaker effects on G6PD than NP did, and showed a tendency to increase the G6PD activity whereas PP did not affect the G6PD activity. These results show that BPA and NP have the effect of enhancing G6PD activities in MCF-7 cells. In an in vivo GPx assay, both BPA and E2 significantly increased the wet uterus weights in immature female rats. Treatment with NP (500 mg/kg/day) significantly increased wet uterus weights in immature female rats. This study implies that phenolic EDCs have weak estrogenic effects on glycolysis. Oral contraceptives (OC) containing synthetic estrogen e,g. ethinyl estradiol (EE) and progestogens (P) are known to reduce glucose tolerance. It was reported [85] that women who used OC for periods exceeding 10 cycles (12-36 months) showed significantly reduced activity of PFK, a key glycolytic enzyme, by 40% and the levels of lactate by 42% in the erythrocytes compared to controls. These observations in women are consistent with those made in female rats and suggest that impaired glucose tolerance is due to reduced glycolysis through lowering the levels of PFK synthesis by EE and P. In vivo intracellular defects in glucose metabolism were observed in obese patients with non-insulin-dependent diabetes mellitus, characterized by decreased glucose uptake. Consistent with the in vivo metabolic defects, pyruvate dehydrogenase and glycogen synthase activities were decreased in leg muscle biopsies of obese patients [86]. It has been reported [87, 88] that the phytoestrogens, genistein and daidzein, down regulated several glycolytic enzymes including G6Pase and PEPCK, and played important roles in regulation of glucose homeostasis in type 1 diabetic mice. In these cases, EE, P, genistein and daidzein appear to act as anti-estrogen agents.
The insulin resistance of skeletal muscle in glucose-tolerant obese individuals is associated with reduced activity of oxidative enzymes and a disproportionate increase in activity of glycolytic enzymes. Non-insulin-dependent diabetes mellitus (NIDDM) is a disorder characterized by even more severe insulin resistance of skeletal muscle. Many individuals with NIDDM are obese. Simoneau et al. [89] examined whether decreased oxidative and increased glycolytic enzyme activities are present in NIDDM. Percutaneous biopsy of vatus lateralis muscle was obtained in eight lean (L) and eight obese (O) non-diabetic subjects and in eight obese NIDDM subjects, which were assayed for marker enzymes of the glycolytic [e.g. PFK, GAPD, HK], glycogen phosphorylase and creatine kinase, a marker of anaerobic ATP re-synthesis and oxidative pathways e.g. citrate synthase (CS) (Tricarboxyl acid cycle) (see section 4.3.) as well as cytochrome-c oxidase (mitochondrial respiratory chain)(see section 4.4). Activities for PFK, GAPD and HK were highest in subjects with NIDDM, following the order of NIDDM > O > L, whereas maximum rate for CS, cytochrome-c oxidase was the lowest in subjects with NIDDM. The ratio of glycolytic/oxidative enzyme activities within skeletal muscle correlated negatively with insulin sensitivity. The HK/CS ratio had the strongest correlation with insulin sensitivity. An imbalance between glycolytic and oxidative enzyme capacities is present in NIDDM subjects and is more severe than in obese or lean glucose-tolerant subjects. The altered ratio between glycolytic and oxidative enzyme activities found in skeletal muscle of individuals with NIDDM suggests that a deregulation between mitochondrial oxidative capacity and capacity for glycolysis is an important component of the expression of insulin resistance[89]. These observations indicate that disturbances of glycolysis by EDCs could be related to obesity and other metabolic diseases.
4. 3. Regulation of Tricarboxylic Acid(TCA) Cycle by E2/ERs and Potential Disturbance by EDCs
4. 3. 1. Regulation of TCA Cycle by E2/ERs
TCA cycle (also called Citric acid cycle) is a biochemical pathway (Fig. 3), which, together with MRC electron transport/oxidative phosphorylation (see section 4.4), plays a pivotal role in cellular respiration. TCA cycle has multiple interconnections with other pathways and provides intermediate substrates for inter-conversion of numerous metabolites. The enzymes and proteins involved in this pathway are shown in Fig. 3.
Figure 3. Regulation of TCA Cycle By Estrogens/ERs.
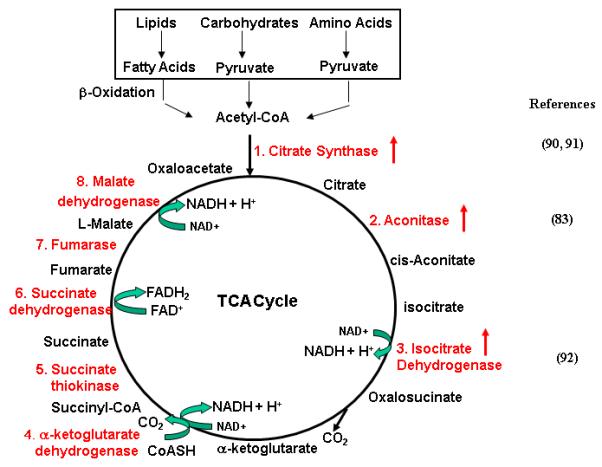
Biochemical reactions of TCA cycle and corresponding enzymes are shown (indicated by red color). Red arrows point to the enzymes whose expression/activity are regulated by E2/ERs.
Estrogens are involved in the regulation of several key enzymes in TCA cycle. The first reaction of TCA cycle is a condensation between the acetyl group of acetyl CoA and oxaloacetate to form citrate. This reaction, a key step in TCA cycle, is catalyzed by citrate synthase (CS). Beckett et al. [90] observed that the activity of CS in skeletal muscle of Ovx rats treated with E2 for 16 days was significantly higher than that in both Ovx and Sham rats. Enhanced CS activity was also observed in cerebral blood vessels of Ovx rats treated with E2 [91]. Pastrorelli et al. [83] identified the enhanced expression of mitochondrial aconitase 2 in Ovx mice treated with E2, as compared with Ovx mice and Sham mice. Yadave [92] observed that treatment Ovx female rats with E2 enhanced activities of NAD- and NADP-linked isocitrate dehydrogenase (ICDH) in the brain, liver and kidney cortex. Yan and Vatner [84] observed that the protein levels of 2-oxoglutarate dehydrogenase were lower in left ventricular (LV) samples of old male monkeys than those in LV samples of old female monkey, probably due to the different E2 levels between male and female monkeys. Gupta et al. [93] investigated the physiological role of E2 in the regulation of energy metabolism of epididymis of rhesus monkey. They measured the activity of succinate dehydrogenase and malate dehydrogenase in these two organs of (a) castrated estrogen treated and (b) castrated estrogen + dihydrotestosterone (DHT) treated animals, and compared with those in castrated and castrated + DHT treated animals. Their results indicated that DHT alone stimulated the activities of all these enzymes whereas E2 failed to stimulate any of the enzymes in castrated animals. However, E2 in combination with DHT caused a marked stimulation of the enzymes and the response of the epididymis. Combination treatment was significantly higher than that caused by DHT alone. The results suggest that circulating estrogen in male has a physiological role and acts synergistically with androgen in regulating accessory sex organ function. Nilsen et al. [94] investigated the effects of estrogens on brain mitochondrial proteome in OVX rats using a proteomic approach. They observed that treatment of OVX rats with E2 for 24 hours significantly enhanced the protein levels of several enzymes involved in TCA cycle, including aconitase, PDH E1b, PDHE2, malate dehydrogenase as well as enzymes involved in coupling of TCA cycle with amino acid synthesis such as Glut OxaL2 and glut DH. These observations indicate that estrogens regulate the activity and expression of enzymes involved in TCA cycle and enzymes involved in intermediate pathways coupling with TCA cycle.
4.3.2. Potential disturbance of TCA by EDCs
There is evidence that TCA cycle is disturbed by EDCs and that this could be related to obesity. Baltrus and Melchior [95] examined the effects of DES on aconitase, succinate dehydrogenase and glutamic-oxalacetic transaminase in rat pituitary and liver and observed that in pituitary, aconitase and succinate dehydrogenase were decreased by approximately the same amounts by DES treatment; and in liver, succinate dehydrogenase and glutamic-oxalacetic transaminase were decreased by DES treatment.
It has been shown that the enzymatic activities involved in TCA cycle are reduced in obese patients. Raben et al. [96] examined muscle fiber morphology and activities of four key enzymes, as well as energy metabolism, in nine normal-weight post obese women and nine matched control subjects. They observed no differences in fiber type composition, but a smaller mean fiber area and area of fiber types I and IIb were found in post obese compared with control subjects. The activities of β-hydroxyacyl-CoA dehydrogenase (HADH) and CS were 20% lower in post obese than in control subjects. However, the activities of lactate dehydrogenase and lipoprotein lipase were not significantly different between post obese and control subjects. Basal metabolic rate and respiratory exchange ratio were similar, but maximal oxygen uptake tended to be lower in post obese than in control subjects. These data suggest that smaller fiber areas and lower enzyme activities, i.e., markers of aerobic capacity of skeletal muscle, may be factors predisposing to obesity.
4.4. Regulation of MRC by E2/ERs and potential disturbance by EDCs
4.4.1. Regulation of MRC by E2/ERs
Mitochondria are best known for their ability to generate the majority (>90%) of cellular energy in the form of ATP and reactive oxygen species (ROS) from NADH and FADH using molecular oxygen (O2) via electron transfer/oxidative phosphorylation [97]. Mitochondria also play a central role in the regulation of cellular survival and apoptosis [98, 99]. In addition, they are involved in the modulation of important cellular processes e.g. intracellular calcium homeostasis, various ion channels and transporters.
MRC is a major structural and functional part of mitochondria. It consists of four complexes, namely complexes I, II, III, and IV, and mitochondrial ATP synthase (complex V) (Fig. 4). All but complex II whose subunits are encoded solely by nDNA contain protein subunits encoded by both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA). The mtDNA-encoded subunits include seven subunits of complex I, one subunit of complex III, three subunits of complex IV and two subunits of complex V. These mtDNA-encoded subunits carry out the basic catalytic role of electron transport. The remaining more than 80 protein subunits are encoded by nDNA and are believed to perform regulatory role. In addition, a number of other protein factors are involved in replication, transcription and translation of mtDNA, all of which are encoded by nDNA, synthesized in cytoplasm and targeted to mitochondria. The proper MRC biogenesis depends on the coordinated expression and correct assembly of the nDNA- and mtDNA-encoded proteins, a complex process that requires a variety of well orchestrated regulatory mechanisms between two separated nuclear and mitochondrial compartments [100, 101]. A number of specific nDNA-encoded factors are involved in the regulation of mitochondrial gene expression and complex assembly in response to growth factors and hormone stimuli [101]. Several lines of evidence indicate that MRC is an important target of estrogens and ERs. The effects of E2/ERs on MRC have been reviewed [102]. Several important points are highlighted as follows:
Figure 4A. Regulation of MRC Proteins by E2/ERs.
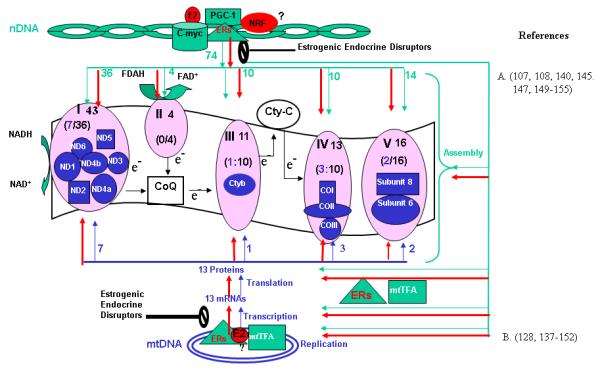
MRC complexes I, II, III, IV and V are shown. Genes encoded by nuclear DNA (nDNA) are indicated by green color; genes encoded by mtDNA are indicated by blue color. The number indicates the number of protein subunits. The red, bold arrows indicate the potential effects of E2 on the expression of nDNA- and mtDNA-genes. Potential targets for estrogenic and anti-estrogenic chemicals are marked by the block symptoms. COI, II, III: cytochrome c oxidase subunits I, II and III; cytb: cytocrome b; cyt c:cytochrome c; CoQ: Coenzyme Q; ND 1to ND6: NADH dehydrogenase subunits 1 to 6; mtTFA: mitochondriual transcription factor A; NRF: Nuclear respiratory factor; PGC-1: subunits 6 & 8: ATP synthase subunits 6 & 8.
A) Estrogens Have Effects on MRC Structure and Functions
The effects of estrogens on mitochondrial structure [103, 104], biogenesis [105, 106], respiration [107, 108], and oxidative phospholylation [109-113] have been documented. There are accumulating evidence supporting the requirement of estrogens and ERs for the preservation and regulation of MRC structure and function [114-124].
B) ERα and ERβ Localize within Mitochondria of E2-target cells
ERβ is present in mitochondria of uterus, ovary [125], rat primary neurons, primary cardiomyocytes, murine hippocampus cell lines [126], the perikarya and proximal dendrites of pyramidal and granule cells of rat coronal hippocampus [127]; human breast cancer MCF-7 [128-130], liver cancer HepG2 cells [128, 129], heart cells[126]; lens epithelial cells [131, 132], osteosarcoma SaOS-2 [133] and sperm [134]. ERα is also detected in mitochondria of HepG2 [128, 129], MCF-7 [128, 130, 135] cells and rat cerebral blood vessels [136].
C) Mitochondrial ERs Play an Important Role in E2-induced mtDNA gene expression
As indicated in Fig. 4, thirteen MRC proteins are encoded by mtDNA. It has been shown that transcript levels of mtDNA genes are enhanced by estrogen in a number of cell types including GH4C1 pituitary tumor cells [137], hippocampus from estrogen-stimulated Ovx female rats[138], the pituitary of Ovx rats [139], female rat liver, cultured hepatocyte and HepG2 cells [140-142], MCF-7 cells [128], rat cerebral blood vessels [91], adipose tissue of mice [143], human breast epithelial cells (MCF-10F) [144], and rat brain [94]. Moreover, it was reported [145] that the levels of mtDNA-encoded 16S RNA were four times higher in mitochondria from female rats than in mitochondria from male rats. In aorta of Ovx mice, the expression of NADH-ubiquinone oxidoreductase subunit 6(ND6)(complex I), two mitochondrial ribosomal RNAs (16S and 12S) and three mitochondrial transfer RNAs was found to be down regulated, principally but not exclusively due to the reduction of estrogens [146]. More importantly, the stimulating effects of E2 on mRNA levels of the mtDNA-encoded genes were significantly inhibited by ICI182780, a pure ER antagonist, and by silencing the expression of ERβ with siRNA, suggesting the involvement of ERs [135, 150]. These known stimulatory effects of E2 on mtDNA transcription and the presence of ERs within mitochondria of target cells indicate that mtDNA could be a major target for E2 acting through the mitochondrial ERs.
Human mtDNA contains estrogen response elements (ERE) (mtEREs) some of which reside in within the D-loop [147, 148], a major regulatory region for mtDNA transcription. The presence of EREs in D-loop raises the possibility that the mitochondrial ER□ and ER□ are involved in regulation of mitochondrial gene expression via their binding to the mtEREs. Using electrophoresis mobility shift assays (EMSAs) and surface plasmon resonance (SPR), Chen et al. [129] demonstrated the binding of rhERα, rhERβ and mitochondrial extracts that contain ERβ to these mtEREs and the enhanced binding by E2 in a dose- and time-dependent manner, indicating that the mitochondrial ERs can interact with mtEREs and may be directly involved in E2 induction of mtDNA transcription.
D) Estrogens Stimulate Expression of nDNA-encoded MRC Proteins and MRC activity
The majority of MRC proteins and other proteins involved in replication, transcription and translation of mtDNA are encoded by nDNA, synthesized in cytoplasm and targeted to mitochondria. Proper MRC biogenesis and function depend on the coordinated expression and correct assembly of both nDNA- and mtDNA-encoded proteins, a complex process that requires a variety of well orchestrated regulatory mechanisms between two physiologically separated nuclear and mitochondrial compartments [100, 101]. Several studies indicate that estrogens induce the expression of nDNA-encoded MRC proteins and stimulate mitochondrial respiration [107, 108, 140, 145, 147, 149-153]. The above observations, together with the observed abnormal mitochondrial cristae in ARKO mice [117, 154] and in cardiocytes of ERαKO mice [115], and the gender differences in mitochondrial morphology and functionality [118, 120], suggest that E2 and ERs are involved in the coordinated expression of (i) mtDNA-encoded subunits; (ii) nDNA-encoded subunits and (iii) nDNA-encoded regulatory/accessory factors required for mtDNA replication, transcription, translation and assembly. However, the molecular mechanisms underlying these estrogen effects on expression of nDNA-encoded MRC proteins are largely unknown and warrant further investigation.
Consistent with the observed effects of estrogens on the expression of mtDNA- and nDNA-genes encoding MRC proteins, it has been observed that treatment of several types of cells with E2 or EE stimulated MRC activity via ERs, as reflected by increased generation of mitochondrial superoxide, ATP and increased oxygen consumption (parameters of MRC activity)[94, 141, 142, 155, 156], associated with inhibition of apoptosis and enhanced distribution of glutasthone in nucleus and mitochondria [141, 142, 157, 158]. It was also observed that treatment of rats with estrogens enhanced the mitochondrial energy efficiency of their cerebral blood vessels[91]. Consistent with these observation, Yan and Vatner [84] found reduced expression and function of proteins involved in electron transport and oxidative phosphorylation in mitochondria only in hearts from old male monkeys, with corresponding decreased oxidation rates with NADH and ascorbate-N,N,N’,N’ ”-tetramethyl-p-phenylenediamine substrates, whereas no such changes were observed in female monkeys. The fact that ovx-induced obesity in mice is associated with a decrease in oxygen consumption indicates repressed energy expenditure in the absence of E2 [159].
E) Regulation of Nuclear-encoded MRC Proteins by c-myc
Several transcription factors including c-myc are involved in the regulation of expression of nuclear-encoded MRC proteins. C-myc is a transcription factor whose expression is stimulated by E2. C-myc plays a central role in the regulation of cell size, cell proliferation and apoptosis. Gene expression analysis suggests that over-expression of c-myc induced nuclear-encoded MRC genes [160-162]. Li et al. [163] determined the effects of ectopic Myc expression or the loss of Myc on mitochondrial biogenesis. They observed that induction of c-Myc in P493-6 cells resulted in increased oxygen consumption and mitochondrial mass and function. Myc null rat fibroblasts have diminished mitochondrial mass and decreased number of normal mitochondria. Reconstitution of c-myc expression in Myc null fibroblasts partially restored mitochondrial mass and function and normal-appearing mitochondria. They also observed in primary hepatocytes that acute deletion of floxed murine Myc by Cre recombinase resulted in diminished mitochondrial mass. The microarray analysis of genes responsive to Myc in human P493-6 B lymphocytes supports a role for Myc in mitochondrial biogenesis, since genes involved in mitochondrial structure and function are over-represented among the Myc-induced genes. In addition to the known direct binding of Myc to many genes involved in mitochondrial structure and function, Myc bound the mitochondrial transcription factor A gene, which encodes a key transcriptional regulator and mitochondrial DNA replication factor, both in P493-6 lymphocytes with high ectopic MYC expression and in serum-stimulated primary human 2091 fibroblasts with induced endogenous MYC. These observations directly established a pivotal role for Myc in regulating mitochondrial biogenesis. The effects of c-myc in the regulation of nuclear-encoded MRC gene expression are mediated via ERs. Cheng et al. [164] demonstrated that c-myc physically interacted with ERαin ERα-responsive promoters and that estrogen enhanced the c-MYC-ERα interactions and facilitated the association of ERα, c-MYC, and the co-activator TRRAP with these EREs, resulting in chromatin remodeling and increased transcription. These results suggest that ERα and c-MYC physically interact to stabilize the ERα-coactivator complex, thereby permitting other signal transduction pathways to fine-tune estrogen-mediated signaling networks. It is likely that c-myc is involved in the regulation of nuclear-encoded MRC genes via its interaction and cooperation with ERs because the promoters of several nuclear-encoded MRC genes contain EREs and sp1 sites.
4.4.2. Potential Disturbance of MRC by EDCs
E2/ER-mediated MRC protein synthesis and oxidative phosphorylation could be disturbed by estrogenic and anti-estrogenic EDCs. As indicated in Fig.1, there are two potentially important targets for EDCs: a) E2/ER-mediated nDNA-encoded MRC proteins and other protein factors/transcription factors involved in the regulation of mtDNA transcription, translation and MRC complex assembly, and b) the E2/Mitochondrial ER-mediated mtDNA-encoded MRC proteins. Ooe et al. [170] investigated the role of DJ-1, an activated ras-dependent oncogene associated with the onset of familial Parkinson’s disease (PD) in humans, in oxidative stresses by BPA, which has been reported to induce oxidative stress in rodents, to male mice and cultured cells. They found that BPA significantly increased the expression level of DJ-1 in the sperm and brain in male mice. In cultured Neuro2a and GC1 cells, BPA induced ROS production and significantly compromised mitochondrial function concomitant with elevated expression and oxidization of DJ-1. DJ-1 was found to maintain the complex I activity against BPA-induced oxidative stress after the localization in mitochondria. These results indicate that BPA affects MRC function and that DJ-1 plays a role in the prevention of BPA-induced mitochondrial injury.
4.5. Regulation of Adenosine Nucleotide Translocator (ANT) by E2/ERs
ANT mediates ATP/ADP shuttle cross mitochondrial membrane, thereby transporting the majority of MRC-generated ATP out of mitochondria in exchange of ADP and Pi (Fig. 5). There are several tissue-specific isoforms of ANT, namely ANT-1, ANT-2, ANT-3 and ANT-4. The expression of the heart-specific ANT isoform (ANT-1) is enhanced by E2 [165]. Interestingly, the anti-apoptotic protein, Bcl-2, is an important component of mitochondrial ANT system (Fig. 5). One of the important functions of Bcl-2 in mitochondria is its involvement in the regulation of ANT operation. Bcl-2 enhances ANT-dependent ADP/ATP exchange activity by direct protein-protein interaction with ANT [166, 167]. Bcl-2 is enhanced by E2 a number of cells [142, 168-173]. In addition to regulating apoptosis, Bcl-2 and members of Bcl-2 family regulate mitochondrial and cell physiology. For instance, t-Bid, a Bcl-2 family member, has been shown to modulate reorganization of mitochondrial cristae. Bcl-2 appears to regulate voltage-dependent anion channel permeability, which has important consequences for mitochondrial transport of adenine nucleotides, Ca2+, and other metabolites. BAD, another member of Bcl-2 family, is required for the binding of glucokinase to a mitochondrial complex. Thus, BAD null mice have altered glucose homeostasis. It has been suggested that Bcl-2 family members may regulate important mitochondrial and cellular functions and serve as sentinels to detect abnormalities in these pathways and, when the abnormalities are severe enough, to initiate or facilitate cell death. Understanding these physiological processes controlled by Bcl-2 and Bcl-2 family will not only be important in understanding cell regulation, and may provide new insights into the regulation of apoptosis [174] but also provide new insights into E2-regulated ANT operation. Because Bcl-2 and t-Bid are involved in ANT-mediated ATP/ADP shuttle and the expression of ANT-1 and Bcl-2 is stimulated by E2, it is likely that estrogen and estrogenic chemicals are capable of enhancing the transport of the majority of MRC-generated ATP out of mitochondria for ATP-dependent cellular processes, stimulating energy expenditure. On the other hand, it has been observed [175] that an anti-estrogen, tamoxifen, decreased mitochondrial ANT content, inhibited the phosphate carrier and induced ATP hydrolysis. It is possible that other EDCs with anti-estrogenic activity are capable of similarly disturbing ANT-mediated ATP/ADP shuttle. Further studies are required to better understand the regulation of ANT by estrogens and anti-estrogenic EDCs.
Figure 5. Regulation of ANT by E2/ERs.
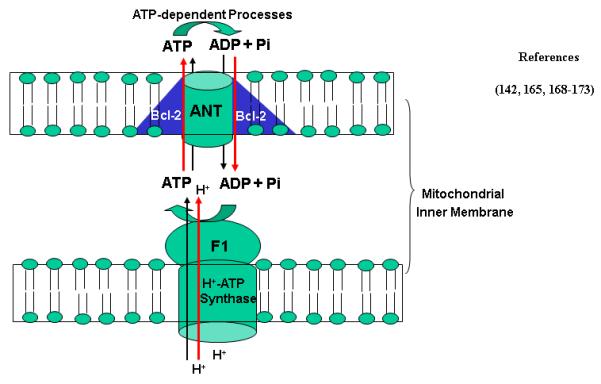
Bcl-2 is an important component of ANT shuttle. The expression of Bcl-2 and some isoforms of ANT is stimulated by E2. Red arrows indicate the stimulating effects of E2. Mitochondrial H+-ATP synthase is shown in lower panel.
4.6. Regulation of Fatty Acid β-Oxidation by E2/ERs and Its Disturbance by EDCs
4.6. 1. Regulation of Fatty Acid β-Oxidation by E2/ERs
Fatty acid (FA) β-oxidation is a sequence of four reactions catalyzed by acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-L-lhydroxyacyl-CoA dehydrogenase and βketoacyl-CoA thiolase. The end product of these reactions is acetyl-CoA, which can either enter TCA cycle or serve as a precursor for FA biosynthesis (Fig. 6).
Figure 6. Regulation of fatty acid β-oxidation by E2/ERs.
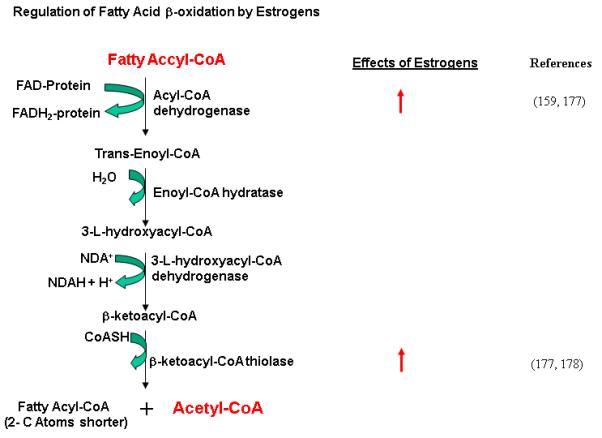
The Biochemical reactions of fatty acid β-oxidation and corresponding enzymes that catalyze these reactions are shown. The enzymes whose expression/activity are stimulated by E2/ERs are indicated by red arrow.
Estrogens play an important role in the regulation of β-oxidation of fatty acids. Postmenopausal women and rodents after Ovx can become obese. It has been reported [176] that myocardial fatty acid utilization was higher in women taking estrogens when compared with men and trended higher when compared with women not receiving E2, suggesting that in postmenopausal women, estrogen use is associated with increased myocardial fatty acid utilization and that estrogens are involved in the regulation of fatty acid metabolism. Toba et al. [177] performed biochemical analysis on the liver of ArKO mice whose endogenous estrogen levels were undetectable. Their results revealed suppression of mRNA expression and activity of enzymes involved in fatty acid metabolism, e.g. very long fatty acyl-CoA synthase (VLACS), medium chain acyl-CoA dehydrogenase, and peroxisomal acy-CoA oxidase (AOX). The impairment was reversed to the wild-type levels by treatment with E2. Kamei et al. [159] examined the expression patterns of genes related to energy expenditure and lipid metabolism in mouse tissues including adipose tissue and skeletal muscle of Ovx mice. They found that in adipose tissue and skeletal muscle, at 2-4 week after Ovs, levels of nuclear receptors and cofactors involved in energy expenditure such as ERR1, PPAR-γ PPARδ, PGC1α and PGC1β were lower than those in control mice. The mRNA levels of their targets, the enzymes for fatty acid β-oxidation (e.g. medium-chain acyl-CoA dehydrogenase and acetyl-CoA oxidase) were lower. In addition, the expression of PPAR-γ and SREBP1, transcription factors important for lipogenesis, was decreased, as well as that of acetyl CoA carboxylase and fatty acid synthase, enzymes for fatty acid synthesis, and diacyl glycerol acetyl transferase 1 and 2, enzymes for triglyceride synthesis. These changes in gene expression are consistent with the obese phenotype in mice after Ovx. Thus, a decrease in the expression of energy expenditure-related genes in adipose tissue and skeletal muscle could, in part, be responsible for obesity after Ovx. Together, these findings indicate a pivotal role of E2 in supporting constitutive hepatic expression of genes involved in fatty acid β-oxidation and in maintaining lipid homeostasis [159, 177, 178].
4.6.2. Potential Disturbance of FA β-Oxidation by EDCs
A number of estrogenic and ant-estrogenic chemicals have been shown to alter FA β-oxidation. For example, it has been reported [87] that phytoestrogens, e.g. genistein and daidzein, prevented diabetes onset by elevating insulin level and reducing hepatic gluconeogenic and FA β-oxidation activity in non-obese diabetic (NOD) mice.
Adjuvant tamoxifen has been widely used for breast cancer treatment. However, hepaticsteatosis is a frequent complication associated with adjuvant tamoxifen treatment. Impaired hepatic FA β-oxidation in peroxisomes, microsomes, and mitochondria results in progression of massive hepatic steatosis in estrogen deficiency, and this impairment is potentially serious because about 3% of the general population in the United States is suffering from nonalcoholic steatohepatitis associated with obesity and hyperlipidemia. Tamoxifen may be involved in impairment of FA β—oxidation in liver, causing hepatic steatosis. Equwa et al. [179] reported that impaired hepatic FA β-oxidation in ArKO mice defective in intrinsic estrogen synthesis could be restored by administering a statin, pitavastatin, to these mice, which significantly restored expression of essential enzymes involved in FA β-oxidation such as very long fatty acyl-CoA synthetase in peroxisome, peroxisomal fatty acyl-CoA oxidase, and medium-chain acyl-CoA dehydrogenase. Severe hepatic steatosis observed in ArKO mice substantially regressed. These findings demonstrate that pitavastatin is capable of restoring impaired FA β-oxidation in vivo via the peroxisome proliferator-activated receptor-alpha-mediated signaling pathway and is potent enough to ameliorate severe hepatic steatosis in mice deficient in intrinsic estrogens.
Adiponectin, which is expressed exclusively in adipose tissues, is able to enhance FA β-oxidation via activation of AMP-activated kinase (AMPK) and phosphorylation of acetyl CoA carboxylase (ACC). ACC phosphorylation and carnitine palmitoyl-transferase-1 (CPT1) activity is rate-limiting factors in FA β-oxidation. Yun et al. [180] observed that treatment of high fat diet-induced obese mice with bisphenol A diglycidyl ether (BADGE) or caffeine for 8 weeks caused a time-depended increase in expression of adiponectin and lipid oxidative enzymes, in association with reduced body weight and epididymal adipose tissue weight and a marked reduction in the number of fatty droplets in the liver.
Bitter melon (Momordica charantia) is one of the favorable vegetables in South China. It has been shown [181-184] that feeding diet-induced obese rats with bitter melon juice reduced adiposity, associated with increased lipid oxidative enzyme activities, uncoupling protein expression, suppression of the visceral fat accumulation, inhibition of adipocyte hypertrophy, and marked down regulation of expressions of lipogenic genes in the adipose. The phytoestrogens and/or other polyphenolic chemicals in bitter melon juice may be responsible for these effects.
Anti-obesity effects of green tea has been recognized [185]. Green tea catechins and epigallocatechin gallate (EGCG) have been demonstrated in cell culture and animal models of obesity to reduce adipocyte differentiation and proliferation, lipogenesis, fat mass, body weight, fat absorption, plasma levels of triglycerides, free fatty acids, cholesterol, glucose, insulin and leptin, as well as to increase beta-oxidation and thermogenesis. Adipose tissue, liver, intestine, and skeletal muscle are target organs of green tea, mediating its anti-obesity effects. Studies conducted with human subjects report reduced body weight and body fat, as well as increased fat oxidation and thermogenesis and thereby confirm findings in cell culture systems and animal models of obesity [185]. It has been shown [186] that EGCG inhibited obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice.
Together, these observations indicate that the endogenous and exogenous chemicals such as tamoxifen, BADGE, caffeine, phytochemicals in bitter melon juice and EGCG in green tea can cause altered expression of adiponectin and lipid oxidative enzymes and correlated with their long-term anti-obesity effects. More studies are needed to investigate their effects on energy metabolism pathways in relation to reduction of obesity.
V. Potential Implications of Disturbance of Energy Metabolism Pathways by EDCs in Obesity
Many lines of evidence indicate that estrogens and estrogenic chemicals are involved in the regulation of energy metabolism pathways from glucose transport → glycolysis → TCA cycle → MRC/Oxidative Phosphorylation→ATP translocation/usage (ANT) (Figs. 1 to 6). Thus, it is likely that the major impact of estrogens, estrogenic and anti-estrogenic EDCs on obesity is that they influence, either stimulate or inhibit, energy metabolism pathways by stimulating or inhibiting the activity and/or expression of key enzymes in these processes. Thus, they are involved in the regulation of energy balance. Estrogens have been shown to stimulate glucose transport and glycolysis in various target organs [63, 74, 75] by altering the expression levels of genes involved in glucose metabolism [65, 70, 71]. Estrogens also stimulate FA β-oxidation by altering hepatic expression of FA metabolizing enzymes[177, 178]. Several key enzymes in TCA Cycle [187-189] and mitochondrial ANT [165] are enhanced by estrogens. It is likely that E2 and ERs enhance the cellular energy generation and expenditure by stimulating MRC function/Oxidative phosphorylation and ANT shuttling.
The energy balance could be altered by up- or down-regulation of the expression and activity of key enzymes involved energy metabolism pathways, and which, in turn, modulate the bodyweight and obesity. For example, it was shown [48] that over-expressing the key enzymes in glycolysis, glucokinase or 6-phospho fructose-2-kinase/fructose-2,6-bisphosphatase, enhanced hepatic glycolysis and reduced bodyweight and adiposity in obese mice. Over-expression of c-myc in liver, the key transcription factors involved in the regulation of enzymes/proteins involved in glycolysis and MRC, prevented obesity and insulin resistance in mice [190]. Estrogens and ERs accelerate metabolic energy expenditure flow by stimulating the activity and/or expression of key enzymes in these processes and thus, enhance energy expenditure. E2 and ERs are important in the regulation of energy balance. On the other hand, deficiency in these effects, due to loss of estrogens in adult animals, e.g. in FSHRKO mice [191], Ovx mice and Ovx Syrian hamster [41, 143, 159, 192] and ARKO mice [16-18], in postmenopausal women or due to the loss of functional ERs, e.g. in ERKO mice and ER mutations, will lead to the inability of cells to cope with E2—induced stimulation of these energy metabolism pathways. As a result, the energy flow will be switched from energy expenditure to energy storage/deposition, and eventually obesity.
In general, any enzymes, particularly for those that catalyze the key steps in energy metabolism pathways, which are regulated by E2/ERs, could be potentially important targets for estrogenic and anti-estrogenic EDCs. Because MRC oxidative phosporylation and ANT shuttle are the rate-limiting pathways in overall cellular energy metabolism, disturbance of these pathways could lead to energy imbalance: inhibition of these pathways will lead to the inability of cells to cope with the continuing E2 stimulation of the upstream glucose translocation, glycolytic and FA β-oxidation pathways, causing accumulation of acetyl CoA, the precursor of FA biosynthesis. As a result, the energy flow will be switched from TCA cycle →MRC→ ATP production/utilization pathways (energy expenditure) to FA biosynthesis/deposition (energy storage), and thus, obesity; On the other hand, inhibition of other key steps in E2-induced energy expenditure pathway could block the energy expenditure pathways, the energy flow is switched toward energy storage pathways, leading to increased triglyceride storage. Thus, exposure to estrogenic or anti-estrogenic EDCs in critical stages of development will disturb E2/ER-mediated MRC energy metabolism, leading to persistent energy imbalance and finally obesity.
Increasing exposure to EDCs appears to be an emerging factor contributing to the current obesity epidemic. To date, only a limited number of EDCs have been used to assess the biochemical and molecular changes at the cellular level. To determine whether exposure to EDCs could lead to obesity and other diseases, we need to have the reliable biomarkers for EDCs and to establish the reliable, sensitive and accurate methods for detecting EDCs at low levels. Currently employed laboratory tests measure the effects solely via the classical nuclear-genomic pathways of EDCs. Because E2, EDCs and ERs can act in nucleus, plasma membrane and mitochondria, there are missing pathways whereby these EDCs operate. The important pathways that could be the disturbed by EDCs are E2/ER-induced energy metabolism pathways, particularly MRC protein synthesis and energy metabolism. These pathways are more directly related to obesity. As indicated in Fig. 1, there are two types of potentially important targets for EDCs. Investigating the effects of EDCs on the synthesis of proteins involved in grucose transport, glycolysis, TCA cycle, MRC, ATN and FA β-oxidationcould could lead to identification of reliable biomarkers for EDCs and to develop unique, accurate, rapid, and cost effective method for assessing the potential EDC activity during development.
VI. Conclusions and Future Perspectives
Evidence is accumulating that indicates that estrogens and ERs are involved in the regulation of energy metabolism pathways from glucose transport to glycolysis to TCA cycle to MRC/Oxidative phosphorylation to ANT shuttle. There might be two parts of energy expenditure: a constitutive flow (Fig. 1, indicated by black arrows) and an E2-induced flow (indicated by the bold, red arrows). The key rate-limiting steps in the energy expenditure pathways could be glucose transport, MRC-mediated electron transport/oxidative phosphorylation and the ANT-mediated ADP/ATP shuttle. Stimulation of glucose transport will increase the amounts of fuels for the entire energy metabolism pathways, whereas stimulation of the last two steps of the energy expenditure flow will create a greater potential, “pulling” a faster energy flow toward ATP utilization. By stimulating of these energy metabolism pathways, E2 and ERs enhance the cellular energy generation and expenditure. Thus, in the absence of E2 and/or ERs, E2-induced energy expenditure flow is not operational, leading to energy accumulation, energy imbalance and thus, obesity. On the other hand, the E2/ER-mediated enhancement of energy metabolism pathways could be disturbed by EDCs, leading to abnormal energy metabolism and energy imbalance. This could be one of the important contributing factors for current increasing trend of obesity epidemic.
Further studies are needed to obtain new insights into the cellular and molecular mechanisms underlying regulation of energy metabolism pathways by estrogens and estrogenic chemicals and potential implications in obesity that are related to increased exposure to EDCs. Specifically, since evidence is accumulating to indicate an potential association between perinatal exposure to endocrine disrupting chemicals, particular those with estrogenic or anti-estrogenic activity, and the development of obesity later in life, further studies to address their effects and the underlying cellular mechanisms of EDCs on energy metabolism pathways during development stages and among inter-generations are urgently needed. Such studies may allow identification of new approaches for effective prevention and/or slowdown the trend of obesity epidemic.
Figure 4B. Role of c-myc in the Regulation of MRC Proteins by E2/ERs.
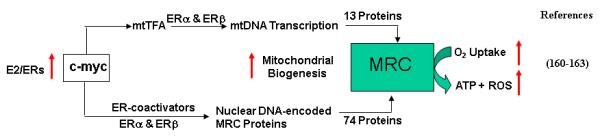
The transcription factor, c-myc, is an E2-regulated protein. It is involved in the stimulation of nDNA-encoded MRC gene expression. It also stimulates the expression of mtTFA, a transcription factors involved in mtDNA transcription. C-myc coordinates the expression of both nDNA-encoded and mtDNA-encoded MRC protein synthesis, and thus enhances mitochondrial biogenesis. Red arrows indicate the stimulating effects by E2.
Acknowledgement
This publication was made possible by the Breast Cancer and the Environment Research Centers grant number U01 ES/CA 12771 from the National Institute of Environmental Health Sciences (NIEHS), and the National Cancer Institute (NCI), NIH, DHHS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NCI, NIH. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work. We also acknowledge Mrs. Ping He of Johns Hopkins School of Medicine for her personal support and encouragement in writing this work.
References
- [1].Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- [2].Calle EE, Thun MJ. Obesity and cancer. Oncogene. 2004;23:6365–6378. doi: 10.1038/sj.onc.1207751. [DOI] [PubMed] [Google Scholar]
- [3].Heindel JJ. Endocrine disruptors and the obesity epidemic. Toxicol Sci. 2003;76:247–249. doi: 10.1093/toxsci/kfg255. [DOI] [PubMed] [Google Scholar]
- [4].Wasan KM, Looije NA. Emerging pharmacological approaches to the treatment of obesity. J Pharm Pharm Sci. 2005;8:259–271. [PubMed] [Google Scholar]
- [5].Menifield CE, Doty N, Fletcher A. Obesity in America. ABNF J. 2008;19:83–88. [PubMed] [Google Scholar]
- [6].Zaninotto P, Head J, Stamatakis E, Wardle H, Mindell J. Trends in obesity among adults in England from 1993 to 2004 by age and social class and projections of prevalence to 2012. J Epidemiol Community Health. 2008 doi: 10.1136/jech.2008.077305. [DOI] [PubMed] [Google Scholar]
- [7].Gu D, Reynolds K, Wu X, Chen J, Duan X, Reynolds RF, Whelton PK, He J. Prevalence of the metabolic syndrome and overweight among adults in China. Lancet. 2005;365:1398–1405. doi: 10.1016/S0140-6736(05)66375-1. [DOI] [PubMed] [Google Scholar]
- [8].Marwaha RK, Tandon N, Singh Y, Aggarwal R, Grewal K, Mani K. A study of growth parameters and prevalence of overweight and obesity in school children from delhi. Indian Pediatr. 2006;43:943–952. [PubMed] [Google Scholar]
- [9].Chlebowski RT. Obesity and early-stage breast cancer. J Clin Oncol. 2005;23:1345–1347. doi: 10.1200/JCO.2005.10.949. [DOI] [PubMed] [Google Scholar]
- [10].Lorincz AM, Sukumar S. Molecular links between obesity and breast cancer. Endocr Relat Cancer. 2006;13:279–292. doi: 10.1677/erc.1.00729. [DOI] [PubMed] [Google Scholar]
- [11].Strom SS, Wang X, Pettaway CA, Logothetis CJ, Yamamura Y, Do KA, Babaian RJ, Troncoso P. Obesity, weight gain, and risk of biochemical failure among prostate cancer patients following prostatectomy. Clin Cancer Res. 2005;11:6889–6894. doi: 10.1158/1078-0432.CCR-04-1977. [DOI] [PubMed] [Google Scholar]
- [12].Damcott CM, Sack P, Shuldiner AR. The genetics of obesity. Endocrinol Metab Clin North Am. 2003;32:761–786. doi: 10.1016/s0889-8529(03)00076-8. [DOI] [PubMed] [Google Scholar]
- [13].Brantley PJ, Myers VH, Roy HJ. Environmental and lifestyle influences on obesity. J La State Med Soc. 2005;157(Spec No 1):S19–27. [PubMed] [Google Scholar]
- [14].Heindel JJ. Role of exposure to environmental chemicals in the developmental basis of disease and dysfunction. Reprod Toxicol. 2007;23:257–259. doi: 10.1016/j.reprotox.2007.01.006. [DOI] [PubMed] [Google Scholar]
- [15].Wade GN, Powers JB. Tamoxifen antagonizes the effects of estradiol on energy balance and estrous behavior in Syrian hamsters. Am J Physiol. 1993;265:R559–562. doi: 10.1152/ajpregu.1993.265.3.R559. [DOI] [PubMed] [Google Scholar]
- [16].Misso ML, Murata Y, Boon WC, Jones ME, Britt KL, Simpson ER. Cellular and molecular characterization of the adipose phenotype of the aromatase-deficient mouse. Endocrinology. 2003;144:1474–1480. doi: 10.1210/en.2002-221123. [DOI] [PubMed] [Google Scholar]
- [17].Misso ML, Hewitt KN, Boon WC, Murata Y, Jones ME, Simpson ER. Cholesterol feeding prevents adiposity in the obese female aromatase knockout (ArKO) mouse. Horm Metab Res. 2005;37:26–31. doi: 10.1055/s-2005-861028. [DOI] [PubMed] [Google Scholar]
- [18].Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, Onishi T, Sugiura T, Shizuta Y. Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol. 2003;176:237–246. doi: 10.1677/joe.0.1760237. [DOI] [PubMed] [Google Scholar]
- [19].Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, Leranth C, Toran-Allerand D, Priest CA, Roberts JL, Gao XB, Mobbs C, Shulman GI, Diano S, Horvath TL. Anorectic estrogen mimics leptin’s effect on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med. 2007;13:89–94. doi: 10.1038/nm1525. [DOI] [PubMed] [Google Scholar]
- [20].Feigelson HS, Jonas CR, Teras LR, Thun MJ, Calle EE. Weight gain, body mass index, hormone replacement therapy, and postmenopausal breast cancer in a large prospective study. Cancer Epidemiol Biomarkers Prev. 2004;13:220–224. doi: 10.1158/1055-9965.epi-03-0301. [DOI] [PubMed] [Google Scholar]
- [21].Demir B, Ozturkoglu E, Solaroglu A, Baskan B, Kandemir O, Karabulut E, Haberal A. The effects of estrogen therapy and estrogen combined with different androgenic progestins on carbohydrate and lipid metabolism in overweight-obese younger postmenopausal women. Gynecol Endocrinol. 2008;24:347–353. doi: 10.1080/01443610802043066. [DOI] [PubMed] [Google Scholar]
- [22].Kristensen K, Pedersen SB, Vestergaard P, Mosekilde L, Richelsen B. Hormone replacement therapy affects body composition and leptin differently in obese and non-obese postmenopausal women. J Endocrinol. 1999;163:55–62. doi: 10.1677/joe.0.1630055. [DOI] [PubMed] [Google Scholar]
- [23].Beato M, Chavez S, Truss M. Transcriptional regulation by steroid hormones. Steroids. 1996;61:240–251. doi: 10.1016/0039-128x(96)00030-x. [DOI] [PubMed] [Google Scholar]
- [24].Beato M, Sanchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. doi: 10.1210/edrv-17-6-587. [DOI] [PubMed] [Google Scholar]
- [25].Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pettersson K, Gustafsson JA. Role of estrogen receptor beta in estrogen action. Annu Rev Physiol. 2001;63:165–192. doi: 10.1146/annurev.physiol.63.1.165. [DOI] [PubMed] [Google Scholar]
- [27].Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- [28].Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol Endocrinol. 2005;19:1951–1959. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Levin ER. Rapid signaling by steroid receptors. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1425–1430. doi: 10.1152/ajpregu.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Levin ER, Pietras RJ. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat. 2008;108:351–361. doi: 10.1007/s10549-007-9618-4. [DOI] [PubMed] [Google Scholar]
- [31].Song RX, Fan P, Yue W, Chen Y, Santen RJ. Role of receptor complexes in the extranuclear actions of estrogen receptor alpha in breast cancer. Endocr Relat Cancer. 2006;13(Suppl 1):S3–13. doi: 10.1677/erc.1.01322. [DOI] [PubMed] [Google Scholar]
- [32].Zhang Z, Maier B, Santen RJ, Song RX. Membrane association of estrogen receptor alpha mediates estrogen effect on MAPK activation. Biochem Biophys Res Commun. 2002;294:926–933. doi: 10.1016/S0006-291X(02)00348-0. [DOI] [PubMed] [Google Scholar]
- [33].Chen JQ, Brown TR, Yager JD. Mechanisms of hormone carcinogenesis: evolution of views, role of mitochondria. Adv Exp Med Biol. 2008;630:1–18. [PubMed] [Google Scholar]
- [34].Chen JQ, Yager JD, Russo J. Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim Biophys Acta. 2005;1746:1–17. doi: 10.1016/j.bbamcr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- [35].Wade GN, Blaustein JD, Gray JM, Meredith JM. ICI 182,780: a pure antiestrogen that affects behaviors and energy balance in rats without acting in the brain. Am J Physiol. 1993;265:R1392–1398. doi: 10.1152/ajpregu.1993.265.6.R1392. [DOI] [PubMed] [Google Scholar]
- [36].Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A. 2000;97:12735–12740. doi: 10.1073/pnas.97.23.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jones ME, Thorburn AW, Britt KL, Hewitt KN, Misso ML, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER. Aromatase-deficient (ArKO) mice accumulate excess adipose tissue. J Steroid Biochem Mol Biol. 2001;79:3–9. doi: 10.1016/s0960-0760(01)00136-4. [DOI] [PubMed] [Google Scholar]
- [38].Ohlsson C, Hellberg N, Parini P, Vidal O, Bohlooly YM, Rudling M, Lindberg MK, Warner M, Angelin B, Gustafsson JA. Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice. Biochem Biophys Res Commun. 2000;278:640–645. doi: 10.1006/bbrc.2000.3827. [DOI] [PubMed] [Google Scholar]
- [39].Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cooke PS, Heine PA, Taylor JA, Lubahn DB. The role of estrogen and estrogen receptor-alpha in male adipose tissue. Mol Cell Endocrinol. 2001;178:147–154. doi: 10.1016/s0303-7207(01)00414-2. [DOI] [PubMed] [Google Scholar]
- [41].Naaz A, Zakroczymski M, Heine P, Taylor J, Saunders P, Lubahn D, Cooke PS. Effect of ovariectomy on adipose tissue of mice in the absence of estrogen receptor alpha (ERalpha): a potential role for estrogen receptor beta (ERbeta) Horm Metab Res. 2002;34:758–763. doi: 10.1055/s-2002-38259. [DOI] [PubMed] [Google Scholar]
- [42].Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, Krikov M, Bhanot S, Barros R, Morani A, Gustafsson JA, Unger T, Kintscher U. Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative crosstalk with PPARgamma. PLoS Genet. 2008;4:e1000108. doi: 10.1371/journal.pgen.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rosenkranz K, Hinney A, Ziegler A, Hermann H, Fichter M, Mayer H, Siegfried W, Young JK, Remschmidt H, Hebebrand J. Systematic mutation screening of the estrogen receptor beta gene in probands of different weight extremes: identification of several genetic variants. J Clin Endocrinol Metab. 1998;83:4524–4527. doi: 10.1210/jcem.83.12.5471. [DOI] [PubMed] [Google Scholar]
- [44].Nilsson M, Naessen S, Dahlman I, Hirschberg A. Linden, Gustafsson JA, Dahlman-Wright K. Association of estrogen receptor beta gene polymorphisms with bulimic disease in women. Mol Psychiatry. 2004;9:28–34. doi: 10.1038/sj.mp.4001402. [DOI] [PubMed] [Google Scholar]
- [45].Liang YQ, Akishita M, Kim S, Ako J, Hashimoto M, Iijima K, Ohike Y, Watanabe T, Sudoh N, Toba K, Yoshizumi M, Ouchi Y. Estrogen receptor beta is involved in the anorectic action of estrogen. Int J Obes Relat Metab Disord. 2002;26:1103–1109. doi: 10.1038/sj.ijo.0802054. [DOI] [PubMed] [Google Scholar]
- [46].Pallottini V, Bulzomi P, Galluzzo P, Martini C, Marino M. Estrogen regulation of adipose tissue functions: involvement of estrogen receptor isoforms. Infect Disord Drug Targets. 2008;8:52–60. doi: 10.2174/187152608784139631. [DOI] [PubMed] [Google Scholar]
- [47].Pedram A, Razandi M, Kim JK, O’Mahony F, Lee EY, Luderer U, Levin ER. Developmental phenotype of a membrane only estrogen receptor alpha (MOER) mouse. J Biol Chem. 2009;284:3488–3495. doi: 10.1074/jbc.M806249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wu C, Kang JE, Peng LJ, Li H, Khan SA, Hillard CJ, Okar DA, Lange AJ. Enhancing hepatic glycolysis reduces obesity: differential effects on lipogenesis depend on site of glycolytic modulation. Cell Metab. 2005;2:131–140. doi: 10.1016/j.cmet.2005.07.003. [DOI] [PubMed] [Google Scholar]
- [49].Bhatia AJ, Wade GN. Energy balance in pregnant hamsters: a role for voluntary exercise? Am J Physiol. 1993;265:R563–567. doi: 10.1152/ajpregu.1993.265.3.R563. [DOI] [PubMed] [Google Scholar]
- [50].Birnbaum LS, Fenton SE. Cancer and developmental exposure to endocrine disruptors. Environ Health Perspect. 2003;111:389–394. doi: 10.1289/ehp.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mead MN. Origins of obesity. Environ Health Perspect. 2004;112:A344. doi: 10.1289/ehp.112-a344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Clevenger WR, Cornwall GA, Carter MW, Bradshaw WS. Diethylstilbestrol-induced perinatal lethality in the rat. I. Relationship to reduced maternal weight gain. Biol Reprod. 1991;44:575–582. doi: 10.1095/biolreprod44.4.575. [DOI] [PubMed] [Google Scholar]
- [53].Zimmerman SA, Clevenger WR, Brimhall BB, Bradshaw WS. Diethylstilbestrol-induced perinatal lethality in the rat. II. Perturbation of parturition. Biol Reprod. 1991;44:583–589. doi: 10.1095/biolreprod44.4.583. [DOI] [PubMed] [Google Scholar]
- [54].Harp JB, Henry SA, DiGirolamo M. Dietary weight loss decreases serum angiotensin-converting enzyme activity in obese adults. Obes Res. 2002;10:985–990. doi: 10.1038/oby.2002.134. [DOI] [PubMed] [Google Scholar]
- [55].Naaz A, Yellayi S, Zakroczymski MA, Bunick D, Doerge DR, Lubahn DB, Helferich WG, Cooke PS. The soy isoflavone genistein decreases adipose deposition in mice. Endocrinology. 2003;144:3315–3320. doi: 10.1210/en.2003-0076. [DOI] [PubMed] [Google Scholar]
- [56].Ema M, Miyawaki E. Effects on development of the reproductive system in male offspring of rats given butyl benzyl phthalate during late pregnancy. Reprod Toxicol. 2002;16:71–76. doi: 10.1016/s0890-6238(01)00200-3. [DOI] [PubMed] [Google Scholar]
- [57].Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:675–680. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kim JC, Shin HC, Cha SW, Koh WS, Chung MK, Han SS. Evaluation of developmental toxicity in rats exposed to the environmental estrogen bisphenol A during pregnancy. Life Sci. 2001;69:2611–2625. doi: 10.1016/s0024-3205(01)01341-8. [DOI] [PubMed] [Google Scholar]
- [59].Morrissey RE, George JD, Price CJ, Tyl RW, Marr MC, Kimmel CA. The developmental toxicity of bisphenol A in rats and mice. Fundam Appl Toxicol. 1987;8:571–582. doi: 10.1016/0272-0590(87)90142-4. [DOI] [PubMed] [Google Scholar]
- [60].Scheepers A, Joost HG, Schurmann A. The glucose transporter families SGLT and GLUT: molecular basis of normal and aberrant function. JPEN J Parenter Enteral Nutr. 2004;28:364–371. doi: 10.1177/0148607104028005364. [DOI] [PubMed] [Google Scholar]
- [61].Neeman M, Degani H. Early estrogen-induced metabolic changes and their inhibition by actinomycin D and cycloheximide in human breast cancer cells: 31P and 13C NMR studies. Proc Natl Acad Sci U S A. 1989;86:5585–5589. doi: 10.1073/pnas.86.14.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Neeman M, Degani H. Metabolic studies of estrogen- and tamoxifen-treated human breast cancer cells by nuclear magnetic resonance spectroscopy. Cancer Res. 1989;49:589–594. [PubMed] [Google Scholar]
- [63].Furman E, Rushkin E, Margalit R, Bendel P, Degani H. Tamoxifen induced changes in MCF7 human breast cancer: in vitro and in vivo studies using nuclear magnetic resonance spectroscopy and imaging. J Steroid Biochem Mol Biol. 1992;43:189–195. doi: 10.1016/0960-0760(92)90207-y. [DOI] [PubMed] [Google Scholar]
- [64].Rivenzon-Segal D, Rushkin E, Polak-Charcon S, Degani H. Glucose transporters and transport kinetics in retinoic acid-differentiated T47D human breast cancer cells. Am J Physiol Endocrinol Metab. 2000;279:E508–519. doi: 10.1152/ajpendo.2000.279.3.E508. [DOI] [PubMed] [Google Scholar]
- [65].Rivenzon-Segal D, Boldin-Adamsky S, Seger D, Seger R, Degani H. Glycolysis and glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int J Cancer. 2003;107:177–182. doi: 10.1002/ijc.11387. [DOI] [PubMed] [Google Scholar]
- [66].Zamora-Leon SP, Golde DW, Concha, Rivas CI, Delgado-Lopez F, Baselga J, Nualart F, Vera JC. Expression of the fructose transporter GLUT5 in human breast cancer. Proc Natl Acad Sci U S A. 1996;93:1847–1852. doi: 10.1073/pnas.93.5.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Smith DE, Gorski J. Extrogen control of uterine glucose metabolism. An analysis based on the transport and phosphorylation of 2-deoxyglucose. J Biol Chem. 1968;243:4169–4174. [PubMed] [Google Scholar]
- [68].Shinkarenko L, Kaye AM, Degani H. C NMR kinetic studies of the rapid stimulation of glucose metabolism by estrogen in immature rat uterus. NMR Biomed. 1994;7:209–217. doi: 10.1002/nbm.1940070503. [DOI] [PubMed] [Google Scholar]
- [69].Kumagai S, Holmang A, Bjorntorp P. The effects of oestrogen and progesterone on insulin sensitivity in female rats. Acta Physiol Scand. 1993;149:91–97. doi: 10.1111/j.1748-1716.1993.tb09596.x. [DOI] [PubMed] [Google Scholar]
- [70].Cheng CM, Cohen M, Wang J, Bondy CA. Estrogen augments glucose transporter and IGF1 expression in primate cerebral cortex. FASEB J. 2001;15:907–915. doi: 10.1096/fj.00-0398com. [DOI] [PubMed] [Google Scholar]
- [71].Welch RD, Gorski J. Regulation of glucose transporters by estradiol in the immature rat uterus. Endocrinology. 1999;140:3602–3608. doi: 10.1210/endo.140.8.6923. [DOI] [PubMed] [Google Scholar]
- [72].Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- [73].Im SS, Kim JW, Kim TH, Song XL, Kim SY, Kim HI, Ahn YH. Identification and characterization of peroxisome proliferator response element in the mouse GLUT2 promoter. Exp Mol Med. 2005;37:101–110. doi: 10.1038/emm.2005.14. [DOI] [PubMed] [Google Scholar]
- [74].Im SS, Kang SY, Kim SY, Kim HI, Kim JW, Kim KS, Ahn YH. Glucose-stimulated upregulation of GLUT2 gene is mediated by sterol response element-binding protein-1c in the hepatocytes. Diabetes. 2005;54:1684–1691. doi: 10.2337/diabetes.54.6.1684. [DOI] [PubMed] [Google Scholar]
- [75].Houston KD, Copland JA, Broaddus RR, Gottardis MM, Fischer SM, Walker CL. Inhibition of proliferation and estrogen receptor signaling by peroxisome proliferator-activated receptor gamma ligands in uterine leiomyoma. Cancer Res. 2003;63:1221–1227. [PubMed] [Google Scholar]
- [76].Wang X, Kilgore MW. Signal cross-talk between estrogen receptor alpha and beta and the peroxisome proliferator-activated receptor gamma1 in MDA-MB-231 and MCF-7 breast cancer cells. Mol Cell Endocrinol. 2002;194:123–133. doi: 10.1016/s0303-7207(02)00154-5. [DOI] [PubMed] [Google Scholar]
- [77].Sakurai K, Kawazuma M, Adachi T, Harigaya T, Saito Y, Hashimoto N, Mori C. Bisphenol A affects glucose transport in mouse 3T3-F442A adipocytes. Br J Pharmacol. 2004;141:209–214. doi: 10.1038/sj.bjp.0705520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bogacka I, Roane DS, Xi X, Zhou J, Li B, Ryan DH, Martin RJ. Expression levels of genes likely involved in glucose-sensing in the obese Zucker rat brain. Nutr Neurosci. 2004;7:67–74. doi: 10.1080/10284150410001710401. [DOI] [PubMed] [Google Scholar]
- [79].Brennan CL, Hoenig M, Ferguson DC. GLUT4 but not GLUT1 expression decreases early in the development of feline obesity. Domest Anim Endocrinol. 2004;26:291–301. doi: 10.1016/j.domaniend.2003.11.003. [DOI] [PubMed] [Google Scholar]
- [80].Minchenko OH, Ochiai A, Opentanova IL, Ogura T, Minchenko DO, Caro J, Komisarenko SV, Esumi H. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4 in the human breast and colon malignant tumors. Biochimie. 2005;87:1005–1010. doi: 10.1016/j.biochi.2005.04.007. [DOI] [PubMed] [Google Scholar]
- [81].Kostanyan A, Nazaryan K. Rat brain glycolysis regulation by estradiol-17 beta. Biochim Biophys Acta. 1992;1133:301–306. doi: 10.1016/0167-4889(92)90051-c. [DOI] [PubMed] [Google Scholar]
- [82].Reiss NA. Ontogeny and estrogen responsiveness of creatine kinase and glycolytic enzymes in brain and uterus of rat. Neurosci Lett. 1988;84:197–202. doi: 10.1016/0304-3940(88)90407-7. [DOI] [PubMed] [Google Scholar]
- [83].Pastorelli R, Carpi D, Airoldi L, Chiabrando C, Bagnati R, Fanelli R, Moverare S, Ohlsson C. Proteome analysis for the identification of in vivo estrogen-regulated proteins in bone. Proteomics. 2005 doi: 10.1002/pmic.200401325. [DOI] [PubMed] [Google Scholar]
- [84].Yan L, Ge H, Li H, Lieber SC, Natividad F, Resuello RR, Kim SJ, Akeju S, Sun A, Loo K, Peppas AP, Rossi F, Lewandowski ED, Thomas AP, Vatner SF, Vatner DE. Gender-specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J Mol Cell Cardiol. 2004;37:921–929. doi: 10.1016/j.yjmcc.2004.06.012. [DOI] [PubMed] [Google Scholar]
- [85].Vijayalakshmi R, Bamji MS, Ramalakshmi BA. Reduced anaerobic glycolysis in oral contraceptive users. Contraception. 1988;38:91–97. doi: 10.1016/0010-7824(88)90098-4. [DOI] [PubMed] [Google Scholar]
- [86].Kelley DE, Mokan M, Mandarino LJ. Intracellular defects in glucose metabolism in obese patients with NIDDM. Diabetes. 1992;41:698–706. doi: 10.2337/diab.41.6.698. [DOI] [PubMed] [Google Scholar]
- [87].Choi MS, Jung UJ, Yeo J, Kim MJ, Lee MK. Genistein and daidzein prevent diabetes onset by elevating insulin level and altering hepatic gluconeogenic and lipogenic enzyme activities in non-obese diabetic (NOD) mice. Diabetes Metab Res Rev. 2008;24:74–81. doi: 10.1002/dmrr.780. [DOI] [PubMed] [Google Scholar]
- [88].Ae Park S, Choi MS, Cho SY, Seo JS, Jung UJ, Kim MJ, Sung MK, Park YB, Lee MK. Genistein and daidzein modulate hepatic glucose and lipid regulating enzyme activities in C57BL/KsJ-db/db mice. Life Sci. 2006;79:1207–1213. doi: 10.1016/j.lfs.2006.03.022. [DOI] [PubMed] [Google Scholar]
- [89].Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997;83:166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- [90].Beckett T, Tchernof A, Toth MJ. Effect of ovariectomy and estradiol replacement on skeletal muscle enzyme activity in female rats. Metabolism. 2002;51:1397–1401. doi: 10.1053/meta.2002.35592. [DOI] [PubMed] [Google Scholar]
- [91].Stirone C, Duckles SP, Krause DN, Procaccio V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005;68:959–965. doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- [92].Yadav RN. Isocitrate dehydrogenase activity and its regulation by estradiol in tissues of rats of various ages. Cell Biochem Funct. 1988;6:197–202. doi: 10.1002/cbf.290060308. [DOI] [PubMed] [Google Scholar]
- [93].Gupta G, Srivastava A, Setty BS. Androgen-estrogen synergy in the regulation of energy metabolism in epididymis and vas deferens of rhesus monkey. Endocr Res. 1991;17:383–394. doi: 10.1080/07435809109106815. [DOI] [PubMed] [Google Scholar]
- [94].Nilsen J, Irwin RW, Gallaher TK, Brinton RD. Estradiol in vivo regulation of brain mitochondrial proteome. J Neurosci. 2007;27:14069–14077. doi: 10.1523/JNEUROSCI.4391-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Baltrus BM, Melchior JB. Effects of administration of estrogens upon enzymes of rat pituitary. II. Aconitase. succinoxidase, and transaminase, Cancer Res. 1961;21:1360–1364. [PubMed] [Google Scholar]
- [96].Raben A, Mygind E, Astrup A. Lower activity of oxidative key enzymes and smaller fiber areas in skeletal muscle of postobese women. Am J Physiol. 1998;275:E487–494. doi: 10.1152/ajpendo.1998.275.3.E487. [DOI] [PubMed] [Google Scholar]
- [97].Goffart S, Wiesner RJ. Regulation and co-ordination of nuclear gene expression during mitochondrial biogenesis. Exp Physiol. 2003;88:33–40. doi: 10.1113/eph8802500. [DOI] [PubMed] [Google Scholar]
- [98].Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- [99].Bossy-Wetzel E, Green DR. Apoptosis: checkpoint at the mitochondrial frontier. Mutat Res. 1999;434:243–251. doi: 10.1016/s0921-8777(99)00032-4. [DOI] [PubMed] [Google Scholar]
- [100].Grivell LA. Nucleo-mitochondrial interactions in mitochondrial gene expression. Crit Rev Biochem Mol Biol. 1995;30:121–164. doi: 10.3109/10409239509085141. [DOI] [PubMed] [Google Scholar]
- [101].Garesse R, Vallejo CG. Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene. 2001;263:1–16. doi: 10.1016/s0378-1119(00)00582-5. [DOI] [PubMed] [Google Scholar]
- [102].Chen JQ, Yager JD, Russo J. Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim Biophys Acta. 2005 doi: 10.1016/j.bbamcr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- [103].Vic P, Vignon F, Derocq D, Rochefort H. Effect of estradiol on the ultrastructure of the MCF7 human breast cancer cells in culture. Cancer Res. 1982;42:667–673. [PubMed] [Google Scholar]
- [104].Alkhalaf M, Chaminadas G, Propper AY, Adessi GL. Ultrastructural changes induced by oestradiol-17 beta, progesterone and oestrone-3-sulphate in guinea-pig endometrial glandular cells grown in primary culture. J Endocrinol. 1989;122:439–444. doi: 10.1677/joe.0.1220439. [DOI] [PubMed] [Google Scholar]
- [105].C C. Tam, Y C. Wong. Ultrastructural study of the effects of 17 beta-oestradiol on the lateral prostate and seminal vesicle of the castrated guinea pig. Acta Anat (Basel) 1991;141:51–62. doi: 10.1159/000147099. [DOI] [PubMed] [Google Scholar]
- [106].Tam CC, Wong YC, White FH, Fowler JP. Morphometric and stereological analysis of the effects of 17 beta-estradiol on the glandular epithelium of the castrated guinea pig lateral prostate. Prostate. 1991;19:279–297. doi: 10.1002/pros.2990190403. [DOI] [PubMed] [Google Scholar]
- [107].Roberts, Szego CM. The influence of steroids on uterine respiration and glycolysis. J Biol Chem. 1953;201:21–30. [PubMed] [Google Scholar]
- [108].Bayliss DA, Cidlowski JA, Millhorn DE. The stimulation of respiration by progesterone in ovariectomized cat is mediated by an estrogen-dependent hypothalamic mechanism requiring gene expression. Endocrinology. 1990;126:519–527. doi: 10.1210/endo-126-1-519. [DOI] [PubMed] [Google Scholar]
- [109].Sergeev PV, Khalilov EM, Obraztsov NV, Seifulla RD. Effect of steroid hormones on the rate of NADPH and NADH oxidation by rat liver microsomes. Farmakol Toksikol. 1975;38:600–603. [PubMed] [Google Scholar]
- [110].Sergeev PV, Seifulla RD, Maiskii AI, Iu P. Denisov, Nikitina ZK. Effect of estrogens on the parameters of oxidative phosphorylation in the liver mitochondria and uterus. Akush Ginekol (Mosk) 1975:63. [PubMed] [Google Scholar]
- [111].Sergeev PV, Seifulla RD, Maiskii AI, Iu P. Denisov, Nikitina ZK. Effect of estrogens on the indicators of oxidative phosphorylation in the mitochondria of the liver and uterine cells. Akush Ginekol (Mosk) 1975:60–61. [PubMed] [Google Scholar]
- [112].Salmony D. The effect of oestrogens and chemically related compounds on the respiration of yeast and on oxidative phosphorylation. Biochem J. 1956;62:411–416. doi: 10.1042/bj0620411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Takenaka S. Studies on the effect of sex-hormones on oxidative phosphorylation. J Jpn Obstet Gynecol Soc. 1965;12:89–93. [PubMed] [Google Scholar]
- [114].Zhai P, Eurell TE, Cotthaus R, Jeffery EH, Bahr JM, Gross DR. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am J Physiol Heart Circ Physiol. 2000;279:H2766–2775. doi: 10.1152/ajpheart.2000.279.6.H2766. [DOI] [PubMed] [Google Scholar]
- [115].Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR. Myocardial ischemiareperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol. 2000;278:H1640–1647. doi: 10.1152/ajpheart.2000.278.5.H1640. [DOI] [PubMed] [Google Scholar]
- [116].Zhai P, Eurell TE, Cotthaus RP, Jeffery EH, Bahr JM, Gross DR. Effects of dietary phytoestrogen on global myocardial ischemia-reperfusion injury in isolated female rat hearts. Am J Physiol Heart Circ Physiol. 2001;281:H1223–1232. doi: 10.1152/ajpheart.2001.281.3.H1223. [DOI] [PubMed] [Google Scholar]
- [117].Toda K, Takeda K, Okada T, Akira S, Saibara T, Kaname T, Yamamura K, Onishi S, Shizuta Y. Targeted disruption of the aromatase P450 gene (Cyp19) in mice and their ovarian and uterine responses to 17beta-oestradiol. J Endocrinol. 2001;170:99–111. doi: 10.1677/joe.0.1700099. [DOI] [PubMed] [Google Scholar]
- [118].Justo R, Frontera M, Pujol E, Rodriguez-Cuenca S, Llado I, Garcia-Palmer FJ, Roca P, Gianotti M. Gender-related differences in morphology and thermogenic capacity of brown adipose tissue mitochondrial subpopulations. Life Sci. 2005;76:1147–1158. doi: 10.1016/j.lfs.2004.08.019. [DOI] [PubMed] [Google Scholar]
- [119].Justo R, Boada J, Frontera M, Oliver J, Bermudez J, Gianotti M. Gender dimorphism in rat liver mitochondrial oxidative metabolism and biogenesis. Am J Physiol Cell Physiol. 2005;289:C372–378. doi: 10.1152/ajpcell.00035.2005. [DOI] [PubMed] [Google Scholar]
- [120].Rodriguez-Cuenca S, Pujol E, Justo R, Frontera M, Oliver J, Gianotti M, Roca P. Sex-dependent thermogenesis. differences in mitochondrial morphology and function, and adrenergic response in brown adipose tissue, J Biol Chem. 2002;277:42958–42963. doi: 10.1074/jbc.M207229200. [DOI] [PubMed] [Google Scholar]
- [121].Valle A, Catala-Niell A, Colom B, Garcia-Palmer FJ, Oliver J, Roca P. Sex-related differences in energy balance in response to caloric restriction. Am J Physiol Endocrinol Metab. 2005;289:E15–22. doi: 10.1152/ajpendo.00553.2004. [DOI] [PubMed] [Google Scholar]
- [122].Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, Gustafsson JA, Mendelsohn ME. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]
- [123].Watson B, Jr., Khan MA, Desmond RA, Bergman S. Mitochondrial DNA mutations in black Americans with hypertension-associated end-stage renal disease. Am J Kidney Dis. 2001;38:529–536. doi: 10.1053/ajkd.2001.26848. [DOI] [PubMed] [Google Scholar]
- [124].Head GA, Obeyesekere VR, Jones ME, Simpson ER, Krozowski ZS. Aromatase-deficient (ArKO) mice have reduced blood pressure and baroreflex sensitivity. Endocrinology. 2004;145:4286–4291. doi: 10.1210/en.2004-0421. [DOI] [PubMed] [Google Scholar]
- [125].Monje P, Boland R. Subcellular distribution of native estrogen receptor alpha and beta isoforms in rabbit uterus and ovary. J Cell Biochem. 2001;82:467–479. doi: 10.1002/jcb.1182. [DOI] [PubMed] [Google Scholar]
- [126].Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr., Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci U S A. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491:81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- [128].Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004;286:E1011–1022. doi: 10.1152/ajpendo.00508.2003. [DOI] [PubMed] [Google Scholar]
- [129].Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial dna estrogen response elements. J Cell Biochem. 2004;93:358. doi: 10.1002/jcb.20178. [DOI] [PubMed] [Google Scholar]
- [130].Pedram A, Razandi M, Wallace DC, Levin ER. Functional Estrogen Receptors in the Mitochondria of Breast Cancer Cells. Mol Biol Cell. 2006 doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Cammarata PR, Chu S, Moor A, Wang Z, Yang SH, Simpkins JW. Subcellular distribution of native estrogen receptor alpha and beta subtypes in cultured human lens epithelial cells. Exp Eye Res. 2004;78:861–871. doi: 10.1016/j.exer.2003.09.027. [DOI] [PubMed] [Google Scholar]
- [132].Cammarata PR, Flynn J, Gottipati S, Chu S, Dimitrijevich S, Younes M, Skliris G, Murphy LC. Differential expression and comparative subcellular localization of estrogen receptor beta isoforms in virally transformed and normal cultured human lens epithelial cells. Exp Eye Res. 2005;81:165–175. doi: 10.1016/j.exer.2005.01.019. [DOI] [PubMed] [Google Scholar]
- [133].Solakidi S, Psarra AM, Sekeris CE. Differential subcellular distribution of estrogen receptor isoforms: Localization of ERalpha in the nucleoli and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochim Biophys Acta. 2005 doi: 10.1016/j.bbamcr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- [134].Solakidi S, Psarra AM, Nikolaropoulos S, Sekeris CE. Estrogen receptors {alpha} and {beta} (ER{alpha} and ER{beta}) and androgen receptor (AR) in human sperm: localization of ER{beta} and AR in mitochondria of the midpiece. Hum Reprod. 2005 doi: 10.1093/humrep/dei267. [DOI] [PubMed] [Google Scholar]
- [135].Chen JQ, Eshete M, Alworth WL, Yager JD. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J Cell Biochem. 2004;93:358–373. doi: 10.1002/jcb.20178. [DOI] [PubMed] [Google Scholar]
- [136].Stirone C, Duckles SP, Krause DN. V. Procaccio, Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol Pharmacol. 2005 doi: 10.1124/mol.105.014662. [DOI] [PubMed] [Google Scholar]
- [137].Van Itallie CM, Dannies PS. Estrogen induces accumulation of the mitochondrial ribonucleic acid for subunit II of cytochrome oxidase in pituitary tumor cells. Mol Endocrinol. 1988;2:332–337. doi: 10.1210/mend-2-4-332. [DOI] [PubMed] [Google Scholar]
- [138].Bettini E, Maggi A. Estrogen induction of cytochrome c oxidase subunit III in rat hippocampus. J Neurochem. 1992;58:1923–1929. doi: 10.1111/j.1471-4159.1992.tb10070.x. [DOI] [PubMed] [Google Scholar]
- [139].Law SW, Apostolakis EM, Samora PJ, O’Malley BW, Clark JH. Hormonal regulation of hypothalamic gene expression: identification of multiple novel estrogen induced genes. J Steroid Biochem Mol Biol. 1994;51:131–136. doi: 10.1016/0960-0760(94)90085-x. [DOI] [PubMed] [Google Scholar]
- [140].Chen J, Schwartz DA, Young TA, Norris JS, Yager JD. Identification of genes whose expression is altered during mitosuppression in livers of ethinyl estradiol-treated female rats. Carcinogenesis. 1996;17:2783–2786. doi: 10.1093/carcin/17.12.2783. [DOI] [PubMed] [Google Scholar]
- [141].Chen J, Gokhale M, Li Y, Trush MA, Yager JD. Enhanced levels of several mitochondrial mRNA transcripts and mitochondrial superoxide production during ethinyl estradiol-induced hepatocarcinogenesis and after estrogen treatment of HepG2 cells. Carcinogenesis. 1998;19:2187–2193. doi: 10.1093/carcin/19.12.2187. [DOI] [PubMed] [Google Scholar]
- [142].Chen J, Delannoy M, Odwin S, He P, Trush MA, Yager JD. Enhanced mitochondrial gene transcript, ATP, bcl-2 protein levels, and altered glutathione distribution in ethinyl estradiol-treated cultured female rat hepatocytes. Toxicol Sci. 2003;75:271–278. doi: 10.1093/toxsci/kfg183. [DOI] [PubMed] [Google Scholar]
- [143].Ye P, Yoshioka M, Gan L, St-Amand J. Regulation of global gene expression by ovariectomy and estrogen in female adipose tissue. Obes Res. 2005;13:1024–1030. doi: 10.1038/oby.2005.120. [DOI] [PubMed] [Google Scholar]
- [144].Chen JQ, Russo PA, Cooke C, Russo IH, Russo J. ERbeta shifts from mitochondria to nucleus during estrogen-induced neoplastic transformation of human breast epithelial cells and is involved in estrogen-induced synthesis of mitochondrial respiratory chain proteins. Biochim Biophys Acta. 2007;1773:1732–1746. doi: 10.1016/j.bbamcr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- [145].Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med. 2003;34:546–552. doi: 10.1016/s0891-5849(02)01356-4. [DOI] [PubMed] [Google Scholar]
- [146].Villablanca AC, Lewis KA, Tham D, Rutledge JC. Differential regulation of gene expression by ovariectomy in mouse aorta. Physiol Genomics. 2003;12:175–185. doi: 10.1152/physiolgenomics.00040.2002. [DOI] [PubMed] [Google Scholar]
- [147].Sekeris CE. The mitochondrial genome: a possible primary site of action of steroid hormones. In Vivo. 1990;4:317–320. [PubMed] [Google Scholar]
- [148].Demonacos CV, Karayanni N, Hatzoglou E, Tsiriyiotis C, Spandidos DA, Sekeris CE. Mitochondrial genes as sites of primary action of steroid hormones. Steroids. 1996;61:226–232. doi: 10.1016/0039-128x(96)00019-0. [DOI] [PubMed] [Google Scholar]
- [149].Roberts S, Szego CM. Steroid interaction in the metabolism of reproductive target organs. Physiol Rev. 1953;33:593–629. doi: 10.1152/physrev.1953.33.4.593. [DOI] [PubMed] [Google Scholar]
- [150].Chau KY, Lam HY, Lee KL. Estrogen treatment induces elevated expression of HMG1 in MCF-7 cells. Exp Cell Res. 1998;241:269–272. doi: 10.1006/excr.1998.4052. [DOI] [PubMed] [Google Scholar]
- [151].Watanabe T, Inoue S, Hiroi H, Orimo A, Kawashima H, Muramatsu M. Isolation of estrogen-responsive genes with a CpG island library. Mol Cell Biol. 1998;18:442–449. doi: 10.1128/mcb.18.1.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Thompson CJ, Tam NN, Joyce JM, Leav I, Ho SM. Gene expression profiling of testosterone and estradiol-17 beta-induced prostatic dysplasia in Noble rats and response to the antiestrogen ICI 182,780. Endocrinology. 2002;143:2093–2105. doi: 10.1210/endo.143.6.8846. [DOI] [PubMed] [Google Scholar]
- [153].Weisz A, Basile W, Scafoglio C, Altucci L, Bresciani F, Facchiano A, Sismondi P, Cicatiello L, De Bortoli M. Molecular identification of ERalpha-positive breast cancer cells by the expression profile of an intrinsic set of estrogen regulated genes. J Cell Physiol. 2004;200:440–450. doi: 10.1002/jcp.20039. [DOI] [PubMed] [Google Scholar]
- [154].Tikkanen MK, Carter DJ, Harris AM, Le HM, Azorsa DO, Meltzer PS, Murdoch FE. Endogenously expressed estrogen receptor and coactivator AIB1 interact in MCF-7 human breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:12536–12540. doi: 10.1073/pnas.220427297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol Endocrinol. 2008;22:609–622. doi: 10.1210/me.2007-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Stirone C, Boroujerdi A, Duckles SP, Krause DN. Estrogen receptor activation of phosphoinositide-3 kinase. akt, and nitric oxide signaling in cerebral blood vessels: rapid and long-term effects, Mol Pharmacol. 2005;67:105–113. doi: 10.1124/mol.104.004465. [DOI] [PubMed] [Google Scholar]
- [157].Chen J, Li Y, Lavigne JA, Trush MA, Yager JD. Increased mitochondrial superoxide production in rat liver mitochondria, rat hepatocytes, and HepG2 cells following ethinyl estradiol treatment. Toxicol Sci. 1999;51:224–235. doi: 10.1093/toxsci/51.2.224. [DOI] [PubMed] [Google Scholar]
- [158].Chen J, Gokhale M, Schofield B, Odwin S, Yager JD. Inhibition of TGF-beta-induced apoptosis by ethinyl estradiol in cultured. precision cut rat liver slices and hepatocytes, Carcinogenesis. 2000;21:1205–1211. [PubMed] [Google Scholar]
- [159].Kamei Y, Suzuki M, Miyazaki H, Tsuboyama-Kasaoka N, Wu J, Ishimi Y, Ezaki O. Ovariectomy in mice decreases lipid metabolism-related gene expression in adipose tissue and skeletal muscle with increased body fat. J Nutr Sci Vitaminol (Tokyo) 2005;51:110–117. doi: 10.3177/jnsv.51.110. [DOI] [PubMed] [Google Scholar]
- [160].Watson JD, Oster SK, Shago M, Khosravi F, Penn LZ. Identifying genes regulated in a Myc-dependent manner. J Biol Chem. 2002;277:36921–36930. doi: 10.1074/jbc.M201493200. [DOI] [PubMed] [Google Scholar]
- [161].Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A. 2002;99:6274–6279. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].O’Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, Sedivy JM. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem. 2003;278:12563–12573. doi: 10.1074/jbc.M210462200. [DOI] [PubMed] [Google Scholar]
- [163].Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Cheng AS, Jin VX, Fan M, Smith LT, Liyanarachchi S, Yan PS, Leu YW, Chan MW, Plass C, Nephew KP, Davuluri RV, Huang TH. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-alpha responsive promoters. Mol Cell. 2006;21:393–404. doi: 10.1016/j.molcel.2005.12.016. [DOI] [PubMed] [Google Scholar]
- [165].Too CK, Giles A, Wilkinson M. Estrogen stimulates expression of adenine nucleotide translocator ANT1 messenger RNA in female rat hearts. Mol Cell Endocrinol. 1999;150:161–167. doi: 10.1016/s0303-7207(99)00002-7. [DOI] [PubMed] [Google Scholar]
- [166].Belzacq AS, Brenner C. The adenine nucleotide translocator: a new potential chemotherapeutic target. Curr Drug Targets. 2003;4:517–524. doi: 10.2174/1389450033490867. [DOI] [PubMed] [Google Scholar]
- [167].Belzacq AS, Vieira HL, Verrier F, Vandecasteele G, Cohen I, Prevost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, Kroemer G. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res. 2003;63:541–546. [PubMed] [Google Scholar]
- [168].Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. Neuroreport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- [169].Wang TT, Phang JM. Effects of estrogen on apoptotic pathways in human breast cancer cell line MCF-7. Cancer Res. 1995;55:2487–2489. [PubMed] [Google Scholar]
- [170].Safe S. Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- [171].Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Li L, Backer J, Wong AS, Schwanke EL, Stewart BG, Pasdar M. Bcl-2 expression decreases cadherin-mediated cell-cell adhesion. J Cell Sci. 2003;116:3687–3700. doi: 10.1242/jcs.00644. [DOI] [PubMed] [Google Scholar]
- [173].Zhao L, Wu TW, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]
- [174].Murphy E, Imahashi K, Steenbergen C. Bcl-2 regulation of mitochondrial energetics. Trends Cardiovasc Med. 2005;15:283–290. doi: 10.1016/j.tcm.2005.09.002. [DOI] [PubMed] [Google Scholar]
- [175].Cardoso CM, Moreno AJ, Almeida LM, Custodio JB. Comparison of the changes in adenine nucleotides of rat liver mitochondria induced by tamoxifen and 4-hydroxytamoxifen. Toxicol In Vitro. 2003;17:663–670. doi: 10.1016/s0887-2333(03)00106-1. [DOI] [PubMed] [Google Scholar]
- [176].Herrero P, Soto PF, Dence CS, Kisrieva-Ware Z, Delano DA, Peterson LR, Gropler RJ. Impact of hormone replacement on myocardial fatty acid metabolism: potential role of estrogen. J Nucl Cardiol. 2005;12:574–581. doi: 10.1016/j.nuclcard.2005.05.009. [DOI] [PubMed] [Google Scholar]
- [177].Toda K, Takeda K, Akira S, Saibara T, Okada T, Onishi S, Shizuta Y. Alternations in hepatic expression of fatty-acid metabolizing enzymes in ArKO mice and their reversal by the treatment with 17beta-estradiol or a peroxisome proliferator. J Steroid Biochem Mol Biol. 2001;79:11–17. doi: 10.1016/s0960-0760(01)00135-2. [DOI] [PubMed] [Google Scholar]
- [178].Nemoto Y, Toda K, Ono M, Fujikawa-Adachi K, Saibara T, Onishi S, Enzan H, Okada T, Shizuta Y. Altered expression of fatty acid-metabolizing enzymes in aromatase-deficient mice. J Clin Invest. 2000;105:1819–1825. doi: 10.1172/JCI9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Egawa T, Toda K, Nemoto Y, Ono M, Akisaw N, Saibara T, Hayashi Y, Hiroi M, Enzan H, Onishi S. Pitavastatin ameliorates severe hepatic steatosis in aromatase-deficient (Ar-/-) mice. Lipids. 2003;38:519–523. doi: 10.1007/s11745-003-1093-x. [DOI] [PubMed] [Google Scholar]
- [180].Yun JW, Shin ES, Cho SY, Kim SH, Kim CW, Lee TR, Kim BH. The effects of BADGE and caffeine on the time-course response of adiponectin and lipid oxidative enzymes in high fat diet-fed C57BL/6J mice: correlation with reduced adiposity and steatosis. Exp Anim. 2008;57:461–469. doi: 10.1538/expanim.57.461. [DOI] [PubMed] [Google Scholar]
- [181].Chan LL, Chen Q, Go AG, Lam EK, Li ET. Reduced adiposity in bitter melon (Momordica charantia)-fed rats is associated with increased lipid oxidative enzyme activities and uncoupling protein expression. J Nutr. 2005;135:2517–2523. doi: 10.1093/jn/135.11.2517. [DOI] [PubMed] [Google Scholar]
- [182].Huang HL, Hong YW, Wong YH, Chen YN, Chyuan JH, Huang CJ, Chao PM. Bitter melon (Momordica charantia L.) inhibits adipocyte hypertrophy and down regulates lipogenic gene expression in adipose tissue of diet-induced obese rats. Br J Nutr. 2008;99:230–239. doi: 10.1017/S0007114507793947. [DOI] [PubMed] [Google Scholar]
- [183].Chen Q, Chan LL, Li ET. Bitter melon (Momordica charantia) reduces adiposity, lowers serum insulin and normalizes glucose tolerance in rats fed a high fat diet. J Nutr. 2003;133:1088–1093. doi: 10.1093/jn/133.4.1088. [DOI] [PubMed] [Google Scholar]
- [184].Chen Q, Li ET. Reduced adiposity in bitter melon (Momordica charantia) fed rats is associated with lower tissue triglyceride and higher plasma catecholamines. Br J Nutr. 2005;93:747–754. doi: 10.1079/bjn20051388. [DOI] [PubMed] [Google Scholar]
- [185].Wolfram S, Wang Y, Thielecke F. Anti-obesity effects of green tea: from bedside to bench. Mol Nutr Food Res. 2006;50:176–187. doi: 10.1002/mnfr.200500102. [DOI] [PubMed] [Google Scholar]
- [186].Bose M, Lambert JD, Ju J, Reuhl KR, Shapses SA, Yang CS. The major green tea polyphenol, (-)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat-fed mice. J Nutr. 2008;138:1677–1683. doi: 10.1093/jn/138.9.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Korsrud GO, Baldwin RL. Effects of endocrinectomy and hormone replacement therapies upon enzyme activities in lactating rat mammary glands. Biol Reprod. 1969;1:21–30. doi: 10.1095/biolreprod1.1.21. [DOI] [PubMed] [Google Scholar]
- [188].Eckstein B, Villee CA. Effect of estradiol on enzymes of carbohydrate metabolism in rat uterus. Endocrinology. 1966;78:409–411. doi: 10.1210/endo-78-2-409. [DOI] [PubMed] [Google Scholar]
- [189].Eckstein B, Shalem M. Influence of oestradiol on citrate accumulation in the uterus and blood of fluoroacetate-treated rats. J Endocrinol. 1966;34:227–231. doi: 10.1677/joe.0.0340227. [DOI] [PubMed] [Google Scholar]
- [190].Riu E, Ferre T, Hidalgo A, Mas A, Franckhauser S, Otaegui P, Bosch F. Overexpression of c-myc in the liver prevents obesity and insulin resistance. Faseb J. 2003;17:1715–1717. doi: 10.1096/fj.02-1163fje. [DOI] [PubMed] [Google Scholar]
- [191].Danilovich N, Babu PS, Xing W, Gerdes M, Krishnamurthy H, Sairam MR. Estrogen deficiency. obesity, and skeletal abnormalities in follicle-stimulating hormone receptor knockout (FORKO) female mice, Endocrinology. 2000;141:4295–4308. doi: 10.1210/endo.141.11.7765. [DOI] [PubMed] [Google Scholar]
- [192].Jones AP, McElroy JF, Crnic L, Wade GN. Effects of ovariectomy on thermogenesis in brown adipose tissue and liver in Syrian hamsters. Physiol Behav. 1991;50:41–45. doi: 10.1016/0031-9384(91)90495-a. [DOI] [PubMed] [Google Scholar]