

Abstract
The Scandinavian wolf population represents one of the genetically most well characterized examples of a severely bottlenecked natural population (with only two founders), and of how the addition of new genetic material (one immigrant) can at least temporarily provide a “genetic rescue”. However, inbreeding depression has been observed in this population and in the absence of additional immigrants, its long-term viability is questioned. To study the effects of inbreeding and selection on genomic diversity, we performed a genomic scan with approximately 250 microsatellite markers distributed across all autosomes and the X chromosome. We found linkage disequilibrium (LD) that extended up to distances of 50 Mb, exceeding that of most outbreeding species studied thus far. LD was particularly pronounced on the X chromosome. Overall levels of observed genomic heterozygosity did not deviate significantly from simulations based on known population history, giving no support for a general selection for heterozygotes. However, we found evidence supporting balancing selection at a number of loci and also evidence suggesting directional selection at other loci. For markers on chromosome 23, the signal of selection was particularly strong indicating that purifying selection against deleterious alleles may have occurred even in this very small population. These data suggest that population genomics allows the exploration of the effects of neutral and non-neutral evolution on a finer scale than what has previously been possible.
Keywords: heterozygosity, genome scan, linkage disequilibrium, inbreeding, conservation genetics
Introduction
When a large number of genetic markers spread across the genome are available, the study of population processes such as the role of selection and random drift in shaping genotypic evolution turns from population genetics to population genomics. There is increasing enthusiasm about the possibility of performing genome wide investigations in natural populations as a means to study, for example, selective sweeps and the genetic background to adaptive population divergence (Luikart et al. 2003; Schlötterer 2003; Beaumont & Balding 2004; Storz 2005). With a well characterized genome at hand, genomic scale studies are not only attractive due to the sheer number of markers used but also because the loci under investigation can be chosen to be more or less evenly distributed across the genome. As a consequence, the possibility to detect signals of selection from rates and patterns of polymorphism is enhanced. At present, most genome-wide approaches are limited to model organisms that have been subject to genome sequencing. However, with the advent of new sequencing technology, large-scale sequence information from non-traditional model organism systems is likely to accumulate rapidly in the near future (Ellegren & Sheldon 2008).
One area in which we foresee that population genomic approaches will become important is in conservation genetics and the associated study of inbreeding depression in small populations (Kohn et al. 2006). The Scandinavian wolf (Canis lupus) is an example of a highly bottlenecked population that has been the focus of extensive studies in conservation genetics (Ellegren et al. 1996; Ellegren 1999; Sundqvist et al. 2001; Flagstad et al. 2003; Vilà et al. 2003a, 2003b; Seddon & Ellegren 2004; Liberg et al. 2005; Seddon et al. 2005, 2006; Bensch et al. 2006). The current population originates from only three founders, and not surprisingly, the offspring of some pairs have inbreeding coefficients as high as 0.3-0.4 (Liberg et al. 2005). This inbreeding has been shown to be negatively correlated with reproductive success, with reduced litter sizes from more related parents, suggesting that the population is in genetic peril in the absence of additional genetic input from new immigrants (Liberg et al. 2005).
The wolf is the wild ancestor of domestic dogs (Canis familiaris) (Vilà et al. 1997). The dog genome has recently been sequenced (Kirkness et al. 2003; Lindblad-Toh et al. 2005) and there is a rich source of polymorphic markers with known location in the genome developed for dogs, including both single nucleotide polymorphisms (Sutter et al. 2004, Lindblad-Toh et al. 2005) and microsatellites (Parker et al. 2004; Sargan et al. 2007). Based on these resources, we report here on a genomic scan of the Scandinavian wolf population using approximately 250 microsatellites evenly distributed across all autosomes and the X chromosome. We show how the genetic make-up of the population has been shaped by inbreeding, but also by selective forces acting on the genome.
Material and methods
Population history
The wolf went extinct as a breeding population in Scandinavia during the second part of the 1960s (Wabakken et al. 2001), though the occasional immigrant strayed into northern Sweden during the 1970s. However, in the early 1980s, an immigrant wolf pair established a territory and started to reproduce in central Sweden. Analyses of autosomal, mitochondrial and Y chromosome markers from individuals born before 1991 show that the population was founded by a single pair in the early 1980s, most likely originating from the neighboring Finnish-Russian population (Sundqvist et al. 2001; Vilà et al. 2003a). As both founders died after a few years, continued reproduction between offspring in this pack of wolves resulted in severe inbreeding (Sundqvist et al. 2001, Vilà et al. 2003a). In 1991, a second pack was established and the detection of a new Y chromosome haplotype among individuals born in 1991 and later revealed that a third founder, a male immigrant, had contributed to the gene pool (Sundqvist et al. 2001, Vilà et al. 2003a). Since then, the population has increased in size, currently harboring at least 18 breeding packs and at least 150 individuals (http://skandulv.nina.no/). Although some additional immigration events have been detected after 1991 (Seddon et al. 2006), close monitoring of mitochondrial and Y-linked markers have shown that these have not entered the breeding population.
Samples
Tissue or blood samples were collected from road kills, legally shot animals, animals found dead or from animals being radio-tagged during 1984-2005 (Table 1) as described in our previous work (e.g. Vilá et al. 2003; Seddon et al. 2005). Genomic DNA was isolated using a standard phenol chloroform protocol, also as described previously. In total, 112 native Scandinavian wolves were analyzed, including one of the three founders, the female. For an analysis of population subdivision, a sample of 24 Russian wolves (from the Tvier and Smoliensk regions) was also genotyped.
Table 1.
Estimated birth year of sampled wolves.
| Birth year | Number of wolves studied |
|---|---|
| 1978 | 1 |
| 1983 | 3 |
| 1987 | 3 |
| 1988 | 1 |
| 1989 | 3 |
| 1990 | 1 |
| 1991 | 4 |
| 1992 | 2 |
| 1994 | 2 |
| 1995 | 2 |
| 1996 | 5 |
| 1997 | 5 |
| 1998 | 9 |
| 1999 | 12 |
| 2000 | 19 |
| 2001 | 6 |
| 2002 | 7 |
| 2003 | 7 |
| 2004 | 18 |
| 2005 | 1 |
Markers and genotyping
Two hundred and fifty eight microsatellite markers were taken from Microsatellite Multiplex Set II and III described by Clark et al. (2004) and Sargan et al. (2007), respectively. They are a combination of di- and tetranucleotide repeats that have been mapped to the canine genome by radiation hybrid and/or linkage mapping methods, with 31 markers coming directly from the genome sequence (Breen et al. 2004; Breen et al. 2001; Sargan et al. 2007). These marker sets have been selected to represent markers evenly distributed across the canine genome; a full list of the markers is provided in the Supplementary Information. The standard PCR profile included (after denaturing 5 min at 95 °C) 10 cycles of 95°C for 30 s, 60°C reducing by 1 degree per cycle to 50°C for 30 s and 72 °C for 30 s. The touchdown cycles were followed by 30 cycles of 95°C for 30 s, 50°C for 30 s and 72°C for 30 s. Genotyping of reaction products was performed by capillary electrophoresis using an Applied Biosystems 3730 instrument and the GENEMAPPER v3.7 software.
All loci were checked to make sure that they conformed to the inferred history of the Scandinavian wolf population, i.e. that a maximum of six (four at X-linked loci) alleles occurred in the population. Specifically, we checked that the original two wolves contributed a maximum of four (for X-linked loci three) alleles and that the third founder contributed a maximum of two (for X-linked loci one) alleles. For loci not conforming to the inferred history of the population, rare alleles (occurring in just one or two individuals) were grouped with the main alleles closest in size. We consider the most likely cause of such rare alleles to either be genotyping error or mutation.
Estimates of linkage disequilibrium
We estimated pair-wise D′ and χ2′ as measures of linkage disequilibrium (LD). Simulation studies have shown that χ2′ is the preferred measure of LD for multi-allelic markers, at least for the purpose of QTL mapping (Zhao et al. 2005). However, for the purpose of this paper the two measures yield similar conclusions and only the more commonly used D′ is presented to facilitate comparisons with other studies.
Haplotypic phase was not known in our dataset and in the absence of detailed pedigree information for the individuals genotyped we could not infer phase. Instead we relied on population haplotype frequency data to infer phase. Haplotype frequencies were first calculated for all genotypes except the double heterozygotes. For the latter we then assigned phase using the haplotype frequencies observed in non-double heterozygotes. Subsequently, the total haplotype frequencies for known and assigned haplotypes were used for estimating LD. The assignment of haplotypic phase to double heterozygotes and the estimation of LD were repeated 1,000 times and we used the average LD values from this iteration. We noticed that using haplotype frequency data from too few wolves skewed the estimates of LD. Therefore, only loci for which the genotypes of more than 25 wolves were available were included in the LD analyses. Even so, estimating haplotype frequencies in this manner will include a risk of not detecting very rare haplotypes.
Simulations
Simulations were based on the known population history of the Scandinavian wolf population (Wabakken et al. 2001; Liberg et al. 2005). For each pack present up until 2002 information on from which pack the alpha female and male descended (the social system of wolves typically involves packs with a single reproducing male and female plus juvenile and sub adult individuals) has been published (Liberg et al. 2005). It is also known in which year the pack was established and when it last bred (Wabakken et al. 2001; Liberg et al. 2005). The exact pedigrees of the wolves genotyped in this study was not known to us. However, information on birth year was available, and using the information published in Wabakken et al. (2001) and Liberg et al. (2005) we could deduce which packs it could have been born in (i.e. which packs were breeding in its birth year). Knowing this we could, using the published data of the ancestries of alpha males and females, further deduce the possible ancestries of each wolf in our dataset and use these for our simulation comparisons
For simulations we first created the three founder individuals with a given number of alleles. For comparisons of overall heterozygosity or LD we wanted to match the number of alleles in our simulations to that observed in our dataset. To do this we identified the number of “founder alleles” of each locus as the number of alleles observed before 1991 (founder alleles from the first two founders) and new alleles observed after 1991 (founder alleles from the third founder). The number of alleles given to the simulated founders were then drawn randomly from the distribution of “founder alleles” we had obtained across all loci in our dataset. The alleles used were randomly assigned to the founder individuals and to the individual's chromosomes. Next we created alpha individuals using the known information about the pedigree, and assigning each individual one chromosome from each of its parents. When simulating loci on the X chromosome male wolves were assigned a Y-chromosome containing no alleles.
For simulation of LD we allowed recombination between two loci on the same chromosome with a frequency ranging from 0 to 50 % (free recombination). Finally we obtained a sample population by creating pups randomly picking an alpha pair among the ones present in a given birth year as parents. Again recombination was allowed when simulating LD. During simulations of LD on the X chromosomes, however, no recombination was allowed in male wolves.
For heterozygosity comparisons we created pups either by randomly choosing a parental alpha pair or by weighting the probability of an alpha pair being chosen as parents by the negative effect introduced by the inbreeding coefficient of the alpha pair given in Liberg et al. (2005). We matched the size of the simulated sample population to that of our data set by, in each year, simulating the same number of pups as individuals with that birth year in our data set. Each simulation was replicated 10,000 times and averages, standard deviations and confidence intervals were obtained from these replicates. As only data at the population history up until 2002 was available all simulation-based comparisons were done without data for wolves born after this year.
To evaluate the quality of our simulation setup we calculated the average inbreeding coefficient of the simulated sample. This was compared to the inbreeding reported for the same sample by Bensch et al. (2006) allowing us to identify systematic deviations of our simulations from the previously established history of the population. The code for simulations is available upon request.
Tests for selection
As the effects of selection and drift on heterozygosity and population subdivision will have developed gradually over the history of the population, only wolves from 1999 or later were used when testing for selection and population subdivision, and only simulation results for these years were used for comparisons. For each locus we compared the observed heterozygosity with that expected for a locus with the same number of founder alleles using the simulation setup described above. We compared the observed heterozygosity at each locus with the confidence interval obtained from the heterozygosity distribution of the 10,000 simulation replications of a locus with the corresponding number of founder alleles. This was repeated with and without Bonferroni correction. As we cannot be sure of the true number of alleles contributed by the two founder males, since they were not sampled, the number of founder alleles may have been underestimated. Our estimates will be conservative for loci with a deficit of heterozygosity, but not so for loci with an excess of heterozygosity. For the latter analysis we therefore repeated the comparisons assuming that founder alleles had been undetected.
We further tested for selection using the Ewens-Watterson neutrality test as our lack of resolution in the Scandinavian wolf pedigree introduces a level of uncertainty to our simulations. The Ewens-Watterson F-test can reveal deviations from a neutral equilibrium model, with excesses or deficits of genetic diversity, given the number of alleles present at a locus. We performed the Ewens-Watterson neutrality test according to Watterson (1977). F is the expected homozygosity under the allele frequencies in the sample (or the sum of all squared allele frequencies). This is compared with the probability given the sample size and the number of alleles.
We also applied the lnRV (Schlötterer, 2002) and lnRH (Schlötterer and Dieringer, 2005) neutrality tests to our data. These tests require the use of a second population and we used data from Russian wolves where this was available. Both the lnRV and lnRH tests should be robust to changes in population size. However, Schlötterer (2002) note that with a recent and strong bottleneck in combination with a low Θ (= 4Neμ), the tests may result in an excess of loci with low variability.
Population subdivision between the Russian and Scandinavian populations was estimated with Wright's FST. We also simulated FST using the simulation setup described above. This time we assigned the simulated founders the actual founder alleles identified at a given locus in the dataset to allow for comparison with the Russian population. This allowed us to obtain a confidence interval for expected FST values under the known population history.
Results
Linkage disequilibrium
The inbred nature of the Scandinavian wolf population suggests that linkage disequilibrium may be extensive. We genotyped 237 evenly distributed microsatellites, representing all autosomes and the X chromosome, in 112 individuals from this population to assess the overall genomic levels of LD. The average distance between autosomal markers was 7.98 Mb and 11.15 Mb between X-linked markers. For pairs of markers located on different autosomes (23,988 pairs of loci), mean D′ is 0.219 (S.D. ± 0.130) which thus forms the background level of LD in this population (Figure 1). Within autosomes (735 pairs of loci), LD is extensive. It decreases with physical distance but does not reach background level until, on average, about 50 Mb (Figure 2). The level of observed LD is remarkable for a natural population of an outbreeding species.
Figure 1.
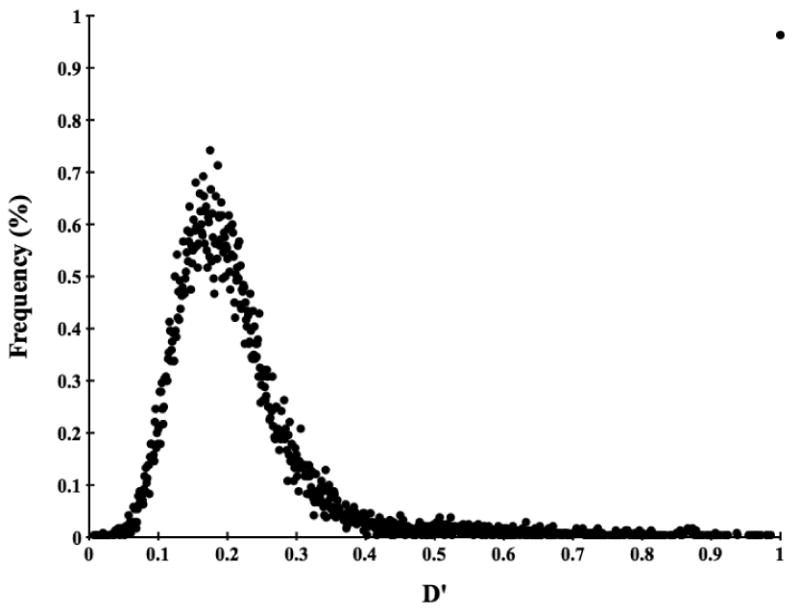
Distribution of D′ for pairs of unlinked loci on different chromosomes. Approximately 1 % (231) of the pairs of loci has a D′ value of 1. However, when LD for these pairs is estimated by χ2′, they have a distribution similar to that of χ2′ for all pair-wise loci, indicating that these high D′ values are artifacts.
Figure 2.
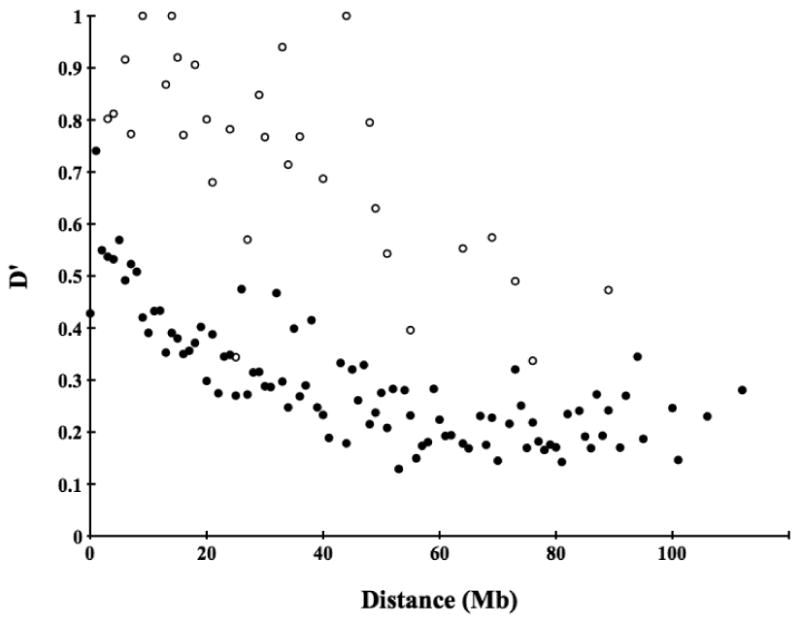
The relationship between LD (D′) and physical distance (Mb) between loci residing on the same chromosome. Mean D′ is given in 1 Mb intervals for autosomes (filled circles) and the X chromosome (open circles).
LD was not uniform on all chromosomes (Table 2). The clearest outlier was the X chromosome with a mean D′ of 0.730 (± 0.200) (Figure 2). From simulations we expected loci on the X chromosome to have about 20 % more LD than the autosomes, but LD on the X chromosome was in many cases twice or more that of autosomal loci separated by the same distance. Six autosomes (CFA1, CFA8, CFA14, CFA23, CFA30, CFA33) showed a mean of more than one standard deviation higher LD than the mean for all pairs of markers separated by corresponding distance.
Table 2.
Average D′ of individual chromosomes.
| Chromosome | Mean marker interval (Mb)a | Mean D′ | S.D. | Expected mean D′b | S.D. |
|---|---|---|---|---|---|
| CFA01 | 44.03 | 0.2976c | 0.1718 | 0.1784 | 0.0583 |
| CFA02 | 27.70 | 0.3201 | 0.1798 | 0.3146 | 0.1753 |
| CFA03 | 40.31 | 0.3114 | 0.1626 | 0.2330 | 0.0905 |
| CFA04 | 36.33 | 0.3109 | 0.1745 | 0.2685 | 0.0838 |
| CFA05 | 32.67 | 0.3456 | 0.1909 | 0.2970 | 0.1943 |
| CFA06 | 16.68 | 0.3060 | 0.2368 | 0.3564 | 0.1784 |
| CFA07 | 9.90 | 0.3002 | 0.0000 | 0.3906 | 0.1709 |
| CFA08 | 39.92 | 0.4803c | 0.2496 | 0.2330 | 0.0905 |
| CFA09 | 26.99 | 0.3395 | 0.1713 | 0.2723 | 0.1262 |
| CFA10 | 27.04 | 0.3878 | 0.1723 | 0.2723 | 0.1262 |
| CFA11 | 26.40 | 0.3475 | 0.1705 | 0.4748 | 0.2268 |
| CFA12 | 10.65 | 0.4982 | 0.1669 | 0.4325 | 0.1605 |
| CFA13 | 25.97 | 0.4541 | 0.2267 | 0.4748 | 0.2268 |
| CFA14 | 19.95 | 0.4461c | 0.1569 | 0.2982 | 0.1318 |
| CFA15 | 31.25 | 0.1998 | 0.0895 | 0.2866 | 0.1361 |
| CFA16 | 16.31 | 0.4343 | 0.1480 | 0.3501 | 0.2026 |
| CFA17 | 24.83 | 0.3525 | 0.1841 | 0.2700 | 0.1191 |
| CFA18 | 19.35 | 0.5530 | 0.2575 | 0.4021 | 0.2163 |
| CFA19 | 24.88 | 0.2756 | 0.2082 | 0.2700 | 0.1191 |
| CFA20 | 20.57 | 0.3321 | 0.1618 | 0.3878 | 0.2114 |
| CFA21 | 14.43 | 0.4165 | 0.2596 | 0.3905 | 0.1864 |
| CFA22 | 28.35 | 0.3651 | 0.1865 | 0.3146 | 0.1753 |
| CFA23 | 20.12 | 0.5937c | 0.3610 | 0.2982 | 0.1318 |
| CFA24 | 17.26 | 0.4273 | 0.2335 | 0.3564 | 0.1784 |
| CFA25 | 25.33 | 0.3077 | 0.0605 | 0.2700 | 0.1191 |
| CFA26 | 14.56 | 0.3124 | 0.1850 | 0.3800 | 0.1635 |
| CFA27 | 19.92 | 0.3687 | 0.2861 | 0.2982 | 0.1318 |
| CFA28 | 18.04 | 0.2818 | 0.2297 | 0.3714 | 0.1646 |
| CFA29 | 12.86 | 0.3700 | 0.0689 | 0.3526 | 0.2314 |
| CFA30 | 19.55 | 0.4849c | 0.2223 | 0.2982 | 0.1318 |
| CFA31 | 18.33 | 0.3157 | 0.2020 | 0.3714 | 0.1646 |
| CFA32 | 11.66 | 0.3511 | 0.2025 | 0.4335 | 0.2005 |
| CFA33 | 14.29 | 0.5794c | 0.3997 | 0.3905 | 0.1864 |
| CFA35 | 13.20 | 0.2985 | 0.1830 | 0.3526 | 0.2314 |
| CFA36 | 11.23 | 0.4561 | 0.2373 | 0.4325 | 0.1605 |
| CFA37 | 14.34 | 0.3712 | 0.2078 | 0.3905 | 0.1864 |
| CFAX | 33.08 | 0.7301c | 0.2002 | 0.2970 | 0.1943 |
Mean interval among all pairs of marker combinations on chromosome
Mean LD for all pairs of markers separated by the same distance as the mean marker interval of the chromosome.
Observed LD more than one standard deviation higher than expected.
Overall genomic levels of diversity
Average heterozygosity across all markers decreased only slightly over the time-span studied (1983-2005), from 64% to 55%. The arrival of the third immigrant around 1990 provided a “genetic rescue” by contributing new alleles and temporarily preventing further inbreeding. When genotype data for wolves with or without the third founder in their ancestry were analyzed separately, the decrease in heterozygosity over time is more marked for wolves not descending from the third founder (Figure 3). There is an approximate drop in heterozygosity from 64% to 45% during the 1980s, before the arrival of the third immigrant. After this, mean heterozygosity decreases from about 60% to 55%.
Figure 3.
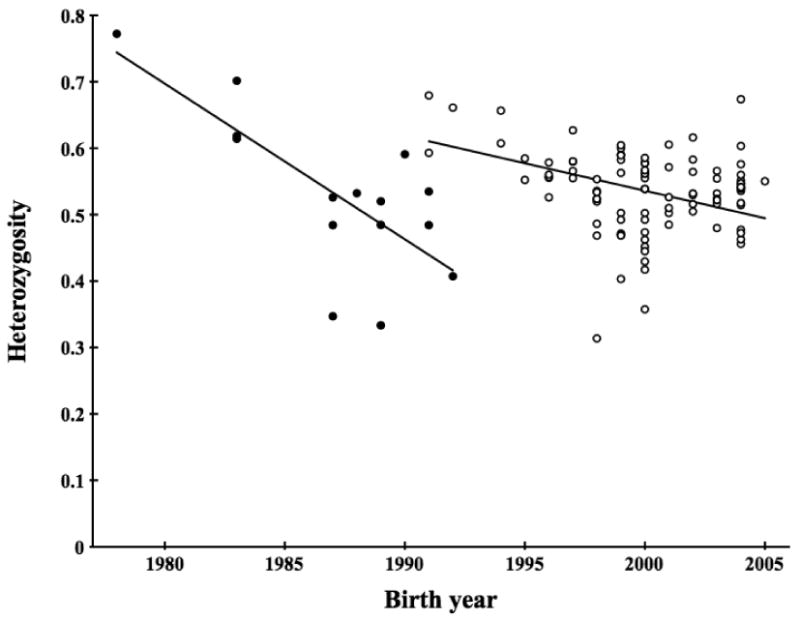
Observed microsatellite heterozygosity for cohorts within the Scandinavian wolf population. Filled circles are animals descending from the initial two founders while open circles are animals with some ancestry also from a third immigrant that first reproduced in 1991. Separate regression lines for the two data sets are given.
We simulated the evolution of heterozygosity based on known population history and compared this with the observed data. There was no clear difference between the simulated and observed values for cohorts, for example, in the form of a heterozygosity excess. If anything, expected heterozygosity tended to be higher than what was observed, with 10 birth years having a higher simulated than observed heterozygosity while only two years showed the opposite pattern (another three years had similar heterozygosity) (Figure 4). As inbreeding has been shown to have a detrimental effect on fitness in this population (Liberg et al. 2005), all packs may not have contributed equally to reproduction. We therefore repeated the simulations allowing the probability of an alpha pair to contribute to the simulation to be weighted by their relatedness. This had limited effect on the simulated heterozygosity, though three birth years showed an increased simulated heterozygosity (data not shown).
Figure 4.
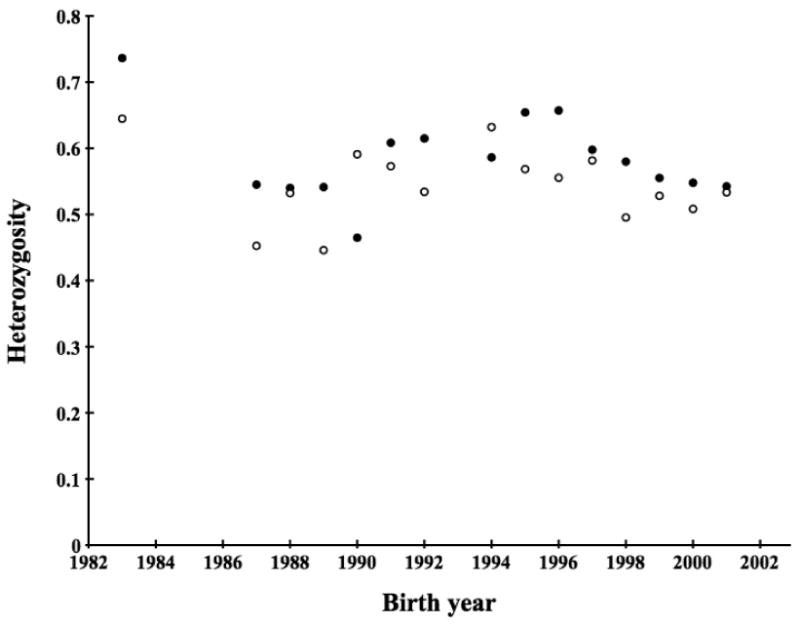
Observed and simulated average heterozygosity for cohorts within the Scandinavian wolf population. Open circles are average heterozygosities per birth year for wolves in the data set. Filled circles are simulated average heterozygosity.
To evaluate the quality of the simulations we simulated inbreeding coefficients in a sample of wolves corresponding to that in our dataset and then compared this with known inbreeding coefficients reported by Bensch et al. (2006) (Figure 5). The two approaches produced similar estimates. A notable exception was the 1993 - 1994 cohort where our simulated inbreeding coefficient markedly exceeded that of the true inbreeding coefficient of sampled wolves. The sampled wolves in this cohort all contained alleles inherited from the third founder and have thus not been sampled randomly, as in the simulations, which probably explains the discrepancy. For two other birth cohorts simulated inbreeding is also significantly higher than the true inbreeding coefficient of the data. There is thus some indication that our simulation set-up tends to overestimate the amount of inbreeding in the population.
Figure 5.
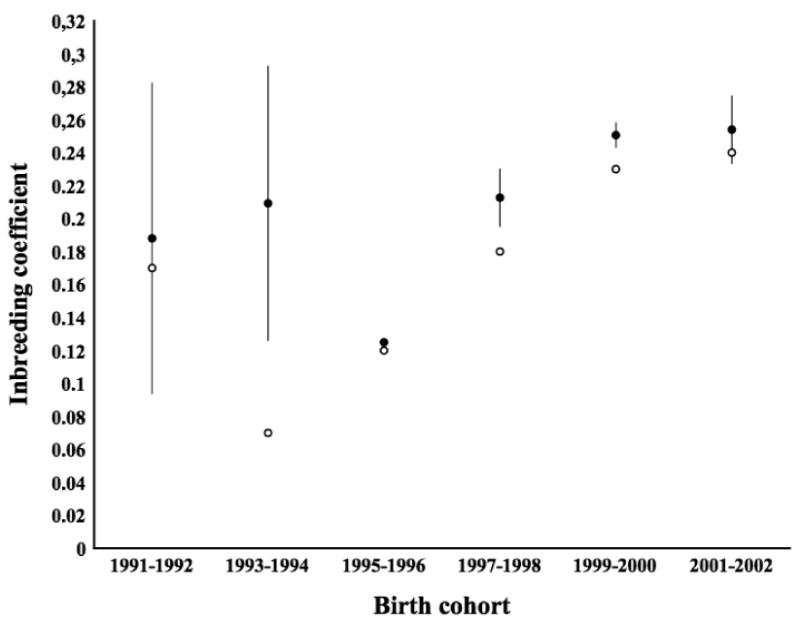
Inbreeding coefficients from Bensch et al. (2006) shown in open circles vs. our simulations shown in filled circles. Error bars show +/- one standard error for simulated values.
Signs of selection
Deviations from Hardy-Weinberg equilibrium could indicate selection. For example, purifying selection resulting from inbreeding depression or balancing selection for heterozygosity could occur. By comparing the observed heterozygosity of individual loci with the level expected from simulations we identified 17 loci showing a significant heterozygosity excess, four of which were significant also after Bonferroni correction (Table 3). These four were located on separate chromosomes (CFA1, CFA2, CFA11 and CFA20). One potential explanation for a heterozygote excess could be that we had been unable to accurately determine the number of alleles contributed by the two unsampled founder males. Assuming that the two founder males contributed two alleles in cases where we just interpreted one, only two loci remained with a significant heterozygote excess (Table 3). No locus had significantly low F values in the Ewens-Watterson homozygosity test after Bonferroni correction, giving little overall support for the action of balancing selection. However, it is possible that this test is not robust to the inclusion of many related individuals.
Table 3.
Loci more heterozygous than expected from simulation data and loci with significantly lower FST than expected. * p<0.05; ** p<0.01; *** p<0.001. Significance levels are after Bonferroni correction.
| Locus | Chromosome | Position | Heterozygosity excess | low FST |
|---|---|---|---|---|
| FH2309 | CFA01 | 85.77 | ***a,c | not tested |
| C02.342 | CFA02 | 21.05 | ***b,c | not tested |
| REN195B08 | CFA04 | 60.34 | ** | |
| FH5126 | CFA04 | 65.25 | * | |
| FH2097 | CFA04 | 71.9 | * | |
| FH3702 | CFA05 | 32.91 | ** | |
| REN204K13 | CFA08 | 15.83 | * | |
| REN287G01 | CFA09 | 63.03 | * | |
| FH4031 | CFA11 | 18.2 | **b | not tested |
| FH5153 | CFA13 | 65.38 | * | |
| FH5161 | CFA17 | 21.25 | * | |
| REN52K18 | CFA18 | 18.68 | * | |
| REN213G21 | CFA19 | 45.8 | * | |
| REN124F16 | CFA20 | 21.93 | ***c | ** |
| FH5166 | CFA20 | 50.52 | * | |
| REN114M19 | CFA20 | 56.66 | * | |
| FH3287 | CFA24 | 44.5 | ** | |
| DGN10 | CFA26 | 26.05 | * | |
| FH4001 | CFA27 | 9.86 | * | |
| REN146G17 | CFA28 | 33.55 | * | |
| CPH2 | CFA32 | 9.65 | * | |
| REN243O23 | CFA34 | 26.76 | * | |
| FH3865 | CFA36 | 23.29 | ** | |
| FH2998 | CFA36 | 28.08 | * |
Also significant with an additional allele contributed by first founder male
No extra allele could have been contributed by first founder male as he had already contributed with two alleles in the dataset.
Also significant with an additional allele contributed by second founder male
By contrast, 45 loci showed a significant deficit of heterozygosity, 15 of which remained significant after Bonferroni correction (Table 4). On chromosome 18, three loci (two of which were immediately adjacent) had lower than expected heterozygosity, and on both chromosomes 1 and 23 two (not neighboring) loci were deficient in heterozygosity. The clearest signal appears to be on chromosome 23 where, before Bonferroni correction, all but two of the seven analyzed loci showed a lack of heterozygosity. Interestingly, chromosome 23 contained the only locus (REN02P03) significant for directional selection with the Ewens-Watterson test (Table 4).
Table 4.
Loci potentially affected by directional selection. * p<0.05; ** p<0.01; *** p<0.001. Significance levels are after Bonferroni correction.
| Locus | Chromosome | Position | Heterozygosity deficit | Ewens-Watterson test | lnRH | lnRV | high FST |
|---|---|---|---|---|---|---|---|
| FH5117 | CFA01 | 30.74 | *** | ||||
| REN143K19 | CFA01 | 121.79 | *** | not tested | not tested | not tested | |
| C03.629 | CFA03 | 24.58 | * | not tested | not tested | not tested | |
| REN204K13 | CFA08 | 15.83 | ** | ||||
| REN147O02 | CFA11 | 61.37 | * | * | |||
| REN213F01 | CFA12 | 16.73 | * | *** | * | ||
| C13.391 | CFA13 | 3.62 | * | * | |||
| REN214L11 | CFA16 | 5.8 | * | * | |||
| REN52K18 | CFA18 | 18.68 | ** | ||||
| REN183B03 | CFA18 | 24.5 | ** | ||||
| REN47J11 | CFA18 | 45.33 | ** | ||||
| REN114M19 | CFA20 | 56.66 | * | ||||
| REN181K04 | CFA23 | 0.1 | ** | ||||
| FH3609 | CFA23 | 35.91 | ** | ** | |||
| REN02P03 | CFA23 | 38.99 | *** | ||||
| REN186B12 | CFA33 | 26.97 | ** | * | |||
| FH2985 | CFAX | 20.84 | *** | ||||
| FH1020 | CFAX | 36.81 | *** | ||||
| FH3027 | CFAX | 41.03 | * | ||||
| REN185C11 | CFAX | 60.85 | *** | ||||
| REN130F03 | CFAX | 70.13 | *** |
A large proportion of the markers (192) were also genotyped in a sample of Russian wolves. Wolves have a continuous distribution throughout northern Eurasia, from Finland and eastwards and although there might be some population substructure within this range, we use the data from Russian wolves as reflecting the source population of Scandinavian wolves (cf. Vila et al. 2003a). Average FST between Scandinavian and Russian wolves across all loci was 0.105 (S.D. ± 0.082), indicating moderate genetic differentiation. The average FST for loci on the X chromosome was higher (0.185, S.D. ± 0.086) though not statistically so. The range of FST-estimates was wide, from 0.001 to 0.496 (Figure 6). When comparing the observed FST values to those expected under simulations thirty-eight loci had significantly lower FST values, twenty-one of which were significant also after Bonferroni correction (Table 3).
Figure 6.
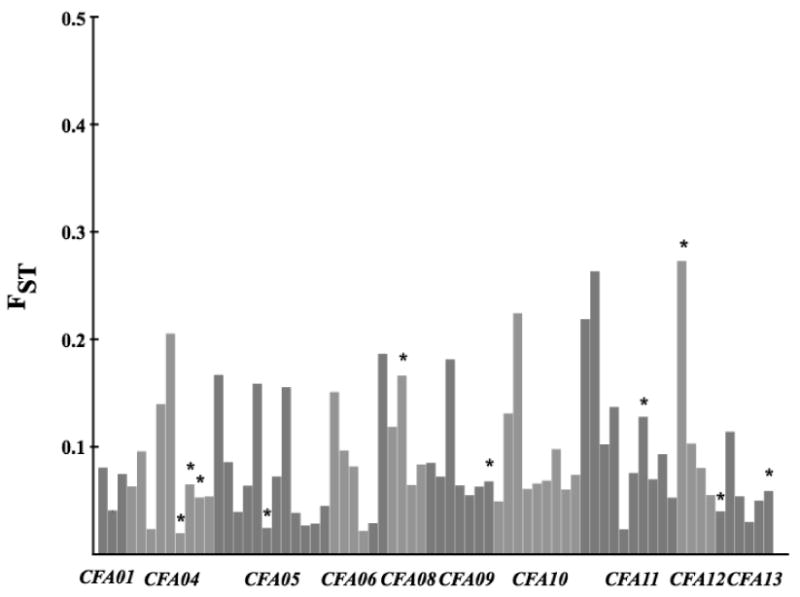
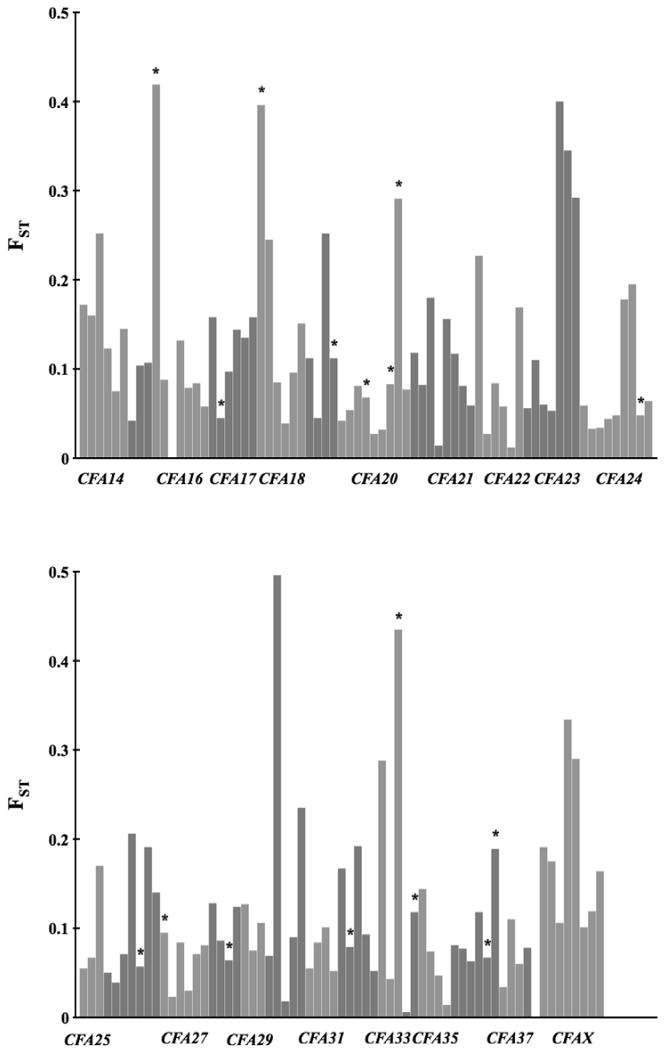
FST for all markers estimated in the comparison of Scandinavian and Russian wolves. Markers from different chromosomes are given in alternating colors. * denotes loci with FST values outside of 95 % confidence intervals from simulated FST values.
Of the 10 autosomal loci with the highest FST values, nine, including three adjacent loci on CFA23 also showed a heterozygosity deficit, indicating selection. However, from simulation of FST-values none of these was significantly higher than expected (Table 4). Another ten loci had significantly higher FST-estimates than expected, five of which remained significant after Bonferroni correction. All these were also lacking in heterozygosity (Table 4).
With genotype data from another population we could also use the lnRH and lnRV tests for detection of selection. Five loci showing a lack of heterozygosity tested significant for directional selection with the lnRH test, six with the lnRV test and three with both lnRH and lnRV (one each on the chromosomes CFA12, CFA23 and CFA33) (Table 4). However, after Bonferroni correction none of the lnRH tests were significant and only two loci with the lnRV test, one of which was located on CFA23.
From all these analyses we conclude that there is some evidence of balancing selection at loci distributed across the genome, but also of directional selection against deleterious alleles exposed during inbreeding in this wolf population. The signal of directional selection seemed strongest on chromosome 23.
Discussion
The history of the Scandinavian wolf population is characterized by a very narrow founding bottleneck followed by two and a half decades of strong inbreeding. Inbreeding depression has been documented in the Scandinavian wolf in the form of decreasing litter size with increasing inbreeding coefficient (Liberg et al. 2005). Moreover, congenital malformations have been found to be more common in this population than in outbred wolf populations in Finland and in Sweden prior to extinction (Räikkönen et al. 2006).
Inbreeding is expected to lead to increased levels of LD between loci. When the duration of a bottleneck is of the same order of magnitude as the bottleneck population size, its effect on patterns of genetic variability, including LD, becomes significant (Nordborg & Tavaré 2002). Depending on the haplotypic phase between selected alleles at different loci, this can reduce the efficacy of selection and lead to difficulties in purging of deleterious alleles. Furthermore, in a small population the effects of selection will be counteracted by genetic drift, augmenting the difficulty of purging deleterious alleles. The Scandinavian wolf population may thus face the problem of reduced fitness from the exposure of deleterious alleles and difficulties in purging these from the gene pool. Our data confirm and extend the observations of Bensch et al. (2006) by revealing extraordinary levels of intra-chromosomal LD, far exceeding that of most outbreeding species studied to date including partially selfing species (e.g. Long et al. 1998; Nordborg et al. 2002; Ingvarsson, 2005; Pardo et al. 2006; Cutter et al. 2006). This means that in some cases LD may extend over whole chromosomes, as many canine autosomes are less than 50 Mb in size. For comparison, LD across dog breeds drops to background levels by 200 kb while within breed LD are close to background levels at 13 Mb (Lindblad-Toh et al. 2005). LD was particularly extensive on the X chromosome, which is not unexpected given that it only recombines in females. Similar observations have been made in humans (Altshuler et al. 2005) and cattle (Sandor et al. 2006).
Loss of heterozygosity can occur at a slower than expected rate in inbred populations if there is selection for heterozygotes. Bensch et al. (2006) have recently studied the relationship between inbreeding coefficient and multi-locus heterozygosity in the Scandinavian wolf population. Using a set of about 30 microsatellite markers, they found that heterozygous wolves were overrepresented among those animals that were recruited to the breeding population. They also observed an overall heterozygosity excess in the population, which could be explained by selection for heterozygotes at recruitment. This is promising for an inbred and endangered population, as it would provide a means to decelerate an on-going loss of heterozygosity. A similar observation and conclusion have been made for a highly bottlenecked island population of mouflon (Kaeuffer et al. 2007).
However, our study fails to replicate the heterozygosity excess for the Scandinavian wolf population reported by Bensch et al. (2006). Generally, the mean genomic level of heterozygosity was very similar to simulated values of what would be expected given the degree of inbreeding in the population. There are several possible explanations for this discrepancy. The present analysis is based on nearly 10 times as many microsatellite markers as the previous study, so the inability to reject an expected model despite greater power may suggest that the previous result was a statistical artifact. On the other hand, Bensch et al. (2006) used a different approach in that they compared observed individual heterozygosities with inbreeding coefficients rather than simulated heterozygosity, something we are unable to do in the absence of access to detailed pedigree information. Also, Bensch et al. (2006) found the excess of heterozygotes when looking specifically at the breeding population while our study does not distinguish between individuals that did or did not contribute to subsequent generation. For future studies it will be important to combine pedigree data with dense genome scans to better understand if selection acts to maintain heterozygosity in this population.
The cause of inbreeding depression is generally attributed to the unmasking of recessive or partly recessive deleterious alleles (Charlesworth & Charlesworth 1999; Dudash & Carr, 1998). The loss of heterozygote advantage is usually thought of as less important although there are some studies going in the opposite direction (Kärkkäinen et al. 1999; Ferreira & Amos 2006). From a conservation point of view, a population suffering inbreeding depression from recessive deleterious alleles will recover in fitness once the deleterious alleles have been purged, while inbreeding depression from loss of heterozygote advantage cannot be restored by purging. In our dataset, many more loci show a deficit of heterozygosity compared to the number showing an excess of heterozygosity. While the markers themselves can be expected to be selectively neutral it is not unreasonable to assume that they reflect events at linked loci, given the amount of LD observed. Taken together this tentatively suggests that recessive deleterious alleles have been an important cause to inbreeding depression in the Scandinavian wolf population.
Linkage disequilibrium is thought to be the cause of heterozygosity - fitness correlations observed in supposedly neutral markers in some species (e.g. Da Silva et al. 2006; Hansson & Westerberg 2002; Slate et al. 2000). With the high levels of LD found in this study and given the high marker density (on average one marker every 8.52 Mb), the effects of selection on genes affecting fitness should be detectable at adjacent microsatellite markers. The markers showing less than expected heterozygosity may thus represent candidate regions for selection. This is particularly so for those regions where there is both a heterozygosity deficit and substantial population subdivision, as indicated by high FST values (cf. Table 4 and Figure 6), or for regions indicating directional selection in multiple tests.
Some regions show particularly strong signals of directional selection. On CFA12, the locus REN213F01 was detected by three different approaches. On CFA23, two adjacent loci both indicated directional selection in two different tests and a third locus on this chromosome had a heterozygosity deficit. The locus REN186B12 on CFA33 had both a heterozygosity deficit and a significantly high FST value, and two adjacent loci on CFA18 (and a third locus on this chromosome) are deficient in heterozygosity.
Of particular interest is chromosome 23 where all but two of the loci showed a depletion of heterozygosity (before correcting for multiple tests). In addition, chromosomal LD was high and several loci tested significant with both Ewens-Watterson and lnRV tests. FST values on this chromosome were among the highest observed. Interestingly, this chromosome region also harbors the one locus where homozygosity had the strongest effect on the likelihood for reproduction in the study of Bensch et al. (2006). We could find no obvious candidate gene from the genome sequence of CFA23 that would suggest an inbreeding defect such as those previously observed in the Scandinavian wolf population (including chryptorchidism and spinal deformities of fusion defects to the vertebrae; Räikkönen et al. 2006). Fine-scale mapping of one or more selected loci on this chromosome may help to identify candidate genes, though the extensive LD observed may obscure genotype – phenotype associations.
A general concern of population genomic approaches for detection of selection is that bottlenecks may mimic the effects of selection and thus produce false positive. This situation could very well apply in this case considering the recent strong population growth in the Scandinavian wolf population. However, the fact that several adjacent loci show signatures of selection is a good hint these are genuine. Another concern in this case is the simulation approach given that we lack detailed pedigree data, something that might reflect the discrepancy between simulated and observed inbreeding coefficients.
Although there are recent examples of the application of single nucleotide polymorphism (SNP) (Seddon et al. 2005) and insertion-deletion markers (Väli et al. 2008) in wolf population genetic studies, most studies thus far have utilized microsatellite markers. With the increased genetic knowledge of the canine genome following in the wake of the canine genome project, the application of dense SNP chips to studies of wolf population genomics would be illuminating. The greater density of markers would allow much finer understanding of the genomic substructure of canid chromosomes in the Scandinavian wolf population and permit more sophisticated analysis of subregions of interest.
Acknowledgments
Financial support was obtained from the Norwegian and Swedish Environmental Protection Agencies. Three anonymous reviewers are acknowledged for helpful comments. J.H. acknowledges Mats Aigner for help with computer programming and the Ewens-Watterson test. E.A.O. and H.G.P. acknowledge the Intramural Program of the National Human Genome Research Institute of the National Institutes of Health.
References
- Altshuler D, Brooks LD, Chakravarti A, et al. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA, Balding DJ. Identifying adaptive genetic divergence among populations from genome scans. Molecular Ecology. 2004;13:969–980. doi: 10.1111/j.1365-294x.2004.02125.x. [DOI] [PubMed] [Google Scholar]
- Bensch S, Andrén H, Hansson B, et al. Selection for heterozygosity gives hope to a wild population of inbred wolves. PLoS ONE. 2006;1:e72. doi: 10.1371/journal.pone.0000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen M, Hitte C, Lorentzen TD, et al. An integrated 4249 marker FISH/RH map of the canine genome. BMC Genomics. 2004;5:65. doi: 10.1186/1471-2164-5-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen M, Jouquand S, Renier C, et al. Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Research. 2001;11:1784–1795. doi: 10.1101/gr.189401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The genetic basis of inbreeding depression. Genetical Research. 1999;74:329–340. doi: 10.1017/s0016672399004152. [DOI] [PubMed] [Google Scholar]
- Clark LA, Tsai KL, Steiner JM, et al. Chromosome-specific microsatellite multiplex sets for linkage studies in the domestic dog. Genomics. 2004;84:550–554. doi: 10.1016/j.ygeno.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Cutter A. Nucleotide polymorphism and linkage disequilibrium in wild populations of the partial selfer Caenorhabditis elegans. Genetics. 2006;172:171–184. doi: 10.1534/genetics.105.048207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva A, Luikart G, Yoccoz NG, Cohas A, Allainé D. Genetic diversity-fitness correlation revealed by microsatellite analyses in European alpine marmots (Marmota marmota) Conservation Genetics. 2006;7:371–382. [Google Scholar]
- Dudash MR, Carr DE. Genetics underlying inbreeding depression in Mimulus with contrasting mating systems. Nature. 1998;393:682–684. [Google Scholar]
- Ellegren H. Inbreeding and relatedness in Scandinavian grey wolves Canis Lupus. Hereditas. 1999;130:239–244. doi: 10.1111/j.1601-5223.1999.00239.x. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Savolainen P, Rosen B. The genetical history of an isolated population of the endangered grey wolf Canis lupus: A study of nuclear and mitochondrial polymorphisms. Philosophical Transactions of the Royal Society: Biological Sciences. 1996;351:1661–1669. doi: 10.1098/rstb.1996.0148. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Sheldon BC. Genetic basis of fitness differences in natural populations. Nature. 2008;452:169–175. doi: 10.1038/nature06737. [DOI] [PubMed] [Google Scholar]
- Ferreira AGA, Amos W. Inbreeding depression and multiple regions showing heterozygote advantage in Drosophila melanogaster exposed to stress. Molecular Ecology. 2006;15:3885–3893. doi: 10.1111/j.1365-294X.2006.03093.x. [DOI] [PubMed] [Google Scholar]
- Flagstad Ø, Walker CW, Vilà C, et al. Two centuries of the Scandinavian wolf population: patterns of genetic variability and migration during an era of dramatic decline. Molecular Ecology. 2003;12:869–880. doi: 10.1046/j.1365-294x.2003.01784.x. [DOI] [PubMed] [Google Scholar]
- Gordon K, Corwin MB, Mellersch CS, Ostrander EA, Ott J. Establishing appropriate genome-wide significance levels for canine linkage analyses. Journal of Heredity. 2003;94:1–7. doi: 10.1093/jhered/esg009. [DOI] [PubMed] [Google Scholar]
- Hansson B, Westerberg L. On the correlation between heterozygosity and fitness in natural populations. Molecular Ecology. 2002;11:2467–2474. doi: 10.1046/j.1365-294x.2002.01644.x. [DOI] [PubMed] [Google Scholar]
- Ingvarsson PK. Nucleotide polymorphism and linkage disequilibrium within and among natural populations of European aspen (Populus tremula L., Salicaceae) Genetics. 2005;169:945–953. doi: 10.1534/genetics.104.034959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeuffer R, Coltman DW, Chapuis JL, Pontier D, Réale D. Unexpected heterozygosity in an island mouflon population founded by a single pair of individuals. Proceedings of the Royal Society London, Series B Biological Sciences. 2007;274:527–533. doi: 10.1098/rspb.2006.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärkkäinen K, Kuittinen H, van Treuren R, Vogl C, Oikarinen S, Savolainen O. Genetic basis of inbreeding depression in Arabis petraea. Evolution. 1999;53:1354–1365. doi: 10.1111/j.1558-5646.1999.tb05400.x. [DOI] [PubMed] [Google Scholar]
- Kirkness EF, Bafna V, Halpern AL, et al. The dog genome: Survey sequencing and comparative analysis. Science. 2003;301:1898–1903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- Kohn MH, Murphy WJ, Ostrander EA, Wayne RK. Genomics and conservation genetics. Trends in Ecology and Evolution. 2006;21:629–637. doi: 10.1016/j.tree.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Liberg O, Andrén H, Pedersen HC, et al. Severe inbreeding depression in a wild wolf (Canis lupus) population. Biology Letters. 2005;1:17–20. doi: 10.1098/rsbl.2004.0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh, Wade CM, Mikkelsen TS, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Long JC, Knowler WC, Hanson RL, et al. Evidence for genetic linkage to alcohol dependence on chromosomes 4 and 11 from an autosome-wide scan in an American Indian population. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 1998;81:216–221. doi: 10.1002/(sici)1096-8628(19980508)81:3<216::aid-ajmg2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nature Reviews Genetics. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Nordborg M, Tavaré S. Linkage disequilibrium: what history has to tell us. Trends in Genetics. 2002;18:83–90. doi: 10.1016/s0168-9525(02)02557-x. [DOI] [PubMed] [Google Scholar]
- Nordborg M, Borevitz JO, Bergelson J, et al. The extent of linkage disequilibrium in Arabidopsis thaliana. Nature Genetics. 2002;30:190–193. doi: 10.1038/ng813. [DOI] [PubMed] [Google Scholar]
- Pardo BG, Hermida M, Fernández C, et al. A set of highly polymorphic microsatellites useful for kinship and population analysis in turbot (Scophthalmus maximus L.) Aquaculture Research. 2006;37:1578–1582. [Google Scholar]
- Parker HG, Kim LV, Sutter NB, et al. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–1164. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- Räikkönen J, Bignert A, Mortensen P, Fernholm B. Congenital defects in a highly inbred wild wolf population (Canis lupus) Mammalian Biology. 2006;71:65–73. [Google Scholar]
- Sandor C, Farnir F, Hansoul S, Coppieters W, Meuwissen T, Georges M. Linkage disequilibrium on the bovine X chromosome: Characterization and use in quantitative trait locus mapping. Genetics. 2006;173:1777–1786. doi: 10.1534/genetics.106.059329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargan DR, Aguirre-Hernandez J, Galibert F, Ostrander EA. An extended microsatellite set for linkage mapping in the domestic dog. Journal of Heredity. 2007;98:221–231. doi: 10.1093/jhered/esm006. [DOI] [PubMed] [Google Scholar]
- Schlötterer C. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics. 2002;160:753–763. doi: 10.1093/genetics/160.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C. Hitchhiking mapping: Functional genomics from the population genetics perspective. Trends in Genetics. 2003;19:32–38. doi: 10.1016/s0168-9525(02)00012-4. [DOI] [PubMed] [Google Scholar]
- Schlötterer C, Dieringer D. Selective Sweep Nurminsky DI. Landes Bioscience; Georgetown, Texas: 2005. A novel test statistic for the identification of local selective sweeps based on microsatellite gene diversity. [Google Scholar]
- Seddon JM, Ellegren H. A temporal analysis shows major histocompatibility complex loci in the Scandinavian wolf population are consistent with neutral evolution. Proceedings of the Royal Society London, Series B Biological Sciences. 2004;271:2283–2291. doi: 10.1098/rspb.2004.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Sundqvist AK, Björnerfeldt S, Ellegren H. Genetic identification of immigrants to the Scandinavian wolf population. Conservation Genetics. 2006;7:225–230. [Google Scholar]
- Seddon JM, Parker HG, Ostrander EA, Ellegren H. SNPs in ecological and conservation studies: a test in the Scandinavian wolf population. Molecular Ecology. 2005;14:503–511. doi: 10.1111/j.1365-294X.2005.02435.x. [DOI] [PubMed] [Google Scholar]
- Slate J, Kruuk LEB, Marshall TC, Pemberton JM, Clutton-Brock TH. Inbreeding depression influences lifetime breeding success in a wild population of red deer (Cervus elaphus) Proceedings of the Royal Society London, Series B Biological Sciences. 2000;267:1657–1662. doi: 10.1098/rspb.2000.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz JF. Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular Ecology. 2005;14:671–688. doi: 10.1111/j.1365-294X.2005.02437.x. [DOI] [PubMed] [Google Scholar]
- Sundqvist AK, Ellegren H, Olivier M, Vilà C. Y chromosome haplotyping in Scandinavian wolves (Canis lupus) based on microsatellite markers. Molecular Ecology. 2001;10:1959–1966. doi: 10.1046/j.1365-294x.2001.01326.x. [DOI] [PubMed] [Google Scholar]
- Sutter NB, Eberle MA, Parker HG, et al. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Research. 2004;14:2388–2396. doi: 10.1101/gr.3147604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilà S, Savolainen P, Maldonado JE, Amorim IR, et al. Multiple and ancient origins of the domestic dog. Science. 1997;276:1687–1689. doi: 10.1126/science.276.5319.1687. [DOI] [PubMed] [Google Scholar]
- Vilà C, Sundqvist AK, Flagstad Ø, et al. Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proceedings of the Royal Society London, Series B Biological Sciences. 2003a;270:91–97. doi: 10.1098/rspb.2002.2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilà C, Walker C, Sundqvist AK, Flagstad Ø, et al. Combined use of maternal, paternal and bi-parental genetic markers for the identification of wolf-dog hybrids. Heredity. 2003b;90:17–24. doi: 10.1038/sj.hdy.6800175. [DOI] [PubMed] [Google Scholar]
- Väli U, Brandström M, Johansson M, Ellegren H. Insertion-deletion polymorphisms (indels) as genetic markers in natural populations. BMC Genetics. 2008;9:8. doi: 10.1186/1471-2156-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wabakken P, Sand H, Liberg O, Bjärvall A. The recovery, distribution, and population dynamics of wolves on the Scandinavian peninsula, 1978-1998. Canadian Journal of Zoology. 2001;79:710–725. [Google Scholar]
- Watterson GA. The homozygosity test of neutrality. Genetics. 1977;88:405–417. doi: 10.1093/genetics/88.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Nettleton D, Soller M, Dekkers JCM. Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genetical Research. 2005;86:77–87. doi: 10.1017/S001667230500769X. [DOI] [PubMed] [Google Scholar]