

Abstract
Recent data indicate that alcohol dependence induces long-term neuroadaptations that recruit a negative emotional state. This leads to excessive alcohol ingestion motivated by relief of negative emotionality. A key mechanism in this transition to negative reinforcement is a recruitment of corticotropin-releasing factor (CRF) signaling within the amygdala. Long term upregulation of CRF1 receptors is observed in the amygdala following a history of dependence, and CRF antagonists selectively block emotionality, excessive alcohol drinking and stress-induced reinstatement of alcohol-seeking in post-dependent animals. Innate upregulation of CRF1 receptor expression mimics the post-dependent phenotype, both with regard to emotional responses and ethanol self-administration. Therefore, the CRF system is emerging as a key element of the neuroadaptive changes driving alcoholism and as a major target for its treatment.
Introduction – a neuroadaptive perspective on alcohol dependence
Alcohol use accounts for 4% of global disease burden [1]. Alcohol dependence, or alcoholism, is a complex disorder in which heritable susceptibility factors contribute 50–60% of the disease risk, and interact with environmental factors to produce and maintain the disease state [2]. Alcoholism is characterized by uncontrolled heavy drinking and a chronic relapsing course [3]. Relapse, that is, return to heavy drinking after intervals of sobriety, is key to this process. Reduction of heavy drinking and relapse prevention are therefore main therapeutic objectives in alcoholism treatment.
Alcoholism evolves over 5–10 years [4]. A central thesis of this paper is that over that time, its motivational and neural substrates undergo a major shift. Early stages of excessive alcohol use are characterized by impulsive drinking, that is, drinking to intoxication in binge-like episodes, common among adolescents and young adults in industrialized countries [5,6]. This behavior is positively reinforced by pleasurable alcohol effects, and interspersed by periods of sobriety during which the individual initially returns to a neutral emotional and motivational state. During this stage, environmental stimuli become associated with pleasurable alcohol effects, setting the scene for ‘reward craving’ [7].
By contrast, following a prolonged history of dependence, low mood, elevated anxiety and increased sensitivity to stress become dominant. At this stage, alcohol use becomes primarily negatively reinforced, that is, motivated by the ability of the drug to eliminate what can collectively be labeled ‘a negative emotional state’. This sets the scene for craving motivated by relief produced through renewed drug intake, or ‘relief craving’ (Figure 1).
Figure 1.

The progression of alcohol dependence over time. The schematic illustrates the shift in underlying motivational mechanisms. From initial, positively reinforcing, pleasurable alcohol effects, the addictive process progresses over time to being maintained by negatively reinforcing relief from a negative emotional state. Data presented in this article suggest that neuroadaptations encompassing a recruitment of extrahypothalamic CRF systems are key to this shift.
This shift is a prime example of ‘reward allostasis’, that is, establishing reward equilibrium at a new, pathological level of functioning following a prolonged period of environmental load [8–10]. The concept of ‘anti-reward’ postulates the existence of brain mechanisms that over time limit drug reward, through neuroadaptations recruited in response to repeated drug taking. Neuroadaptations can be ‘within systems’ mediating drug reward themselves, or occur through activation of systems that oppose those (‘between systems’) [11]. A key proposition of this paper is that between-systems neuroadaptations, represented by recruitment of an over-active corticotropin-releasing factor (CRF) system, mediate stress sensitivity and negative emotions in later stages of alcoholism. Understanding the pathology of this circuitry offers attractive novel targets for alcoholism treatment.
History of dependence and the post-dependent phenotype
Laboratory rodents do not voluntarily consume alcohol to intoxication, in part because of taste aversion similar to what humans experience when they first sample alcohol. Higher levels of consumption can be achieved by masking the taste of alcohol with a sweetener, which is faded out as alcohol concentrations are increased. However, even using fading procedures, rats that have not been bred for high alcohol preference will rarely consume in excess of 2 g alcohol/kg/day, and blood alcohol concentrations (BACs) required for dependence will rarely be achieved [12,13]. The predictive validity of consumption at this level for human alcohol dependence is unclear [12].
By contrast, prolonged periods of forced brain exposure to BACs commonly occurring in human alcoholism (~150–250 mg/dL) will trigger long-term neuroadaptations leading to markedly increased voluntary alcohol intake (Figure 2), and other consequences relevant for clinical alcoholism. This syndrome can collectively be labeled ‘the post-dependent state’ (Box 1). Forced intoxication can be induced by offering an alcohol-containing liquid diet as the sole source of food, or by intragastric alcohol gavage. However, alcohol vapor inhalation offers numerous practical advantages for this purpose [14]. Compared with control conditions without alcohol exposure, vapor exposure produces markedly upregulated levels of alcohol self-administration that persist long beyond acute withdrawal [15], and results in long-term dysregulation of neuronal gene expression profiles [16]. Repeated cycles of forced intoxication and withdrawal, paralleling the clinical course of alcoholism, are more effective in inducing the post-dependent state than constant exposure [16–20]. This might be due to progressively increasing activation of glutamatergic transmission over repeated withdrawals [21]. Duration of exposure is also critical, and persistent phenotypic changes are obtained after longer exposure duration [22]. Excessive self-administration or consumption induced by a history of dependence is qualitatively different from basal intake, and probably of particular relevance for human alcoholism. Some of the key evidence for this comes from studies on the CRF system reviewed below.
Figure 2.

Following a history of ethanol dependence induced by exposure to ethanol vapor, long-lasting neuroadaptations lead to the induction of excessive levels of voluntary alcohol consumption. (a) Post-dependent neuroadaptations can be induced by intermittently exposing animals to intoxicating alcohol levels over 7 weeks, and then allowing them to recover for 3 weeks to eliminate acute withdrawal. (b) A behavioral hallmark of the post-dependent phenotype is a marked and persistent upregulation of voluntary ethanol intake compared with animals that have been exposed to air only. Data taken from Ref. [22].
Box 1. The post-dependent state
The term ‘post-dependent’ has been introduced to reflect the sum of neuroadaptations that are induced as an individual becomes dependent on alcohol, and that remain for extended periods of time thereafter. These neuroadaptations can be maintained by continued brain alcohol exposure, but one of their key characteristics is that they remain even in its absence. Based on genetic susceptibility and other factors, the extent and duration of post-dependent neuroadaptations show considerable individual variability. In some individuals, the neuroadapted, post-dependent state is hypothesized to remain indefinitely (‘once an alcoholic, always an alcoholic’), while in others it remits.
CRF, behavioral stress responses and emotionality in the post-dependent state
CRF is a 41 amino acid polypeptide with a wide distribution throughout the brain. The highest densities of CRF-positive neurons are found within the paraventricular nucleus of the hypothalamus, but CRF-positive cells are also present in extrahypothalamic structures, including the central nucleus of the amygdala (CeA) and bed nucleus of stria terminalis (BNST), two components of the extended amygdala, and the brainstem [23,24]. CRF was discovered as the hypothalamic releasing factor for adrenocorticotropic hormone (ACTH) [23], but was subsequently also found to mediate a broad range of behavioral stress and anxiety responses [25]. CRF actions are mediated through two categories of G protein-coupled receptors, CRF1 and CRF2, which are primarily Gs-coupled in neurons [26]. Hypothalamic CRF neurons mediate endocrine stress responses through activation of pituitary CRF1 receptors. By contrast, behavioral stress responses are largely mediated by extrahypothamic CRF1 receptors, primarily in the amygdala and BNST. Effects of CRF2 activation are less clear, but are commonly opposite to those of CRF1 [27,28]. CRF1 signaling mediating behavioral stress responses is quiescent under a wide range of conditions, but becomes activated in the presence of uncontrollable stress [29,30]. This illustrates the principle that neuropeptides are commonly released at high neuronal firing frequencies, acting as ‘alarm systems’ [31].
Elevated anxiety is a hallmark of alcohol withdrawal, and is CRF mediated, because it is blocked by CRF antagonism in the CeA [32,33], while CRF levels or release are increased both in CeA [34] and BNST [35] during this phase. More importantly, in animals with a history of dependence, stress sensitivity and a negative emotional state remain upregulated long after acute withdrawal has subsided. This represents a shift in responsiveness rather than a shift of baseline. No overt differences in anxiety-like behavior were observed in a standard anxiety model, the elevated plus-maze, during protracted abstinence, but when animals were challenged by restraint stress, anxiety-like behavior was markedly accentuated in the post-dependent group. This accentuated anxiety was blocked by the non-selective peptide CRF antagonist D-Phe CRF12–41, while the antagonist was without effect in the control group that had not been previously exposed to alcohol [36]. Similarly, following a history of dependence, enhanced fear-induced suppression of behavior was found in another classical anxiety model, the Vogel conflict test, where it persisted for extensive periods of time (6–12 weeks) after exposure to alcohol. Notably, the Vogel test is inherently a stressor, because of the foot shock used to create the conflict. The increase in anxiety-like behavior in this model following a history of dependence was also fully reversed by systemic treatment with a selective CRF1 antagonist, in this case 3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine (MTIP) [37].
Similar results have been obtained using other means to induce the post-dependent state. Rats subjected to three cycles of withdrawal from an alcohol diet, but not to a single withdrawal, showed increased anxiety-like behavior during early abstinence [38]. Although this acute withdrawal effect subsided within 48h, a remaining recruitment of systems mediating a negative emotional state was demonstrated by the observation that, following a history of three withdrawal episodes, a single further withdrawal from re-exposure to chronic ethanol led to an increased anxiogenic-like effect. In this model, administration of a CRF antagonist during the first two cycles of withdrawal blocked the increase in anxiety-like behavior [39,40]. Furthermore, rats with a history of dependence obtained through a liquid diet protocol with multiple withdrawals were behaviorally more sensitive to a preceding restraint stress. The potentiated anxiogenic effect of this stressor was blocked in post-dependent animals by systemic administration of the CRF1 antagonist CP-154 526 [18]. Based on these and other observations, a ‘kindling/stress’ hypothesis was proposed [19]. It states that repeated cycles of withdrawal drive a progression to a sensitized stress response, and that activation of CRF1 receptors plays a major role in this process.
CRF and dependence-induced excessive drinking
Experiments with the CRF system demonstrate that excessive post-dependent self-administration or intake of alcohol is fundamentally different from basal levels. Post-dependent animals tested two hours into withdrawal exhibited markedly elevated rates of self-administration. These were consistently brought down to non-dependent levels by systemic treatment with three different non-peptide, CRF1 selective antagonists: antalarmin, MJL-1–109–2 or R121919 (Figure 3). None of the antagonists affected self-administration in non-dependent animals [41]. In a follow-up study, the non-selective peptide CRF antagonist D-Phe CRF12–41 microinjected into the CeA blocked excessive post-dependent self-administration rates, while microinjections into BNST or the nucleus accumbens shell were ineffective. Furthermore, CeA injections of D-Phe CRF12–41 in animals without a history of dependence were also ineffective, once again demonstrating that the CRF system, presumably within the amygdala, is recruited to drive excessive alcohol self-administration in the post-dependent state [42].
Figure 3.
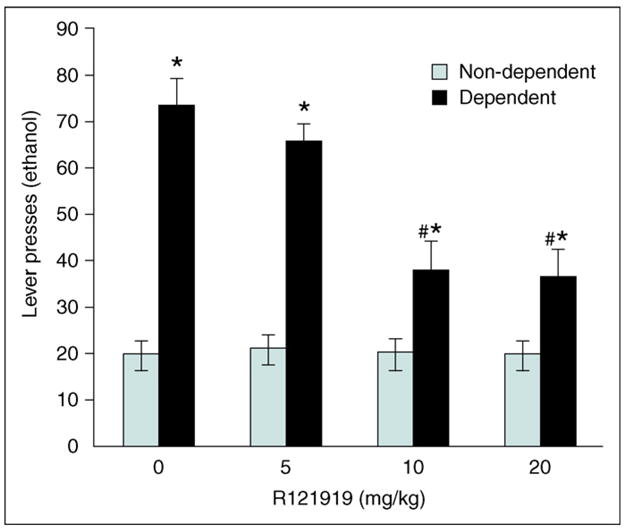
A hallmark of all CRF1 receptor antagonists tested to date is their ability to block selectively excessive rates of operant alcohol self-administration, or excessive two-bottle free-choice alcohol consumption in post-dependent animals at doses that do not affect these behaviors in animals without a history of dependence [41,44]. This is illustrated here by the prototypical non-peptide CRF1 antagonist, R121919 (*P < 0.001 compared with the same drug dose in non-dependent animals; #P < 0.0001 compared with vehicle treatment in dependent animals). Clinical development of R121919 for major depression was discontinued, but similar data have been obtained with other non-peptide CRF1 antagonists that might have a potential for clinical development, such as antalarmin [41,57] or MTIP [44]. Redrawn, with permission, from Ref. [41].
These effects of CRF antagonism were observed during acute withdrawal, but excessive voluntary alcohol drinking and self-administration has also been found long after forced alcohol exposure [16,43,44]. D-Phe CRF12–41 injected intracerebroventricularly blocked the increased ethanol drinking observed during both acute withdrawal and protracted abstinence, while this antagonist was inactive in animals without a history of dependence [43]. Similarly, an established procedure for dependence induction using gastric gavage [44], followed by cycles of self-administration and imposed deprivation periods, also resulted in excessive self-administration. After several weeks, the novel selective non-peptide CRF1 antagonist MTIP suppressed alcohol self-administration in post-dependent animals to non-dependent levels, while the same doses of MTIP were inactive in animals without a history of dependence [44]. In summary, recruitment of CRF signaling drives excessive alcohol self-administration and consumption in post-dependent animals, both during withdrawal and long after withdrawal has subsided.
CRF and stress-induced relapse to alcohol-seeking
Three categories of environmental stimuli are known to trigger relapse in alcohol-dependent individuals [45,46]: small, ‘priming’ alcohol doses; conditioned cues associated with prior availability of alcohol; and stress. The relapse process can be modeled in experimental animals using reinstatement of alcohol-seeking by any of these stimuli (Box 2). Both the non-selective D-Phe CRF12–41 and the CRF1 selective antagonist CP-154 526 blocked stress-induced reinstatement [47]. A subsequent study in post-dependent animals demonstrated a dissociation between cue- and stress-induced reinstatement [48]. The former, but not the latter, was blocked by the opioid receptor antagonist naltrexone, used clinically to treat alcoholism. Conversely, CRF antagonism using D-Phe CRF12–41 blocked stress-induced reinstatement, while leaving cue-induced reinstatement unaffected. Post-dependent animals display a markedly increased sensitivity to blockade of stress-induced reinstatement by CRF antagonism [44]. The selective CRF1 antagonist MTIP entirely blocked this behavior at 10mg/kg, a dose at which no effect was seen in animals without a history of dependence. Taken together, these data show that CRF1 receptors mediate stress-induced reinstatement, and that a recruitment of the CRF system in the post-dependent state renders animals preferentially sensitive to blockade of relapse-like behavior by CRF1 antagonism.
Box 2. Animal models of alcohol consumption, self-administration and relapse
Voluntary alcohol consumption can be assessed by simply providing free-choice access to two bottles, one containing an alcohol solution and the other a non-alcoholic fluid. Consumption in this type of model might, however, be driven by a variety of factors unrelated to pharmacological actions of alcohol, such as taste and calories.
Self-administration approaches require an operant response, most commonly a lever-press, to be emitted for alcohol reward. These models are generally thought to gauge better the motivation to obtain alcohol. For CRF1 antagonists, however, this distinction appears less important, and results with voluntary consumption and self-administration are the same.
Excessive dependence-induced versus basal consumption and self-administration appears instead to be the key distinction for CRF1 antagonists. The former, but not the latter, is consistently blocked by this class of compounds. Excessive self-administration and consumption can result from a prolonged history of forced brain alcohol exposure using alcohol vapor, liquid diet or gastric gavage, or from genetic selection for high alcohol preference. Forced exposure paradigms might bypass some mechanisms involved in voluntary initiation of alcohol use, but growing evidence supports their predictive validity for medications development [13].
Reinstatement of alcohol seeking has in recent years been established as an animal model of relapse-like behavior. In this model, operant self-administration is first established, and during this time an association can also be formed between a discrete cue and delivery of alcohol. Lever-pressing on the alcohol lever is subsequently extinguished by removing the reinforcer. Finally, to evaluate motivation for alcohol-seeking induced by a priming dose, an alcohol-associated cue or a foot-shock stress, the respective stimulus is introduced, and lever-pressing on the previously alcohol-associated lever is measured without actually delivering alcohol [70,71].
Similarly to other CRF-mediated behavioral stress responses, adrenalectomy has no effect on stress-induced reinstatement of alcohol-seeking, demonstrating mediation by extrahypothalamic CRF systems [47]. However, the neurocircuitry through which CRF mediates reinstatement in the post-dependent state is not fully understood. A suggestive parallel might be provided by work on stress-induced reinstatement of cocaine-seeking, which showed a recruitment of CRF control over ventral tegmental area (VTA) dopamine output following a history of cocaine dependence, mediated by an upregulation of VTA glutamate release [49]. This finding maps well onto the neuronal network described for stress-induced reinstatement of cocaine-seeking [50], but it is unclear whether the circuitry is the same for reinstatement of alcohol-seeking. The α2-adrenergic antagonist yohimbine, a pharmacological stressor that can substitute for foot-shock to reinstate alcohol-seeking [51], has recently been shown to upregulate CRF expression in CeA [52], another structure within this ‘reinstatement network’. However, to date, only one antagonist microinjection study has attempted to localize directly CRF mediation of stress-induced reinstatement of alcohol-seeking, and this study reported blockade by injections into the median raphe [53], a structure that falls outside the proposed cocaine reinstatement circuitry. Multiple CRF pathways might be involved in different addictions, and also act in concert to mediate different alcohol-related behaviors.
Neural substrates of the post-dependent behavioral phenotype – the CRF system
Recruitment of CRF signaling within the extended amygdala is a major factor behind increased stress sensitivity, excessive self-administration and relapse in the post-dependent state. The mechanisms through which this occurs are beginning to emerge. During acute alcohol withdrawal, release of CRF is increased in the amygdala [34]. Presumably as a reflection of this, decreased tissue levels of CRF were seen within this structure in early withdrawal [42,54]. Six weeks after last alcohol exposure, amygdala CRF had not only recovered, but also increased to supra-normal levels [54]. Elevated tissue content of CRF peptide in the amygdala could either reflect increased synthesis or decreased utilization. The finding of increased CRF transcript levels in the CeA during the post-dependent state supports increased synthesis in CeA in this condition [37].
A major contribution, however, comes from an upregulation of CRF1 receptor expression and binding within the amygdala. This is consistent with the left-shifted dose–response curve for CRF1 antagonists observed in the post-dependent state. Perhaps the best demonstration that CRF1 upregulation produces the characteristics of the post-dependent phenotype was obtained in the genetically selected, alcohol preferring Marchigian-Sardinian Preferring (msP) rat [55]. These animals were found essentially to represent a behavioral phenocopy of post-dependent rats, with which they share increased stress sensitivity, excessive self-administration of alcohol and increased propensity for relapse-like behavior. A screen for differential gene expression in the msP rat showed a marked upregulation of the transcript encoding the CRF1 receptor within the amygdala complex. This was linked to an allele at the Crhr1 locus (that encodes the CRF1 receptor) unique to msP rats, characterized by two single nucleotide polymorphisms (SNPs) in the promoter region. Genetic variation at the Crhr1 locus as a susceptibility factor for excessive alcohol drinking might have parallels in humans, where a similar association was recently reported [56]. In msP rats, the selective CRF1 antagonist antalarmin reduced alcohol self-administration to non-dependent levels and blocked stress-induced reinstatement of alcohol-seeking at doses that did not affect non-selected rats without a history of dependence [57]. This is a further parallel to the post-dependent phenotype. Interestingly, when msP animals were given ad lib access to alcohol, the ensuing consumption was sufficient to downregulate the receptor transcript to normal levels [58].
Following up on the msP findings, a similar upregulation of CRF1 expression was found in genetically non-selected, post-dependent rats (Figure 4) [37]. This upregulation persisted three months after ethanol exposure, reflecting a long term neuroadaptation rather than acute withdrawal. Similar to the msP findings, receptor upregulation was most pronounced in the basolateral (BLA) and medial amygdala (MeA), and was found in CeA only to a lesser extent (W. Sommer et al., unpublished). The contribution of amygdalar subnuclei to the post-dependent state remains to be established. Microinjections of a CRF antagonist in CeA blocked post-dependent excessive self-administration, but the BLA or MeA were not tested [42]. Amygdalar nuclei are interconnected, and receptors expressed in BLA or MeA neurons could be inserted into terminals in other amygdala regions. Furthermore, CRF antagonists microinjected into the brain could act at multiple, serially connected CRF sites within this structure.
Figure 4.

A long-term upregulation of the Crhr1 transcript, encoding the CRF1 receptor, is found within the amygdala complex in the post-dependent state, and is likely to mediate excessive alcohol consumption. (a,b) Illustrative sections of (a) control rats and (b) post-dependent animals that have been intermittently exposed to intoxicating alcohol levels over 7 weeks, and then allowed to recover for 3 weeks to eliminate acute withdrawal. (c) Quantitative analysis of in situ hybridization data from the two groups. Basolateral amygdala (BLA) and medial amygdala (MeA) are particularly affected. These data parallel, and provide a biological substrate for, observations that CRF1 receptor antagonists can block the elevated voluntary drinking and increased behavioral sensitivity to stress found in post-dependent animals. Data taken from Ref. [37].
The results from post-dependent and msP rats differ from those previously obtained in mutant mice lacking the CRF1 receptor [59]. Similar to what we found in post-dependent animals with upregulated Crhr1 expression [37], the Crhr1 mutants showed a paradoxical, delayed increase in voluntary alcohol consumption in response to forced swim stress. This study employed constitutive mutants, in which deletion of Crhr1 might lead to compensatory developmental effects, and altered glutamatergic function was indeed reported in this model. Also, these animals were not dependent on alcohol. Together, msP and post-dependent data indicate that upregulation of CRF1 receptors is sufficient to produce the post-dependent phenotype, and that blockade of this receptor can normalize post-dependent behavior.
Contribution of other neurotransmitter systems to the post-dependent phenotype
Expression of the astroglial glutamate transporter GLAST is elevated both in post-dependent rats [16] and brains of human alcoholics [60]. GLAST removes extracellular glutamate [61], and its upregulation is presumably compensatory to elevated glutamate release in the post-dependent state [21]. Given an established role for glutamate in stress pathology [62], functional glutamate antagonists might be able to suppress excessive drinking by reducing negative reinforcement by alcohol. Acamprosate, which is approved for the clinical treatment of alcoholism, might exemplify this, and suppresses post-dependent excessive drinking while leaving basal consumption in non-dependent animals unaffected [16].
Neuropeptide Y (NPY), or antagonism at presynaptic NPY–Y2 receptors that drives increased release of endogenous NPY, also selectively suppress alcohol self-administration and drinking in post-dependent animals [63–66]. Importantly, in non-dependent animals, NPY is either inactive, or can increase intake of alcohol, presumably through hypothalamic stimulation of appetite [67]. More than a decade ago, we proposed that NPY counteracts effects of CRF within the amygdala [68]. We note that the selective suppression by NPY of excessive post-dependent drinking is exactly what would be predicted by that model.
Summary and translational perspective
Recent data demonstrate a recruitment of extrahypothalamic CRF systems following a history of alcohol dependence, or owing to genetic selection for high alcohol preference. This does not necessarily confer an overt phenotype, but when faced with a stressor, individuals with a hyperactive CRF system have exaggerated emotional responses. Furthermore, either post-dependent or innate upregulation of the CRF system gives rise to excessive rates of alcohol self-administration, presumably through negative reinforcement of alcohol intake. Finally, susceptibility to stress-induced relapse-like behavior is also upregulated under these conditions. Data from post-dependent and genetically selected animals converge, and point to an upregulation of CRF1 receptors within the amygdala as a major factor behind the behavioral phenotype described here. Together, these data predict that blocking hyperactive signaling at CRF1 receptors in individuals with a history of dependence or innate susceptibility will inhibit heavy drinking and reduce the risk of relapse. As CRF1 antagonism selectively blocks stress- but not alcohol-cue associated relapse, a combination of naltrexone and a CRF1 antagonist might act synergistically to prevent relapse.
No treatments yet exist that effectively target the phenomena of negative emotional state, stress-sensitivity and relief-craving in alcoholism. Targeting pathological negative emotional states and drug-seeking motivated by relief craving is an attractive addition to the treatment armamentarium. Complying with medications that act through this kind of mechanism is inherently reinforced. The lack of effect under non-pathological conditions, combined with an ability to inhibit heavy drinking and relapse, and an attractive profile from the perspective of the patient offer considerable promise that CRF1 antagonists might represent a major step forward for the treatment of alcoholism, and indeed, as has been suggested previously, perhaps also other addictions [69].
Outstanding questions
Recruitment of the CRF system is not the only long-term neuroadaptation that occurs with a history of dependence. A key phenomenon that seems to evolve in parallel is a recruitment of a hyperglutamatergic state, which shifts central excitation–inhibition balance towards excitation [21]. An important outstanding question is whether a link exists between neurodapatations affecting CRF and glutamate systems, or whether they independently contribute to the post-dependent phenotype.
Acknowledgments
Work by M.H. has been supported by intramural funding from the National Institute on Alcohol Abuse and Alcoholism (NIAAA), and the Swedish Medical Research Council. Part of this work has been carried out under a collaborative research and development agreement (CRADA) with Eli Lilly and Co. Work by G.F.K. has been supported by funding from the Pearson Center for Alcoholism and Addiction Research, and by NIH grants AA08459 and AA06420 from the NIAAA, and DK26741 (G.F.K.) from the National Institute of Diabetes and Digestive and Kidney Diseases.
References
- 1.Ezzati M, et al. Selected major risk factors and global and regional burden of disease. Lancet. 2002;360:1347–1360. doi: 10.1016/S0140-6736(02)11403-6. [DOI] [PubMed] [Google Scholar]
- 2.Goldman D, et al. The genetics of addictions: uncovering the genes. Nat Rev Genet. 2005;6:521–532. doi: 10.1038/nrg1635. [DOI] [PubMed] [Google Scholar]
- 3.Dackis C, O’Brien CP. Neurobiology of addiction: treatment and public policy ramifications. Nat Neurosci. 2005;8:1431–1436. doi: 10.1038/nn1105-1431. [DOI] [PubMed] [Google Scholar]
- 4.Åmark C. A study in alcoholism; clinical, social-psychiatric and genetic investigations. Acta Psychiatr Neurol Scand. 1951;70 (Supplementum):1–283. [PubMed] [Google Scholar]
- 5.McCarty CA, et al. Continuity of binge and harmful drinking from late adolescence to early adulthood. Pediatrics. 2004;114:714–719. doi: 10.1542/peds.2003-0864-L. [DOI] [PubMed] [Google Scholar]
- 6.Dawson DA, et al. Another look at heavy episodic drinking and alcohol use disorders among college and noncollege youth. J Stud Alcohol. 2004;65:477–488. doi: 10.15288/jsa.2004.65.477. [DOI] [PubMed] [Google Scholar]
- 7.Heinz A, et al. Reward craving and withdrawal relief craving: assessment of different motivational pathways to alcohol intake. Alcohol Alcohol. 2003;38:35–39. doi: 10.1093/alcalc/agg005. [DOI] [PubMed] [Google Scholar]
- 8.Sterling P, Eyer J. Allostasis: a new paradigm to explain arousal pathology. In: Fisher R, Reason J, editors. Handbook of Life Stress, Cognition and Health. John Wiley & Sons; 1988. pp. 629–649. [Google Scholar]
- 9.McEwen BS. Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- 10.Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 11.Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- 12.Heilig M, Egli M. Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharmacol Ther. 2006;111:855–876. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 13.Egli M. Can experimental paradigms and animal models be used to discover clinically effective medications for alcoholism? Addict Biol. 2005;10:309–319. doi: 10.1080/13556210500314550. [DOI] [PubMed] [Google Scholar]
- 14.Rogers J, et al. Long-term ethanol administration methods for rats: advantages of inhalation over intubation or liquid diets. Behav Neural Biol. 1979;27:466–486. doi: 10.1016/s0163-1047(79)92061-2. [DOI] [PubMed] [Google Scholar]
- 15.Roberts AJ, et al. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- 16.Rimondini R, et al. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- 17.O’Dell LE, et al. Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res. 2004;28:1676–1682. doi: 10.1097/01.alc.0000145781.11923.4e. [DOI] [PubMed] [Google Scholar]
- 18.Breese GR, et al. Prior multiple ethanol withdrawals enhance stress-induced anxiety-like behavior: inhibition by CRF1- and benzodiazepine-receptor antagonists and a 5-HT1a-receptor agonist. Neuropsychopharmacology. 2005;30:1662–1669. doi: 10.1038/sj.npp.1300706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breese GR, et al. Conceptual framework for the etiology of alcoholism: a ‘kindling’/stress hypothesis. Psychopharmacology (Berl) 2005;178:367–380. doi: 10.1007/s00213-004-2016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–1838. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- 21.De Witte P, et al. Alcohol and withdrawal: from animal research to clinical issues. Neurosci Biobehav Rev. 2003;27:189–197. doi: 10.1016/s0149-7634(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 22.Rimondini R, et al. A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J Stud Alcohol. 2003;64:445–449. doi: 10.15288/jsa.2003.64.445. [DOI] [PubMed] [Google Scholar]
- 23.Vale W, et al. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 24.Swanson LW, et al. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 25.Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther. 2004;311:427–440. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- 26.Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–77. doi: 10.1016/s0165-6147(02)01946-6. [DOI] [PubMed] [Google Scholar]
- 27.Muller MB, Wurst W. Getting closer to affective disorders: the role of CRH receptor systems. Trends Mol Med. 2004;10:409–415. doi: 10.1016/j.molmed.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Makino S, et al. Multiple feedback mechanisms activating corticotropin-releasing hormone system in the brain during stress. Pharmacol Biochem Behav. 2002;73:147–158. doi: 10.1016/s0091-3057(02)00791-8. [DOI] [PubMed] [Google Scholar]
- 29.Griebel G, et al. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1, 3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor1 receptor antagonist. II Characterization in rodent models of stress-related disorders. J Pharmacol Exp Ther. 2002;301:333–345. doi: 10.1124/jpet.301.1.333. [DOI] [PubMed] [Google Scholar]
- 30.Gully D, et al. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A): a potent and selective corticotrophin-releasing factor1 receptor antagonist. I Biochemical and pharmacological characterization. J Pharmacol Exp Ther. 2002;301:322–332. doi: 10.1124/jpet.301.1.322. [DOI] [PubMed] [Google Scholar]
- 31.Hokfelt T, et al. Chemical anatomy of the brain. Science. 1984;225:1326–1334. doi: 10.1126/science.6147896. [DOI] [PubMed] [Google Scholar]
- 32.Baldwin HA, et al. CRF antagonist reverses the ‘anxiogenic’ response to ethanol withdrawal in the rat. Psychopharmacology (Berl) 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- 33.Rassnick S, et al. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- 34.Merlo Pich E, et al. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olive MF, et al. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 2002;72:213–220. doi: 10.1016/s0091-3057(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 36.Valdez GR, et al. Antagonism of corticotropin-releasing factor attenuates the enhanced responsiveness to stress observed during protracted ethanol abstinence. Alcohol. 2003;29:55–60. doi: 10.1016/s0741-8329(03)00020-x. [DOI] [PubMed] [Google Scholar]
- 37.Sommer WH, et al. Up-regulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala Crhr1 expression following a history of dependence. Biol Psychiatry. doi: 10.1016/j.biopsych.2007.01.010. in press. [DOI] [PubMed] [Google Scholar]
- 38.Overstreet DH, et al. Accentuated decrease in social interaction in rats subjected to repeated ethanol withdrawals. Alcohol Clin Exp Res. 2002;26:1259–1268. doi: 10.1097/01.ALC.0000023983.10615.D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knapp DJ, et al. SB242084, flumazenil, and CRA1000 block ethanol withdrawal-induced anxiety in rats. Alcohol. 2004;32:101–111. doi: 10.1016/j.alcohol.2003.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overstreet DH, et al. Modulation of multiple ethanol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav. 2004;77:405–413. doi: 10.1016/j.pbb.2003.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Funk CK, et al. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61:78–86. doi: 10.1016/j.biopsych.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Funk CK, et al. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol self-administration in withdrawn, ethanol-dependent rats. J Neurosci. 2006;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valdez GR, et al. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- 44.Gehlert DR, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27:2718–2726. doi: 10.1523/JNEUROSCI.4985-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brownell KD, et al. Understanding and preventing relapse. Am Psychol. 1986;41:765–782. doi: 10.1037//0003-066x.41.7.765. [DOI] [PubMed] [Google Scholar]
- 46.Shaham Y, et al. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 47.Le AD, et al. The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology (Berl) 2000;150:317–324. doi: 10.1007/s002130000411. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Weiss F. Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci. 2002;22:7856–7861. doi: 10.1523/JNEUROSCI.22-18-07856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang B, et al. Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci. 2005;25:5389–5396. doi: 10.1523/JNEUROSCI.0955-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McFarland K, et al. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le AD, et al. Role of alpha-2 adrenoceptors in stress-induced reinstatement of alcohol seeking and alcohol self-administration in rats. Psychopharmacology (Berl) 2005;179:366–373. doi: 10.1007/s00213-004-2036-y. [DOI] [PubMed] [Google Scholar]
- 52.Funk D, et al. Effects of environmental and pharmacological stressors on c-fos and corticotropin-releasing factor mRNA in rat brain: relationship to the reinstatement of alcohol seeking. Neuroscience. 2006;138:235–243. doi: 10.1016/j.neuroscience.2005.10.062. [DOI] [PubMed] [Google Scholar]
- 53.Le AD, et al. The role of corticotropin-releasing factor in the median raphe nucleus in relapse to alcohol. J Neurosci. 2002;22:7844–7849. doi: 10.1523/JNEUROSCI.22-18-07844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zorrilla EP, et al. Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology (Berl) 2001;158:374–381. doi: 10.1007/s002130100773. [DOI] [PubMed] [Google Scholar]
- 55.Ciccocioppo R, et al. Genetically selected Marchigian Sardinian alcohol-preferring (msP) rats: an animal model to study the neurobiology of alcoholism. Addict Biol. 2006;11:339–355. doi: 10.1111/j.1369-1600.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Treutlein J, et al. Genetic association of the human corticotropin releasing hormone receptor 1 (CRHR1) with binge drinking and alcohol intake patterns in two independent samples. Mol Psychiatry. 2006;11:594–602. doi: 10.1038/sj.mp.4001813. [DOI] [PubMed] [Google Scholar]
- 57.Hansson AC, et al. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci U S A. 2006;103:15236–15241. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansson AC, et al. Region-specific down regulation of Crhr1 gene expression in alcohol preferring msP rats following ad lib access to alcohol. Addict Biol. 2007;12:30–34. doi: 10.1111/j.1369-1600.2007.00050.x. [DOI] [PubMed] [Google Scholar]
- 59.Sillaber I, et al. Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRH1 receptors. Science. 2002;296:931–933. doi: 10.1126/science.1069836. [DOI] [PubMed] [Google Scholar]
- 60.Flatscher-Bader T, Wilce PA. Chronic smoking and alcoholism change expression of selective genes in the human prefrontal cortex. Alcohol Clin Exp Res. 2006;30:908–915. doi: 10.1111/j.1530-0277.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- 61.Rothstein JD, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 62.Swanson CJ, et al. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4:131–144. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 63.Thorsell A, et al. NPY in alcoholism and psychiatric disorders. EXS. 2006;95:183–192. doi: 10.1007/3-7643-7417-9_14. [DOI] [PubMed] [Google Scholar]
- 64.Rimondini R, et al. Suppression of ethanol self-administration by the neuropeptide Y (NPY) Y2 receptor antagonist BIIE0246: evidence for sensitization in rats with a history of dependence. Neurosci Lett. 2005;375:129–133. doi: 10.1016/j.neulet.2004.10.084. [DOI] [PubMed] [Google Scholar]
- 65.Thorsell A, et al. Effects of neuropeptide Y and corticotropin-releasing factor on ethanol intake in Wistar rats: interaction with chronic ethanol exposure. Behav Brain Res. 2005;161:133–140. doi: 10.1016/j.bbr.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 66.Thorsell A, et al. Effects of neuropeptide Y on appetitive and consummatory behaviors associated with alcohol drinking in wistar rats with a history of ethanol exposure. Alcohol Clin Exp Res. 2005;29:584–590. doi: 10.1097/01.alc.0000160084.13148.02. [DOI] [PubMed] [Google Scholar]
- 67.Kelley SP, et al. Neuropeptide-Y in the paraventricular nucleus increases ethanol self-administration. Peptides. 2001;22:515–522. doi: 10.1016/s0196-9781(01)00361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heilig M, et al. Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci. 1994;17:80–85. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 69.Sarnyai Z, et al. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev. 2001;53:209–243. [PubMed] [Google Scholar]
- 70.Katner SN, et al. Reinstatement of alcohol-seeking behavior by drug-associated discriminative stimuli after prolonged extinction in the rat. Neuropsychopharmacology. 1999;20:471–479. doi: 10.1016/S0893-133X(98)00084-0. [DOI] [PubMed] [Google Scholar]
- 71.Le AD, et al. Reinstatement of alcohol-seeking by priming injections of alcohol and exposure to stress in rats. Psychopharmacology (Berl) 1998;135:169–174. doi: 10.1007/s002130050498. [DOI] [PubMed] [Google Scholar]