

Abstract
The Epstein-Barr virus protein, LMP1, is a functional mimic of the cellular receptor CD40, but signals to B lymphocytes in an amplified and sustained manner compared to CD40. LMP1 contributes to the development of B cell lymphoma in immunosuppressed patients, and may exacerbate flares of certain autoimmune diseases. The cytoplasmic (CY) domain of LMP1 binds the signaling adaptor TRAF2 with lower avidity than the CY domain of CD40, and TRAF2 is needed for CD40-mediated degradation of TRAFs 2 and 3. LMP1 doesn’t induce TRAF degradation, and employs TRAF3 as a positive mediator of cell signaling, whereas CD40 signals are inhibited by TRAF3. We thus tested the hypothesis that relative affinity for TRAF2, and/or distinct sequence differences in the TRAF2/3 binding sites of CD40 vs. LMP1, controls the disparate ways in which CD40 and LMP1 use TRAFs 2 and 3, and their distinct signaling characteristics. CD40 and LMP1 mutants in which the TRAF binding site sequences were swapped were examined, testing TRAF binding and degradation, and induction of B cell activation. Results revealed that TRAF binding affinity and TRAF binding site sequence dictate a distinct subset of CD40 vs. LMP1 signaling properties. Examination of TRAF binding, degradation, cytokine production, IgM secretion, and the activation of c-Jun kinase and NF-κB revealed that some events are dictated by TRAF binding site sequences, others partially regulated, and still others are independent of the TRAF binding site sequence.
Keywords: B Cells, Viral, Cytokines, Antibodies, Cell Activation
Introduction
Epstein-Barr virus (EBV), the causative agent of infectious mononucleosis, has been clearly linked to lymphoproliferative diseases in the immunosuppressed (1). EBV is associated with several human malignancies including Burkitt’s and Hodgkin’s lymphoma, and nasopharyngeal carcinoma (2–5). LMP1 is the major oncoprotein of EBV, and is required for EBV-mediated transformation of human B cells in vitro (6). LMP1 is also expressed by and strongly implicated in the pathogenesis of most EBV-associated lymphomas (3). In addition to its role in malignancy, LMP1 expression has been associated with human autoimmune disease. Synovial cells from rheumatoid arthritis (RA) patients express LMP1 (7), and LMP1 has been shown to be re-expressed by B cells in flares of systemic lupus erythematosus (SLE), which may exacerbate morbidity associated with the flare (8). Consistent with these findings, mice expressing a transgene with the external domain of mouse CD40 (mCD40) and the CY domain of LMP1 have elevated levels of pro-inflammatory cytokines and autoantibodies (9), and exacerbated disease in a mouse RA model (10).
LMP1 is a functional mimic of the TNF receptor (TNFR) superfamily member, CD40 (11). When expressed in B cells, LMP1 induces the production of Ab and cytokines, upregulation of adhesion and costimulatory molecules, and protection from apoptosis (12). LMP1 can substitute for CD40 to induce a T-dependent humoral response in transgenic mice (13), and only the LMP1 carboxyl (COOH) CY domain is necessary to replace CD40 in mediating an Ab response that includes isotype switching and affinity maturation (9). We and others have shown that the COOH CY domain is both necessary and sufficient to mediate LMP1-mediated B cell activation (13–16). In contrast to CD40, however, LMP1 signaling to B cells is amplified and sustained (14), leading to enhanced B cell activation (14). Consistent with this finding, transgenic expression of a chimeric CD40-LMP1 molecule in mice leads to B cell hyperactivation, autoreactivity, and abnormal lymphoid architecture in secondary lymphoid organs (9). Thus, LMP1’s exaggerated signaling properties give it the ability to promote B cell-mediated disorders. Understanding the molecular basis for LMP1’s aberrant signaling is of considerable interest, both for understanding how these signaling pathways are regulated, and for potential application of this information to design therapies that target LMP1 function.
LMP1 consists of a short N-terminal and long COOH CY domain, separated by 6 membrane-spanning domains, which aggregate to initiate ligand-independent signaling (3). LMP1 and CD40 share a short COOH CY domain motif which allows binding to members of the TRAF family of signaling adaptor proteins. Binding of TRAFs 1, 2, 3, and 5 is mediated by the general motif PxQxT, commonly referred to as the TRAF binding site (TBS) (Fig. 1)(17). Each TRAF binds the TBS in a distinct but overlapping manner (18). The TBS of CD40 is considered a ‘major’ TRAF2-binding motif of PVQETL, while the TBS of LMP1 has been called a ‘minor’ TRAF2 binding motif (PQQATD) (19). CD40 and LMP1 associate with the same binding crevice of TRAF3, but LMP1 has additional binding contacts that may contribute to its more robust association with TRAF3 (17, 20). For both CD40 and LMP1, the TBS influences NF-κB and JNK activation, surface molecule upregulation, and IgM secretion (21–32).
Figure 1.
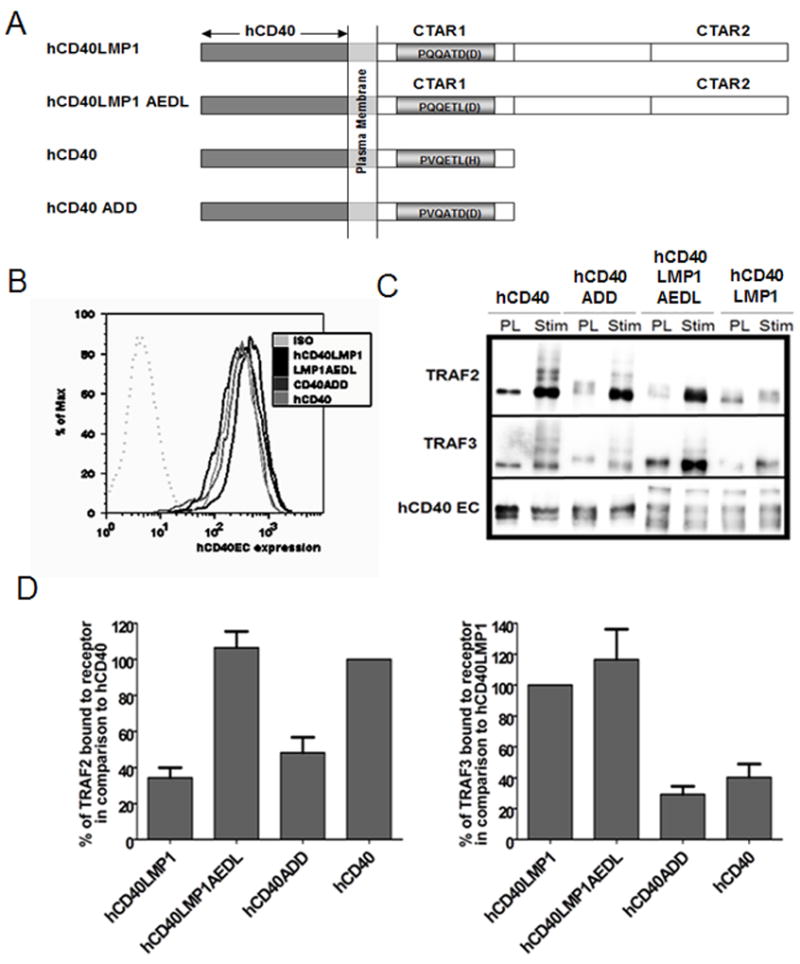
Recruitment of TRAF2 and TRAF3 by hCD40LMP1AEDL and hCD40ADD in B cells. A, Domain composition of hCD40LMP1, hCD40LMP1AEDL, hCD40, and hCD40ADD. The LMP1 chimeric receptors are composed of the extracellular and transmembrane domains of hCD40 and either the full length CY domain of LMP1 (aa 187–386 of LMP1) or a TBS mutant version of this LMP1 CY domain (hCD40LMP1AEDL) where the sequence of the TBS has been changed from PQQATDD to PQQETLD (the CD40 TBS sequence). hCD40 is WT hCD40 while hCD40ADD has had its TBS mutated from that of CD40 (PVQETLH) to that of LMP1 (PVQATDD). B, M12 and M12 B cell clones were expression matched into sets expressing similar levels of hCD40, hCD40LMP1, hCD40LMP1AEDL, and hCD40ADD as determined by immunofluorescence flow cytometry. Similar results were obtained for CH12.LX subclones (not shown). C, M12.4.1 B cells were stably transfected with hCD40LMP1, hCD40, hCD40ADD, or hCD40LMP1AEDL and stimulated with 10 μl of Dynabeads coated with anti-hCD40 Ab for 15 min. The post lysis (PL) control sample was lysed before addition of anti-hCD40 Ab coated Dynabeads as in Methods. Samples were blotted for TRAF2, TRAF3, and hCD40. D, Quantification of TRAF binding in B cells was performed by measuring the bands with a low-light imaging system. The desitometric quantification of the TRAF band was normalized to the desitometric quantification of the corresponding CD40 band. The normalized densitometric quantification of TRAFs co-immunoprecipitated with a receptor following cell lysis (“PL”) was subtracted from the normalized value of the stimulated (“Stim”) samples. The normalized values for hCD40 ADD and hCD40LMP1AEDL are represented graphically as percentages of the normalized values of hCD40 and hCD40LMP1. Data shown are representative of six or more independent experiments utilizing two or more clones of each transfectant.
TRAFs interact with LMP1 in several unexpected and different ways, compared to their interactions with CD40 (17). TRAFs 1 and 2 cooperate to promote a subset of CD40-mediated signals, while deficiencies of either or both these TRAFs have no major effect on in vitro LMP1 signaling to B cells (33). Conversely, TRAF3 is a negative regulator of CD40-induced B cell activation, but an important positive element of LMP1-induced signaling (25, 32, 34). TRAF2 recruitment to CD40 induces TRAF2 dependent polyubiquitination and proteasome-dependent degradation of both itself and TRAF3, but this doesn’t occur upon LMP1 signaling (14, 34–36). Differential TRAF usage and regulation by LMP1 in comparison to CD40 contributes to its unique signaling nature.
To further our understanding of how CD40 and LMP1 differentially regulate the TRAFs, we devised a novel and complementary approach to build upon mutational analysis of the TBS. This approach retains the normal sequence of each TBS while placing it in the context of the CY domain of the complementary receptor. This allows a direct comparison of the contributions of the TBS in signaling by CD40 and LMP1 to B lymphocytes, while retaining the normal overall structure of each receptor’s CY domain. To this end, we created recombinant human CD40 and chimeric hCD40LMP1 molecules in which the major TRAF binding site of CD40 (PVQETLH) and the minor TRAF binding motif of LMP1 (PQQATDD) were swapped. The signaling characteristics and TRAF binding potentials of each of these molecules compared to their WT counterparts were examined. Results reveal that the nature of the TRAF binding site strongly regulates some aspects of CD40 vs. LMP1 signaling, partially controls others, and plays no apparent role in a third category of functions.
Materials and Methods
Cells
The mouse B cell lines M12.4.1 and CH12.LX have been previously described (37–39). B cell lines were maintained in RPMI 1640, 10 μM 2–mercaptoethanol (GIBCO, Grand Island, NY) with 10% heat-inactivated FCS (Atlanta Biologicals, Lawrenceville, GA) and antibiotics (medium referred to as BCM-10). Cells transfected with either WT human CD40 (hCD40), hCD40LMP1, hCD40LMP1AEDL, or hCD40ADD were maintained in 400 μg/mL G418 disulfate (Research Products International, Mt. Prospect, IL). All stably transfected subclones were generated by electroporation as previously described (40).
Antibodies
Rabbit anti-TRAF2 Ab was purchased from Medical and Biological Laboratories Co. Ltd. (Nagoya, Japan). Rabbit anti-TRAF3 (H122) and rabbit anti-hCD40 (H-120) Abs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-actin mAb (MAB150R) was purchased from Chemicon Int. (Temecula, CA). Rabbit anti-IκBα and anti-pJNK1/2 Abs were purchased from Cell Signaling Technology, Inc. (Beverly, MA). B cells were stimulated through hCD40 using the anti-hCD40 mAb G28.5 (mouse IgG1) produced from a hybridoma obtained from the ATCC (Manassas, VA). The isotype control mAb (iso) for G28.5, MOPC31c, was purchased from Sigma Chemical Co. (St. Louis, MO). The anti-mouse (m)CD40 mAb, 1C10, was produced from a hybridoma provided by Dr. Frances Lund (Trudeau Institute, Saranac Lake, NY). The isotype control mAb EM95 (rat IgG2a) was provided by Dr. Thomas Waldschmidt (University of Iowa, Iowa City, IA).
Co-immunoprecipitation
Ten × 106 B cells were stimulated with G28.5 (10μg/10 ul Dynal beads, Invitrogen) for 15 minutes and receptors immunoprecipitated (IP) as previously described (41, 42). Amounts of TRAF2 and TRAF3 co-immunoprecipitated with the various receptors were detected by Western blotting as described below.
DNA constructs
WT-hCD40 and the hCD40LMP1 chimeric DNA constructs have been previously described (14). The hCD40ADD and hCD40LMP1AEDL molecules were created from the WT-hCD40 and hCD40LMP1 constructs by PCR SOEing (43). The primers for the hCD40LMP1AEDL joint were 5′-caagagaccttagattctggc and 5′-aatctaaggtctcttgttgagg. The hCD40ADD mutation was made with the primer 5′-gtgcaggcgactgatgat using a construct containing the WT hCD40 CY domain into which a noncoding change had been engineered to create a new SacI restriction enzyme recognition sequence. This construct (hCD40TSS) allows removal of just the CY domain of CD40. The WT hCD40 CY domain in the hCD40TSS construct was then replaced with the mutated ADD CY sequence via standard cloning techniques.
TRAF degradation
The TRAF degradation assay has been described previously (35, 44, 45). Briefly, 3 × 106 cells were washed in RPMI 1640, resuspended in 2ml of BCM-10 and added to a 6 well tissue culture plate. The cells were stimulated with 10ug/ml G28.5, 1C10, or isotype control mAbs (EM95 + MOPC31c), then incubated for the indicated time periods at 37°C. After chilling plates to 4 °C, cells and medium were transferred into 1.5 ml Eppendorf tubes and centrifuged at 4°C for 2 min at 200 × g. Whole cell lysates were prepared by removing the supernatant and adding 200 μl of 2X SDS-PAGE loading dye to the pellet. The lysates were sonicated with 15 pulses at 90% duty cycle, output 1.5. The samples were boiled for 5 min at 95°C and kept on ice prior to gel loading.
JNK phosphorylation
1 × 106 cells were washed in RPMI 1640, resuspended in 1ml of BCM-10 in 1.5ml Eppendorf tubes, and rested for 1hr. at 37°C. The cells were then stimulated for 5, 10, 15, 30, 45, or 75 minutes with α-hCD40 (G28.5), α-mCD40 (1C10), isotype control (EM95 + MOPC31c) mAbs, or medium alone. Whole cell lysates were prepared as described for the TRAF degradation assay. The presence of phospho-JNK (pJNK) in lysates was detected using a phospho-specific JNK Ab on Western blots of samples subjected to SDS-PAGE, as described (46).
Western blots
5–10 μL of sample were resolved on 10% SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore, Bedford, MA), and membranes were blocked with 10% non-fat dried milk in 20mM TRIS buffered saline with 0.1 % TWEEN (TBST) for 1 hour. The membranes were washed 3 times in TBST and incubated overnight at 4°C with one of the above Abs. Blots were incubated with secondary Abs for 1 hour and developed with an enhanced chemiluminescence system (Supersignal West Pico; Pierce Biotechnology, Rockford, IL). To accurately compare and quantify the amount of protein analyzed, Western blot chemiluminescence was read on a low-light digital camera (LAS-1000 or LAS-3000, Fujifilm Medical Systems USA, Stamford, CT), using the Image Gauge program (Fujifilm Medical Systems).
IgM secretion
Quantitation of IgM-secreting transfected CH12.LX cells stimulated through WT hCD40, hCD40LMP1, hCD40LMP1AEDL, or hCD40ADD was accomplished as previously described (34, 47). Briefly, CH12.LX transfected subclones were cultured in 96-well plates (1.5 × 103/well) with various stimuli. Anti-CD40 and isotype control mAbs were used at a final concentration of 2μg/ml. SRBC (Elmira Biologicals, Iowa City, IA) at a final concentration of 0.1% were used as a source of the Ag for which the Ig of CH12.LX is specific (phosphatidylcholine) (48). Triplicate cultures were incubated for 72 h, and viable cells were counted by Trypan blue exclusion. IgM-secreting cells were enumerated as SRBC hemolytic plaques/million recovered viable cells, as previously described (47).
Cytokine production
To quantify IL-6 production, transfected subclones of CH12.LX cells (1 × 105 cells/ml, 1ml total volume in a 24 well plate) were co-cultured with anti-CD40 or isotype control mAbs (2μg/ml) for 48 h in BCM-10, and the supernatants were examined for IL-6 by ELISA as previously described (49). To measure TNF-α production, transfected subclones of M12.4.1 B cells were resuspended in BCM-10 (5 × 105 cells/ml, 200μl/well) and placed in an anti-TNF-coated 96-well flat-bottom plate with anti-CD40 or isotype control Abs. We have found that this ‘in plate’ assay is necessary because B cells rapidly bind the TNFα that they secrete (31). Cells were stimulated with the various Ab for 4h, after which culture supernatants were assayed for TNF-α by ELISA as previously described (31).
NF-κB Luciferase Reporter Assay
The NF-κB Luciferase Reporter Assay has been described previously (25). M12.4.1 B cell subclones (2 × 107 cells) expressing hCD40, hCD40LMP1, hCD40LMP1AEDL, or hCD40ADD were electroporated at 225 V and 50 mS with 38 μg4X NF-κB firefly luciferase (a gift from Dr. Edward Clark, Universityof Washington, Seattle, WA) and 2 μg renilla (null) luciferase reporter plasmids (Promega). After transfection, cells wererested in medium containing 15% FCS overnight at 37°C. Cellswere washed and resuspended in BCM-10, aliquoted into 24-wellplates (2 ml/well), and stimulated with 10 μg of anti-mCD40,anti-hCD40, or isotype control mAbs for 6 h at 37°C. Celllysates were analyzed for the firefly and renilla luciferase activities with the Dual Luciferase Reporter Assay kit (Promega)on a TD-20/20 Luminometer (Turner Designs) following the manufacturer’s protocol.
Results
Effects of differences in CD40 vs. LMP1 TRAF binding site sequence on TRAF2/3 recruitment
TRAF2 and TRAF3 associate with CD40 via a PxQxT motif located in a region separate from the TRAF6 binding site in the CD40 CY domain (50). These TRAFs associate with LMP1 in a region of similar sequence necessary for LMP1-mediated B cell transformation, commonly referred to as CY activating region 1 (CTAR1) (21, 26). However, the specific amino acid residues within and immediately flanking this site differ between CD40 and LMP1 (19). The major TBS of hCD40 binds more TRAF2 than hCD40LMP1, whereas hCD40LMP1’s minor TBS binds more TRAF3 than hCD40 (Fig. 1) (19, 24, 25).
To begin this study, we examined whether these particular TBS sequences dictate preferential binding of hCD40 or hCD40LMP1 to TRAF2 and/or TRAF3. To do so, we expression matched multiple sets of clones in two different cell lines by immunofluorescence flow cytometry. A “set” is a grouping of four cell line clones expressing either hCD40, hCD40LMP1, hCD40ADD, or hCD40LMP1AEDL at similar levels. To begin a study, all experimental comparisons were performed within one set of clones. Subsequent experiments using additional expression matched sets of clones were used to confirm initial findings. Comparisons of each receptor were made within additional expression matched sets and not between (an example of an expression matched set of M12 B cell clones can be viewed in Fig 1B). Abs specific for the hCD40 extracellular domains of each molecule were used to immunoprecipitate the receptors from cell lysates 15 minutes post-stimulation with agonistic Ab. Western blotting of precipitates subject to SDS-PAGE was employed to detect the relative amounts of TRAF2/3 co-immunoprecipitated with each receptor. Conversion of the sequence of the hCD40 TBS to that of hCD40LMP1 (hCD40ADD) reduced by approximately two-fold the ability of hCD40ADD to bind TRAF2, compared to hCD40 (Fig. 1). However, the binding of hCD40ADD to TRAF3 was unchanged. Conversely, converting the TBS of hCD40LMP1 to that of hCD40 gave hCD40LMP1AEDL the ability to bind comparatively increased amounts of TRAF2. Similar to the hCD40ADD molecule, the ability of hCD40LMP1AEDL to bind TRAF3 was unaltered from that of its parent receptor. Thus, the TBS sequences of both CD40 and LMP1 predicted the relative ability to bind TRAF2 but not TRAF3.
Effects of differences in CD40 vs. LMP1 TRAF binding site sequence on TRAF2/3 degradation
TRAF2 and 3 association with CD40 but not LMP1 induces their polyubiquitination and proteasome-dependent degradation (44). TRAF degradation is dependent upon the RING domain of TRAF2 (34, 35, 46). We thus asked if hCD40LMP1 acquires TRAF-degrading ability if its binding site is altered to allow more robust TRAF2 binding. The increased TRAF2 binding ability of hCD40LMP1AEDL (Fig. 1) correlated with the ability of hCD40LMP1 to induce TRAF2, but not TRAF3 degradation upon signaling (Fig. 2). However, the decreased TRAF2 binding ability of hCD40ADD did not preclude this molecule’s induction of TRAF2 and TRAF3 degradation (Fig. 2). There was a trend towards lower ultimate hCD40ADD-induced TRAF2 degradation in comparison to hCD40 after 3 hrs of stimulation, but degradation still occurred (Fig. 2 and data not shown). Interestingly, hCD40ADD signaling seemed more efficient at inducing TRAF3 degradation compared to hCD40. cIAP1/2 molecules have been reported to be necessary for CD40 signaling induced TRAF2/3 degradation (51). However, we cannot reliably detect association of endogenous levels of the cIAPs with hCD40 or hCD40LMP1 in B cells (data not shown), so could not assess any impact of the TBS mutations in this regard. Taken together, these results suggest that the TBS of CD40 can positively influence the ability to induce TRAF2 degradation. However, CD40 uses regions outside of the TBS to mediate signaling induced TRAF3 degradation.
Figure 2.
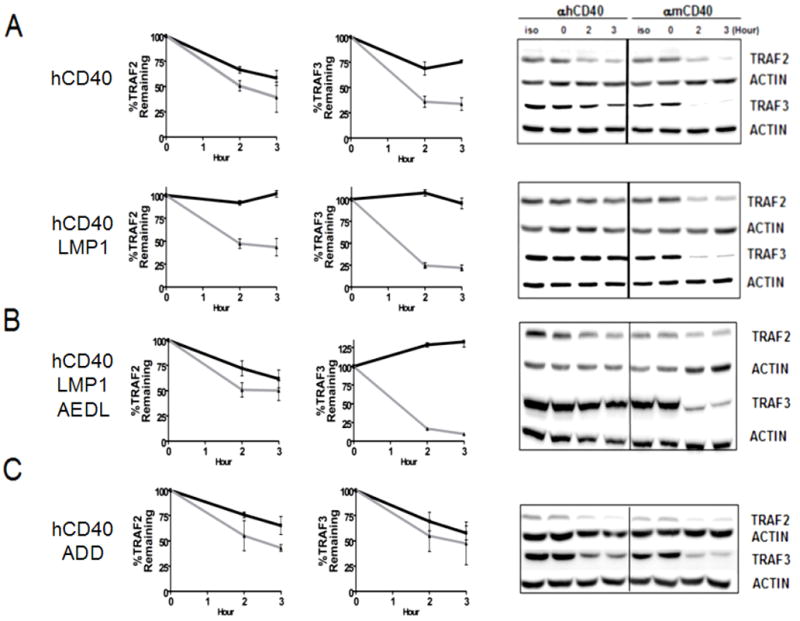
Differential abilities of hCD40, hCD40LMP1, hCD40ADD, and hCD40LMP1AEDL CY domains to induce TRAF degradation. Whole cell lysates from M12.4.1 B cells transfected with hCD40 or hCD40LMP1 (A), hCD40LMP1AEDL (B) or hCD40ADD (C) and stimulated with isotype control Ab, anti-mCD40 Ab, or anti-hCD40 Ab were analyzed for TRAF2 and TRAF3 degradation. Quantification of TRAFs (A–C) was performed by measuring the intensities of TRAF2, TRAF3, and actin bands as in Methods. The amount of each TRAF band was normalized to the corresponding actin band. These values were then normalized to the 0 hour time, which was set as 100% of either TRAF2 or TRAF3. Black solid lines represent TRAF degradation induced by hCD40 agonistic mAb, grey lines represent TRAF degradation induced by anti-mCD40 agonistic mAb, and serves as an endogenous control for each transfected subclone. Data shown are representative of three or more experiments performed with two or more clones of each transfectant.
Effects of differences in CD40 vs. LMP1 TRAF binding site sequence on Ig, IL-6, and TNF-α secretion
The CY domain of LMP1 mediates amplified production of Ig, IL-6, and TNF-α, compared to that of CD40 (10, 14). hCD40LMP1AEDL-induced Ig secretion was reduced to CD40-like levels, whereas that induced by hCD40ADD signaling was increased to levels similar to hCD40LMP1 (Fig. 3C). However, TNF-α secretion and CD23/CD80 upregulation induced by hCD40ADD and hCD40LMP1AEDL were not altered from the levels mediated by their parental counterparts (Fig. 3A and data not shown). These findings indicate that induction of Ig is principally regulated by the TBS sequence, but additional or alternate factors regulate differences in TNFα production and CD23/CD80 upregulation.
Figure 3.
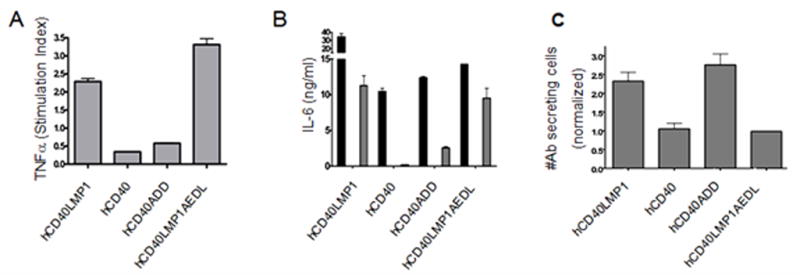
Effects of TBS mutation on TNF-α, IL-6, and IgM production. A, M12.4.1 B cells stably transfected with hCD40LMP1, hCD40, hCD40ADD, or hCD40LMP1AEDL were stimulated for 4 hours with agonistic mouse or human CD40-specific mAbs or isotype control mAbs. TNF-α secreted in response to stimulation was assayed from cell culture supernatants by ELISA. Data are presented as hCD40-induced TNF-α normalized to mCD40-induced TNF-α. TNF-α secreted in response to isotype control mAbs was subtracted from CD40 stimulated groups prior to normalization. B, CH12.LX B cells stably transfected with hCD40LMP1, hCD40, hCD40ADD, or hCD40LMP1AEDL were stimulated for 2 days with Hi5 insect cells expressing hCD154, agonistic anti-mouse or human anti-CD40 mAbs, or isotype control mAbs. IL-6 was assayed from cell culture supernatants by ELISA. Black bars represent cells stimulated by Hi5 insect cells expressing hCD154, open bars represent cells stimulated by agonistic anti-mCD40 mAb, gray bars represent cells stimulated by agonistic anti-hCD40 mAb. Open bars are generally below the level of detection because anti-CD40 mAb does not induce IL-6 production via hCD40 (49). IL-6 secreted in cultures containing isotype control mAbs or untransformed Hi5 insect cells was subtracted from values obtained from cultures stimulated via hCD40 extracellular domains. C, CH12.LX B cells stably transfected with hCD40LMP1, hCD40, hCD40ADD, or hCD40LMP1AEDL were stimulated for 3 days with agonistic mouse or human anti-CD40 mAbs or isotype control mAbs. Data are presented as the mean number of Ab secreting cells ± S.E. of replicate cultures induced in response to hCD40 stimulation normalized to the number of Ab secreting cells induced in response to mCD40 stimulation. Ab secreted by cells cultured with isotype control mAbs was subtracted from CD40 stimulated groups prior to normalization. Data shown are representative of three or more experiments performed with two or more clones of each transfectant.
CD40 signaling induced by agonistic anti-CD40 Ab is insufficient to induce B cells to secrete IL-6; membrane bound CD154 stimulation is required for this event (49). Signaling initiated by agonistic Ab to hCD40LMP1, however, is sufficient to induce B cells to secrete IL-6 (9). Therefore, we wished to determine if the TBS sequence is relevant to the more robust stimulation required by CD40 to induce IL-6 secretion. hCD40LMP1AEDL signaling induced IL-6 secretion in response to agonistic Ab. Thus, this ability was not lost by converting its TBS to that of CD40. However, hCD40ADD gained the ability to induce IL-6 production in response to anti-CD40 Ab, similar to molecules with a LMP1 CY domain (Fig. 3B). Like TRAF degradation, regulation of IL-6 production was partially, but not solely, regulated by the TBS sequence.
Effects of differences in CD40 vs. LMP1 TRAF binding site sequence on early signaling events
JNK Activation
TRAF2 is necessary for CD40-mediated, but not LMP1-mediated, JNK activation in B cells (45). In contrast, whereas TRAF3−/− mouse B cells are markedly defective in hCD40LMP1-mediated JNK activation, TRAF3 deficiency results in an increase in CD40-mediated JNK activation (34). TRAF3 has also been shown to negatively regulate CD40-mediated Ig production and CD40-BCR synergy (45, 52), while promoting LMP1-mediated Ig production (34). Thus, JNK is a very important early signaling pathway connected to multiple downstream effector functions of CD40 and LMP1. Similar to NF-κB activation (Fig. 5), the pattern of JNK activation differs between hCD40 and hCD40LMP1, with hCD40LMP1 mediating a slower but more sustained activation (Fig. 4) (24, 25, 44). Fig. 4 shows that hCD40LMP1AEDL-induced JNK activation converted to the rapid, transient pattern mediated by hCD40. hCD40ADD-mediated JNK activation showed a pattern intermediate to that of hCD40LMP1 and hCD40. Like hCD40, hCD40ADD signaling induced an early peak of JNK activation. However hCD40ADD-mediated JNK activation was also sustained, similar to hCD40LMP1. These results show that the TBS played important roles in the nature of both hCD40 and hCD40LMP1 mediated JNK activation.
Figure 5.
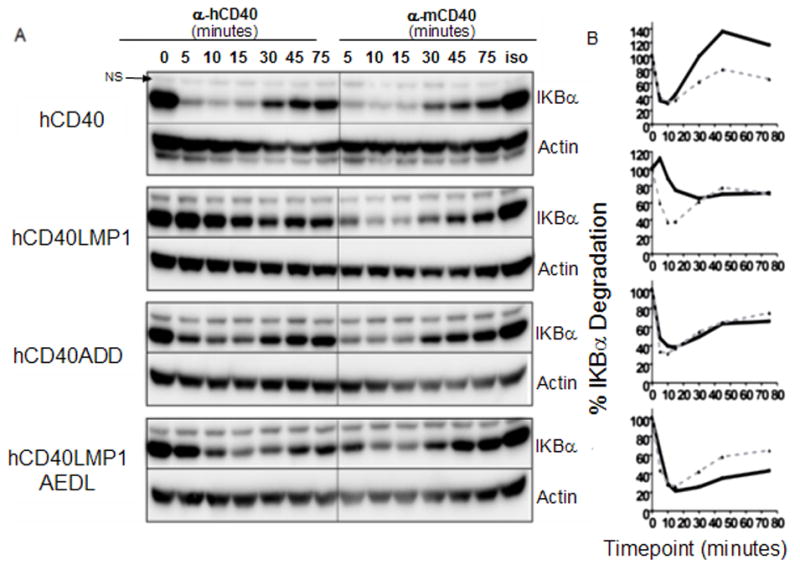
Effect of TBS mutationd on canonical NF-κB activation. A, Whole cell lysates from M12.4.1 B cell transfectants stimulated for indicated times with agonistic anti-mouse or human CD40 mAb or isotype control mAbs were analyzed by immunoblotting for total IκBα or actin. B. Quantification of IκBα degradation was performed by measuring total IκBα and actin bands with a low-light imaging system. The amount of IκBα was normalized to the corresponding actin band. These values were then normalized to the 0 time (0 min. IκBα/actin), which was set as 100%. Black solid lines represent IκBα degradation induced by anti-hCD40 agonistic mAb, grey dashed lines represent IκBα degradation induced by anti-mCD40 agonistic mAb. Data shown are representative of three or more experiments performed with two or more clones of each transfectant.
Figure 4.
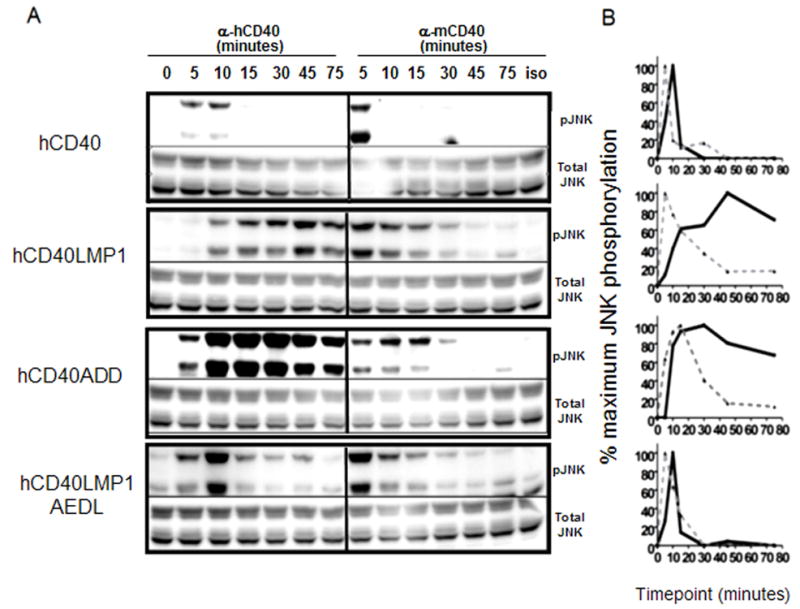
Effect of TBS mutation on JNK activation. A, Whole cell lysates from M12.4.1 B cell transfectants stimulated for the indicated times with agonistic anti-mouse or human CD40 mAbs or isotype control mAbs were analyzed by immunoblotting for phosphorylated JNK (pJNK) or total JNK. B. Quantification of JNK activation was performed by measuring pJNK and the corresponding total JNK bands with a low-light imaging system. The amount of each pJNK band was normalized to the corresponding total JNK band. These values were then normalized to the point of maximal JNK activation (greatest pJNK/total JNK), which was set as 100%. Black solid lines represent JNK activation induced by anti-hCD40 agonistic mAb, grey dashed lines represent JNK activation induced by anti-mCD40 agonistic mAb. Data shown are representative of three or more experiments performed with two or more clones of each transfectant.
NF-κB activation
CD40 and LMP1 both activate the canonical and non-canonical NF-κB pathways (53, 54). However, hCD40LMP1-mediated canonical pathway induction is slower and more sustained than hCD40 (Fig. 5) (44). Fig. 5 shows that both hCD40LMP1AEDL and hCD40ADD induced the degradation of IκBα with the same kinetics as stimulation through endogenous mCD40 and hCD40. These results indicate that regions additional to the TBS of CD40 were important in regulating the process of IκBα degradation, but the delayed and sustained pattern of canonical NF-κB activation typical of hCD40LMP1 depended upon its unique TBS.
Fig. 6 illustrates that the sum of activation of NF-κB pathways at 6 hours post-stimulation is greater with both hCD40LMP1AEDL and hCD40LMP1 than in the CD40 counterparts. Whereas the hCD40 and hCD40ADD molecules induced roughly the same amount of NF-κB activation, hCD40LMP1AEDL signaling trended toward higher levels of NF-κB activation than did hCD40LMP1 signaling (Fig. 6). These results show that the sum of later activation of combined canonical and non-canonical NF-κB pathways is primarily dictated by regions outside of the TRAF binding site, although early activation of the canonical pathway is sensitive to TBS differences (Fig. 5).
Figure 6.
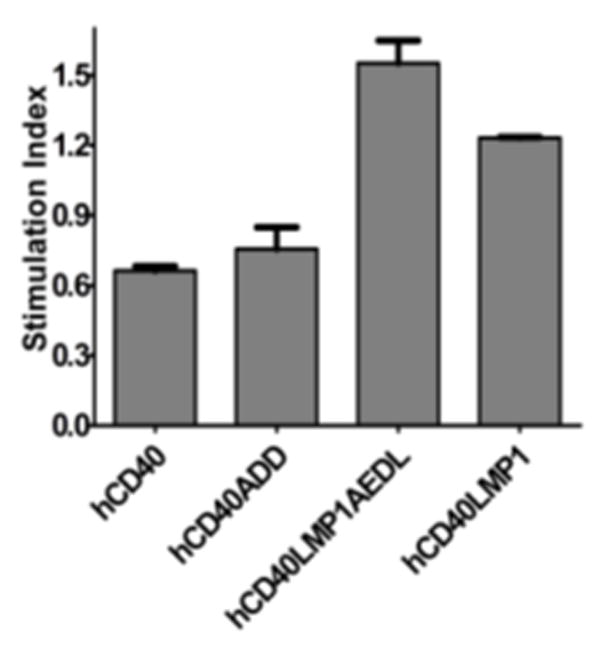
Effect of TBS mutation on total NF-κB activation. M12 B cells expressing either hCD40, hCD40LMP1, hCD40LMP1AEDL, or hCD40ADD were transiently transfected with a NF-κB firefly reporter plasmid and a renilla control plasmid, stimulated with α-mouse or human CD40 or isotype control Abs for 6h. Values from each sample were normalized for transfection efficiency by dividing the renilla luciferase activity by the firefly luciferase activity in each sample. The stimulation index for each receptor was determined by dividing the normalized values of each sample as: (αhCD40 mAb stimulated- isotype control mAb stimulated)/(αmCD40 mAb stimulated- isotype control mAb stimulated). Data shown are representative of three or more experiments performed with two or more clones of each transfectant.
Discussion
Although LMP1 is a functional mimic of CD40, these receptors use the TRAFs in distinct and sometimes contrasting ways (25, 34, 42). The present findings further indicate that physical association with LMP1 regulates the TRAF molecules differently than association with CD40. Here we demonstrate that the TBS of CD40 and hCD40LMP1 differentially regulated association with TRAFs and certain downstream functions.
Some studies suggest that CD40 and LMP1 signaling is regulated by cooperation of the TRAF2/3 binding site with the TRAF6 binding site, and cooperation between the CTAR1 and 2 regions, respectively (24, 30, 55). Investigations of CD40 and LMP1 molecules with mutated TBS clearly demonstrate the importance of the TBS in signaling (22, 27, 28). However, interpretation of these results is complex because mutations disrupting the integrity of the TBS may have either or both direct effects on proteins associating with the TBS, and indirect effects on signaling pathways which rely on cooperativity between different structural regions. For CD40 signaling, cooperation between the TRAF2/3 binding site and the TRAF6 binding site has been suggested to be important in regulating TNF-α secretion (30). Cooperation between the CTAR1 and 2 regions of LMP1 is important for TRAF3 binding and CD80 upregulation (24, 26). Here we show that hCD40 and hCD40LMP1-induced TNF-α secretion, CD80 upregulation, and TRAF3 binding were unaltered by TBS swapping. This indicates that any cooperative pathways emanating from hCD40 and hCD40LMP1 are intact in hCD40ADD and hCD40LMP1AEDL molecules. This work extends mutational analysis studies of the TBS in LMP1 and CD40 signaling by removing variables introduced by disruption of cooperative pathways.
Interestingly, the respective TBS of CD40 and LMP1 regulated the strength of TRAF2 but not TRAF3 binding (Fig. 1) (19, 21, 24). Our findings are consistent with other reports and further implicate regions outside of the TBS as important in TRAF3 binding (28, 56). This information is crucial for the design of potential therapeutics that might target LMP1-TRAF3 association. Most likely, a COOH-terminal portion of LMP1 called CTAR2 is influencing TRAF3 binding. Previous findings demonstrate cooperation between CTAR1 and 2 in LMP1 signaling and TRAF association (24, 26, 55). These studies show that mutation of either CTAR1 or 2 significantly alters TRAF association (24, 26). While the TBS in CTAR1 exhibits similar preference for TRAF1, 2, and 3, the CTAR2 region either directly influences preferential TRAF3 binding or interacts with additional factors which influence TRAF3 association (24). Further studies will be needed to examine these two possibilities.
The present report illustrates that a receptor’s ability to recruit TRAF2 cannot fully explain whether TRAF2/3 degradation will occur upon signaling. Despite lowering the TRAF2 binding of hCD40ADD compared to hCD40, hCD40ADD retained its ability to induce TRAF2/3 degradation, although TRAF2 degradation was at a lower level than that induced by hCD40 at later times. This result, together with the finding that hCD40LMP1AEDL initiated TRAF2 degradation, suggests that the CD40 TBS is most important in enabling TRAF2 but not TRAF3 degradation. CD40 signaling-induced TRAF2/3 degradation is thought to be induced by TRAF2-dependent recruitment of ubiquitin ligases (51). Results presented here suggest that the TBS sequence of CD40 imparts to TRAF2 the ability to recruit a subset of these ubiquitin ligases, but other regions of CD40 may be required to recruit all of the participants necessary to promote maximal TRAF2 degradation and initiate TRAF3 degradation. The rate of TRAF3 degradation was improved by the replacement of the hCD40 TBS with that of hCD40LMP1. Perhaps factors which associate via the CD40 TBS compete for space with TRAF3 degradation factors that associate via other regions of CD40 within the signaling complex. By removing the CD40 TBS and the ability of TBS-associated factors to bind CD40, TBS-independent factors may be able to more efficiently bind CD40 and mediate TRAF3 degradation. It should prove interesting to determine what other regions and factors influence TRAF2 and 3 degradation. These results also illustrate differences in the way the TBS sequences of CD40 and LMP1 interact with and regulate TRAFs for signaling. While both CD40 and LMP1 bind TRAF2 via their respective TBS, the sequence of the CD40 TBS induces TRAF2 to perform distinct functions compared to the TBS of LMP1.
The TBS of LMP1 clearly influences the exaggerated signaling nature of hCD40LMP1. The TBS of hCD40LMP1 imparted to hCD40ADD the ability to induce IL-6 in response to agonistic Ab, in contrast to hCD40 (14, 49). This ability was not merely the result of loss of a negative regulatory mechanism associated with the TBS of CD40, because hCD40LMP1AEDL retained the ability to induce IL-6 secretion in response to agonistic Ab.
In the case of signals activating the canonical NF-κB pathway, an interesting intermediate pattern of TBS influence emerged for both hCD40ADD and hCD40LMP1AEDL (Fig. 5). This suggests that, like IL-6 production, both TBS properties as well as additional factors regulate the ability to activate the canonical NF-κB pathway. Despite the effects of the TBS on activation of the canonical NF-κB pathway, the overall sum of both NF-κB activation pathways at later time points correlated with regions outside of the TBS in both hCD40 and hCD40LMP1 (Fig. 6). hCD40LMP1AEDL’s maintenance of an hCD40LMP1-like NF-κB activation profile is most likely due to its ability to maintain IκBα degradation at later times, similar to hCD40LMP1. Further, regions outside of the TBS may be important in mediating non-canonical NF-κB activation. Inasmuch as these regions were not effected by switching of the TBS between hCD40 and hCD40LMP1, the mutant versions of these molecules would be able to activate the non-canonical pathway to a similar degree as their WT counterparts.
The activation of JNK showed a stronger dependence upon the distinct TBS, and this was also reflected in a downstream function of hCD40ADD, IL-6 production, heavily influenced by the JNK pathway (57). hCD40LMP1AEDL signaling, however, maintained the hCD40LMP1-like ability of inducing IL-6 secretion in response to agonistic Ab despite its inability to activate the JNK pathway for prolonged periods of time. This suggests that other LMP1 TBS-independent pathways are capable of a compensatory contribution to this signaling outcome. Future studies comparing recruitment of factors important for JNK activation to hCD40ADD and hCD40LMP1AEDL in comparison to their parental counterparts should aid in determining whether differential association with positive or negative regulatory mechanisms, or both, contribute to differences in JNK activation.
LMP1 utilizes TRAF3 as a positive regulator of several of its signaling pathways, while TRAF3 is a negative regulator of CD40 (25). Although there is strong structural similarity in each receptor’s proposed binding to TRAF3, several additional contacts are found for LMP1 (17). This indicates that there may be differences in the way TRAF3 associates with each TBS. TRAF2 appears to be functionally more critical for hCD40 than hCD40LMP1 signaling (24, 25, 34), highlighting another difference in TRAF utilization by CD40 and its viral mimic. The present study provides additional insights into which aspects of TRAF2/3 association and hCD40LMP1 signals are regulated by the TBS, and to what extent. This information has strong potential value for designing small molecule therapies that interrupt key pathogenic aspects of LMP1 signaling while avoiding disruption of CD40 signals, or if desired, therapies that would disrupt both signaling pathways.
Acknowledgments
This material is the result of work supported in part with resources and the use of facilities at the Iowa City VA Medical Center, Iowa City, Iowa.
Abbreviations in this paper
- TNF-R
TNF-receptor
- TRAF
TNF receptor-associated factor
- LMP1
Latent membrane protein 1
- CY
cytoplasmic
- EBV
Epstein Barr Virus
- RA
Rheumatoid Arthritis
- SLE
Systemic lupus erythematosus
- Ab
Antibodies
- COOH
carboxy
- TBS
TRAF2/3 binding site
- CTAR1&2
Cytoplasmic activating region 1&2
Footnotes
These studies were supported by NIH grants CA099997 and AI49993 to G.A.B. J.P.G. received support from NIH grant T32 AI007485. C.R.M. received support from NIH grant T32 HL07638-18.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Lyons SF, Liebowitz DN. The roles of human viruses in the pathogenesis of lymphoma. Sem Oncol. 1998;25:461–475. [PubMed] [Google Scholar]
- 2.Thorley-Lawson DA, Miyashita EM, Khan G. EBV and the B cell: that’s all it takes. Trends Microbiol. 1996;4:204–208. doi: 10.1016/s0966-842x(96)90020-7. [DOI] [PubMed] [Google Scholar]
- 3.Bishop GA, Busch LK. Molecular mechanisms of B lymphocyte transformation by Epstein-Barr virus. Microbes Infec. 2002;4:853–857. doi: 10.1016/s1286-4579(02)01605-2. [DOI] [PubMed] [Google Scholar]
- 4.Cahir McFarland ED, Izumi KM, Mosialos G. Epstein-barr virus transformation: involvement of latent membrane protein 1-mediated activation of NF-kappaB. Oncogene. 1999;18:6959–6964. doi: 10.1038/sj.onc.1203217. [DOI] [PubMed] [Google Scholar]
- 5.Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Semin Cancer Biol. 2001;11:435–444. doi: 10.1006/scbi.2001.0410. [DOI] [PubMed] [Google Scholar]
- 6.Kaye KM, Izumi KM, Kieff E. EBV LMP1 is essential for B-lymphocyte growth transformation: EBV strategy in normal and neoplastic B cells. Proc Natl Acad Sci (USA) 1993;90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takei M, Mitamura K, Fujiwara S, Horie T, Ryu J, Osaka S, Yoshino S, Sawada S. Detection of EBV-encoded small RNA 1 and LMP1 in synovial lining cells from rheumatoid arthritis patients. Int Immunol. 1997;9:739–743. doi: 10.1093/intimm/9.5.739. [DOI] [PubMed] [Google Scholar]
- 8.Gross AJ, Hochberg D, Rand WM, Thorley-Lawson DA. EBV and SLE: A new perspective. J Immunol. 2005;174:6599–6607. doi: 10.4049/jimmunol.174.11.6599. [DOI] [PubMed] [Google Scholar]
- 9.Stunz LL, Busch LK, Munroe ME, Tygrett L, Sigmund C, Waldschmidt TW, Bishop GA. Expression of the LMP1 cytoplasmic tail in mice induces hyperactivation of B lymphocytes and disordered lymphoid architecture. Immunity. 2004;21:255–266. doi: 10.1016/j.immuni.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Munroe ME, Arbiser JL, Bishop GA. Honokiol, a natural plant product, inhibits inflammatory signals and alleviates inflammatory arthritis. J Immunol. 2007;179:753–763. doi: 10.4049/jimmunol.179.2.753. [DOI] [PubMed] [Google Scholar]
- 11.Bishop GA, Hostager BS. Signaling by CD40 and its mimics in B cell activation. Immunol Res. 2001;24:97–109. doi: 10.1385/IR:24:2:097. [DOI] [PubMed] [Google Scholar]
- 12.Busch LK, Bishop GA. The EBV transforming protein, LMP1, mimics and cooperates with CD40 signaling in B lymphocytes. J Immunol. 1999;162:2555–2561. [PubMed] [Google Scholar]
- 13.Uchida J, Yasui T, Takaoka-Shichijo Y, Muraoka M, Kulwichit W, Raab-Traub N, Kikutani H. Mimicry of CD40 signals by EBV LMP1 in B lymphocyte responses. Science. 1999;286:300–303. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 14.Brown KD, Hostager BS, Bishop GA. Differential signaling and TRAF degradation by CD40 and the EBV oncoprotein LMP1. J Exp Med. 2001;193:943–954. doi: 10.1084/jem.193.8.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert SL, Martinez OM. LMP1 of EBV activates PI3-K to induce production of IL-10. J Immunol. 2007;179:8225–8234. doi: 10.4049/jimmunol.179.12.8225. [DOI] [PubMed] [Google Scholar]
- 16.Mosialos G, Birkenback M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The EBV transforming protein LMP1 engages signaling proteins for the TNF-R family. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 17.Wu S, Xie P, Welsh K, Li C, Ni C, Zhu X, Reed JC, Satterthwait AC, Bishop GA, Ely KR. LMP1 protein from EBV is a structural decoy in B lymphocytes for binding to TRAF3. J Biol Chem. 2005;280:33620–33626. doi: 10.1074/jbc.M502511200. [DOI] [PubMed] [Google Scholar]
- 18.Bishop GA, Hostager BS, Brown KD. Mechanisms of tumor necrosis factor receptor associated factor (TRAF) regulation in B lymphocytes. J Leuk Biol. 2002;72:19–23. [PubMed] [Google Scholar]
- 19.Ye H, Park YC, Kreishman M, Kieff E, Wu H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Molecular Cell. 1999;4:321–330. doi: 10.1016/s1097-2765(00)80334-2. [DOI] [PubMed] [Google Scholar]
- 20.Li C, Norriss PS, Ni CZ, Havert ML, Chiong EM, Tran BR, Cabezas E, Reed JC, Satterthwait AC, Ware CF, Ely KR. Structurally distinct recognition motifs in LTbR and CD40 for TRAF-mediated signaling. J Biol Chem. 2003;278:50523–50529. doi: 10.1074/jbc.M309381200. [DOI] [PubMed] [Google Scholar]
- 21.Devergne O, Hatzivassiliou E, Izumi KM, Kaye KM, Kleijnen MF, Kieff E, Mosialos G. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for B-lymphocyte transformation: role in NF-kappaB activation. Mol Cell Biol. 1996;16:7098–7108. doi: 10.1128/mcb.16.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devergne O, Cahir McFarland ED, Mosialos G, Izumi KM, Ware CF, Kieff E. Role of the TRAF binding site and NF-kappaB activation in Epstein-Barr virus latent membrane protein 1-induced cell gene expression. J Virol. 1998;72:7900–7908. doi: 10.1128/jvi.72.10.7900-7908.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huen DS, Henderson SA, Croom-Carter D, Rowe M. The EBV LMP1 mediates activation of NF-κB and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene. 1995;4:549–560. [PubMed] [Google Scholar]
- 24.Xie P, Bishop GA. Roles of TRAF3 in signaling to B lymphocytes by CTAR regions 1 and 2 of the EBV-encoded oncoprotein LMP1. J Immunol. 2004;173:5546–5555. doi: 10.4049/jimmunol.173.9.5546. [DOI] [PubMed] [Google Scholar]
- 25.Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in signaling by LMP1, but not CD40, in B lymphocytes. J Exp Med. 2004;199:661–671. doi: 10.1084/jem.20031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Busch LK, Bishop GA. Multiple carboxyl-terminal regions of the EBV oncoprotein, LMP1, cooperatively regulate signaling to B lymphocytes via TRAF-dependent and TRAF-independent mechanisms. J Immunol. 2001;167:5805–5813. doi: 10.4049/jimmunol.167.10.5805. [DOI] [PubMed] [Google Scholar]
- 27.Sutherland CL, Krebs DL, Gold MR. An 11-amino acid sequence in the cytoplasmic domain of CD40 is sufficient for activation of JNK, activation of MAPKAP kinase-2, phosphorylation of IκBα, and protection of WEHI-231 cells from anti-IgM-induced growth arrest. J Immunol. 1999;162:4720–4730. [PubMed] [Google Scholar]
- 28.Pullen SS, Dang TTA, Crute JJ, Kehry MR. CD40 signaling through TRAFs. Binding site specificity and activation of downstream pathways by distinct TRAFs. J Biol Chem. 1999;274:14246–14254. doi: 10.1074/jbc.274.20.14246. [DOI] [PubMed] [Google Scholar]
- 29.Leo E, Welsh K, Matsuzawa S, Zapata JM, Kitada S, Mitchell RS, Ely KR, Reed JC. Differential requirements for TRAF family proteins in CD40-mediated induction of NF-κB and JNK activation. J Biol Chem. 1999;274:22414–22422. doi: 10.1074/jbc.274.32.22414. [DOI] [PubMed] [Google Scholar]
- 30.Manning E, Pullen SS, Souza DJ, Kehry M, Noelle RJ. Cellular responses to murine CD40 in a mouse B cell line may be TRAF dependent or independent. Eur J Immunol. 2002;32:39–49. doi: 10.1002/1521-4141(200201)32:1<39::AID-IMMU39>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 31.Hostager BS, Bishop GA. Role of TRAF2 in the activation of IgM secretion by CD40 and CD120b. J Immunol. 2002;168:3318–3322. doi: 10.4049/jimmunol.168.7.3318. [DOI] [PubMed] [Google Scholar]
- 32.Hostager BS, Bishop GA. Cutting Edge: Contrasting roles of TRAF2 and TRAF3 in CD40-mediated B lymphocyte activation. J Immunol. 1999;162:6307–6311. [PubMed] [Google Scholar]
- 33.Xie P, Hostager BS, Munroe ME, Moore CR, Bishop GA. Cooperation between TRAFs 1 and 2 in CD40 signaling. J Immunol. 2006 doi: 10.4049/jimmunol.176.9.5388. in press. [DOI] [PubMed] [Google Scholar]
- 34.Hostager BS, Haxhinasto SA, Rowland SR, Bishop GA. TRAF2-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem. 2003;278:45382–45390. doi: 10.1074/jbc.M306708200. [DOI] [PubMed] [Google Scholar]
- 35.Brown KD, Hostager BS, Bishop GA. Regulation of TRAF2 signaling by self-induced degradation. J Biol Chem. 2002;277:19433–19438. doi: 10.1074/jbc.M111522200. [DOI] [PubMed] [Google Scholar]
- 36.Xie P, Brown L, Stunz L, Bishop G. TRAF3 inhibits signaling by Toll-like receptors in B lymphocytes. J Immunol. 2008 in press. [Google Scholar]
- 37.Hostager BS, Hsing Y, Harms DE, Bishop GA. Different CD40-mediated signaling events require distinct CD40 structural features. J Immunol. 1996;157:1047–1053. [PubMed] [Google Scholar]
- 38.Bishop GA, Haughton G. Induced differentiation of a transformed clone of Ly-1+ B cells by clonal T cells and antigen. Proc Natl Acad Sci (USA) 1986;83:7410–7414. doi: 10.1073/pnas.83.19.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bishop GA, Ramirez LM, Koretzky GA. Growth inhibition of a B cell clone mediated by ligation of IL-4 receptors or membrane IgM. J Immunol. 1993;150:2565–2574. [PubMed] [Google Scholar]
- 40.Bishop GA, Frelinger JA. Haplotype-specific differences in signaling by transfected class II molecules to a Ly-1+ B-cell clone. Proc Natl Acad Sci (USA) 1989;86:5933–5937. doi: 10.1073/pnas.86.15.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in Signaling by LMP1 But Not CD40 in B Lymphocytes. J Exp Med. 2004;199:661–671. doi: 10.1084/jem.20031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowland SR, Tremblay ML, Ellison JM, Stunz LL, Bishop GA, Hostager BS. A novel mechanism for TRAF6-dependent CD40 signaling. J Immunol. 2007;179:4645–4653. doi: 10.4049/jimmunol.179.7.4645. [DOI] [PubMed] [Google Scholar]
- 43.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 44.Brown KD, Hostager BS, Bishop GA. Differential signaling and tumor necrosis factor receptor-associated factor (TRAF) degradation mediated by CD40 and the Epstein-Barr virus oncoprotein latent membrane protein 1 (LMP1) J Exp Med. 2001;193:943–954. doi: 10.1084/jem.193.8.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hostager BS, Haxhinasto SA, Rowland SL, Bishop GA. Tumor Necrosis Factor Receptor-associated Factor 2 (TRAF2)-deficient B Lymphocytes Reveal Novel Roles for TRAF2 in CD40 Signaling. J Biol Chem. 2003;278:45382–45390. doi: 10.1074/jbc.M306708200. [DOI] [PubMed] [Google Scholar]
- 46.Moore CR, Bishop GA. Differential regulation of CD40-mediated TRAF degradation in B lymphocytes. J Immunol. 2005;175:3780–3789. doi: 10.4049/jimmunol.175.6.3780. [DOI] [PubMed] [Google Scholar]
- 47.Bishop GA. Requirements of class II-mediated B cell differentiation for class II crosslinking and cAMP. J Immunol. 1991;147:1107–1114. [PubMed] [Google Scholar]
- 48.Mercolino TJ, Arnold LW, Haughton G. Phosphatidyl choline is recognized by a series of Ly-1+ murine B cell lymphomas specific for erythrocyte membranes. J Exp Med. 1986;163:155–165. doi: 10.1084/jem.163.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baccam M, Bishop GA. Membrane-bound CD154, but not anti-CD40 mAbs, induces NF-κB independent B cell IL-6 production. Eur J Immunol. 1999;29:3855–3866. doi: 10.1002/(SICI)1521-4141(199912)29:12<3855::AID-IMMU3855>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 50.Bishop GA, Moore CR, Xie P, Stunz LL, Kraus ZJ. TRAF proteins in CD40 signaling. In: Wu H, editor. TRAFs. Landes Biosciences; New York: 2007. in press. [DOI] [PubMed] [Google Scholar]
- 51.Matsuzawa A, Tseng PH, Vallabhapurapu S, Luo JL, Zhang W, Wang H, Vignali DA, Gallagher E, Karin M. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haxhinasto SA, Hostager BS, Bishop GA. Cutting Edge: Molecular mechanisms of synergy between CD40 and the BCR: Role for TRAF2 in receptor interaction. J Immunol. 2002;169:1145–1149. doi: 10.4049/jimmunol.169.3.1145. [DOI] [PubMed] [Google Scholar]
- 53.Bishop GA. The multifaceted roles of TRAFs in the regulation of B cell function. Nat Rev Immunol. 2004;4:775–786. doi: 10.1038/nri1462. [DOI] [PubMed] [Google Scholar]
- 54.Middeldorp JM, Pegtel DM. Multiple roles of LMP1 in Epstein-Barr virus induced immune escape. Semin Cancer Biol. 2008;18:388–396. doi: 10.1016/j.semcancer.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 55.Floettmann JE, Eliopoulos AG, Jones M, Young LS, Rowe M. EBV LMP1 signalling is distinct from CD40 and involves physical cooperation of its two C-terminus functional regions. Oncogene. 1998;17:2383–2392. doi: 10.1038/sj.onc.1202144. [DOI] [PubMed] [Google Scholar]
- 56.Pullen SS, Miller HG, Everdeen DS, Dang TTA, Crute JJ, Kehry MR. CD40-TRAF interactions: Regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochem. 1998;37:11836–11845. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- 57.Vanden Bush T, Bishop GA. TLR7 and CD40 cooperate in IL-6 production via enhanced JNK and AP-1 activation. Eur J Immunol. 2008;38:400–409. doi: 10.1002/eji.200737602. [DOI] [PMC free article] [PubMed] [Google Scholar]