

Abstract
The role of CX3CR1 in regulating the function of monocytes and microglia was examined in mice in which CX3CR1 had been replaced by green fluorescent protein (GFP). Induction of experimental autoimmune uveitis (EAU) in these mice resulted in increased disease severity at day 23 postimmunization with uveitogenic peptide when compared with CX3CR1-positive mice and increased apoptosis of neuronal cells in the inner nuclear layer. Resident microglia within the retina were activated equally as EAU developed in mice with or without CX3CR1, as determined by changes in morphology, suggesting that the microglial cell response did not account for the differences. Although the inflammatory infiltrate had increased in mice without CX3CR1 at day 23 postimmunization, the percentage of natural killer cells in the infiltrate was not changed in these mice. Similarly, increased disease severity at this stage was not associated with an overall increased percentage of macrophages in the retinal inflammatory infiltrate or in increased activation of these cells. The increased recruitment of monocytes to the retina in response to EAU induction in CX3CR1GFP/GFP mice compared with CX3CR1GFP/+ mice was not reflected in increased migration away from vessels, leading to marked clustering of GFP+ cells around veins and venules in these mice. It is possible that this monocyte/macrophage clustering leads to the increased severity of disease seen in the mice by focusing and so intensifying the inflammatory response.
Keywords: autoimmunity, cell trafficking, chemokine receptor, chemokines, inflammation, monocytes
Introduction
Mononuclear phagocytes are generated in the bone marrow and under steady-state conditions they recirculate in the blood for only a few days before a limited number are recruited into the central nervous system (CNS). Here they form a network of phagocytic potential antigen-presenting cells, some of which differentiate into long-lived, highly specialized cells, the microglia, which are the mononuclear phagocytes of the CNS.1 In an inflammatory situation monocytes are recruited to the inflammatory site and can differentiate into macrophages, often becoming the main effector cells and responsible for many of the damaging effects of inflammation. Trafficking of mononuclear phagocytes and the differential control and relationship of monocytes/macrophages and microglia are still poorly understood and likely to differ depending on the inflammatory site and stimulus.
Chemokines have wide-ranging roles in the positioning of specific leucocytes.2 Mononuclear phagocytes are heterogeneous in terms of their expression of an extensive variety of different markers including chemokine receptors.3,4 Two chemokine receptors in particular have been identified as important for their trafficking, CCR2 and CX3CR1.5 It has been suggested that CCR2 is more important for trafficking of monocytes to an inflammatory site, for example in peritonitis,6 autoimmune encephalitis7 and tuberculosis,8 whereas CX3CR1 is more important in immune surveillance and the movement of mononuclear phagocytes in the steady-state situation.5
However, evidence suggests that the situation is likely to be more complex than this paradigm suggests. CCR2 is also involved in monocyte emigration from the bone marrow9,10 and although CCR2-positive monocytes may be recruited preferentially to an inflammatory site, CCR2-negative monocytes are also able to traffic to sites of inflammation.9,11,12 In addition, CX3CR1 has been implicated in the recruitment of monocytes in inflammatory situations; for example, in atherosclerosis,13,14 crescentic glomerulonephritis15 and in cerebral ischaemia.16 Recent studies have shown that CX3CR1-positive monocytes patrol the endothelium in the steady state but provide a rapidly infiltrating population at the first signs of infection.17 In a model of atherosclerosis, although CX3CR1-positive monocytes entered atherosclerotic plaques, CX3CR1 was not essential whereas CCR2-positive monocytes, also expressing substantial levels of CX3CR1,5 required CX3CR1, in addition to CCR2, to accumulate.11 It is not known whether this is the case in other inflammatory situations such as in the CNS.
Apart from recruitment, it has been suggested that CX3CL1 is important for microglial regulation and communication between microglia and neurons. In the CNS, neurons are responsible for much of the production of CX3CL118 and microglia express CX3CR1.19In vitro studies have shown that CX3CL1 reduces the production of nitric oxide, interleukin-6 and tumour necrosis factor-α by activated microglia20,21 and suppresses neuronal cell death.20,22 However, other reports suggest that CX3CL1 can induce microglia activation,23 increase production of matrix metalloproteinase 924 and is important for microglial survival by inhibiting Fas-mediated apoptosis.25
Recently it has been shown in vivo, in murine models of neurotoxicity in the brain and CNS, that CX3CL1 may protect neurons from the damaging effects of activated microglia.19 Similarly, in a murine model of retinal ageing and laser-induced choroidal neovascularization, the lack of CX3CR1 in mice caused pronounced accumulation of activated microglia subretinally leading to retinal degeneration and neovascularization.26 However, the stimulus may be critical because after facial nerve axotomy27 and laser-induced injury,28 microglial activation in CX3CR1−/− mice remains normal.
CX3CR1 may have both a proinflammatory role in recruiting monocytes to the inflammatory site and an anti-inflammatory role in damping microglial activation. The aim was therefore to examine the effect of CX3CR1 deficiency on both microglial behaviour and monocyte recruitment as inflammatory disease develops. We have used a model of human posterior ocular inflammation, experimental autoimmune uveitis (EAU), in mice in which monocytes/macrophages are important effector cells, crossing a disrupted blood–retina barrier (BRB), infiltrating the retina and causing sight-threatening damage.29
Materials and methods
Animals and the retinal inflammation model
Eight- to sixteen-week-old C57BL/6, CX3CR1GFP/GFP and CX3CR1GFP/+ mice27 were bred and maintained in the Medical Research Facility (University of Aberdeen, Aberdeen, UK). CX3CR1 had been replaced by the enhanced green fluorescent protein gene EGFP and was on a full C57BL/6 background. As there is biallelic expression of the CX3CR1 locus all surface CX3CR1-positive cells in heterozygous mice express green fluorescent protein (GFP).27 Procedures were approved according to the Home Office Regulations for Animal Experimentation (UK).
The animals were immunized with human interphotoreceptor retinoid binding protein (IRBP) peptide 1–20 (GPTHLFQPSLVLDMAKVLLD, 10 mg/ml; Sigma-Genosys, Cambridge, UK) emulsified 1 : 1 in complete Freund’s adjuvant – H37Ra (Difco Laboratories, Detroit, MI) with an additional 2·5 mg/ml Mycobacterium tuberculosis– H37Ra (Difco Laboratories). Fifty microlitres were injected subcutaneously in each thigh (500 μg peptide per animal). An additional 100 μl Bordetella pertussis toxin (Health Protection Agency, Salisbury, UK) was administered intraperitoneally at a concentration of 10 μg/ml. In this model, the first signs of retinal inflammation are observed between day 10 and day 15 postimmunization (p.i.). Most mice show peak disease between day 23 and day 26 which is reduced by day 32 p.i.30
Histological evaluation of EAU
Eyes from animals were harvested on day 23 and day 28 p.i. Eyes were snap frozen in OCT™ (Tissue Tek, Sakura, Zoeterwoude, the Netherlands) using isopentane and dry ice. Cryostat sections (8-μm thick) were prepared and air-dried for 48 hr. The dried sections were stained with haematoxylin and eosin. Histological evaluation of EAU was performed using our customized version of the grading system.31 This method assigns a score of up to five both for cellular infiltration, by counting infiltrating cells and granulomas in different areas of the posterior chamber and assessing the degree of vasculitis, perivascular cuffing and choroidal thickening, and for structural abnormalities, by assessing the degree of loss of rod outer segments and neuronal layers and the number of retinal folds and retinal detachment. Two independent ‘blind’ examiners determined the scores by observing the haematoxylin and eosin-stained sections under × 20 objective lens. For each eye, six sections were graded.
Quantification of apoptosis in inner and outer nuclear layers of retina
Apoptosis was detected by TdT-mediated biotin–dUTP nick end labelling (TUNEL) staining using a Klenow FragEL™ DNA fragmentation detection kit according to the manufacturer’s instructions (Calbiochem; Merck Biosciences, Nottingham, UK). Cryostat sections, prepared as above from mice day 23 p.i., were fixed in 4% (volume/volume) formaldehyde. Stained and unstained cells were counted throughout the inner and outer nuclear layers.
Immunohistochemistry
Eyes were frozen and 8-μm cryostat sections were cut, fixed and stained as described previously.32 Briefly, sections were incubated with primary antibody, rat monoclonal antibody immunoglobulin G2b (IgG2b) to F4/80 (1 : 100; Serotec Ltd, Oxford, UK) or mouse monoclonal antibody IgG2a to inducible nitric oxide synthase (iNOS, 1 : 50; Transduction Laboratories, Affiniti Research Products, Exeter, UK) or pan-natural killer (NK) antibody D X 5 (rat monoclonal antibody IgM, 1 : 50; Serotec Ltd) for 1 hr at room temperature. Rabbit anti-mouse F(ab′)2 fragments were used to block non-specific binding of iNOS antibody. Isotype control antibodies were obtained from Serotec. Samples were then incubated for 1 hr, first with biotinylated secondary antibody and second with a streptavidin–biotinylated alkaline phosphatase complex (both Dako Ltd, Cambridge, UK) before the addition of fast red substrate solution. Six sections per eye and three or four fields of view per section (× 20 objective) were examined and red cells and blue infiltrating cells were counted. Infiltrating cells were determined by their position and morphology.
Microglia in CX3CR1GFP/GFP and CX3CR1GFP/+
Retinal whole-mounts were prepared from CX3CR1GFP/GFP and CX3CR1GFP/+ mice at day 10 p.i. Retinas were removed, washed twice in phosphate-buffered saline for 15 min, spread on clean glass slides and mounted vitreous side up.33 Mounts were examined using a confocal laser scanning microscope, LSM 510 (Zeiss, Göttingen, Germany). The lengths of microglial dendrites were measured using Zeiss LSM Image Examiner (Zeiss). Dendrites were picked at random and measured from the point of attachment to the cell body (three or four per cell). The values were averaged to give a mean value for dendrite length of naïve and EAU microglial cells.
Mean fluorescence intensity (MFI) of individual cells was determined using ImageJ34 in non-immunized and day 10 p.i. CX3CR1GFP/GFP and CX3CR1GFP/+ mice. In addition, Image Examiner was used to determine the MFI of green fluorescent cell clusters accumulating around vessels at different days p.i. by taking measurements 180 μm across retinal veins and venules at three different lengths. Measuring from the optic disc, MFI values for microglial cell clusters at 250, 500 and 750 μm were measured, perpendicular to the length of the vessel. Three vessels on each retina were measured for both naïve and inflamed eyes. Background fluorescence was subtracted by taking the mean intensity values of regions outside the whole-mount. Individual cells at specified distances away from vessels were also counted in each retinal whole-mount.
Data analysis
Probability values were calculated using an unpaired two-tailed Student’s t-test. Groups were compared using a one-way analysis of variance with Tukey multiple comparison test. Probability values of P < 0·05 were considered significant.
Results
Development of EAU in mice lacking CX3CR1
Experimental autoimmune uveitis was induced in both CX3CR1GFP/GFP and CX3CR1GFP/+ mice and disease was graded in terms of inflammatory infiltrate and structural damage at days 23 and 28 p.i. The EAU in the C57BL/6 strain of mice has been shown to peak about day 25 p.i. and to be reduced by day 32 p.i.30 Mice, either CX3CR1GFP/GFP or CX3CR1GFP/+ which had not been immunized with IRBP peptide, showed no infiltrative or structural changes (Fig. 1a). Mice homozygous for the CX3CR1 deletion showed significantly increased disease severity in terms of both infiltrate and structural damage at day 23 p.i. compared with heterozygous mice (P< 0·05) (Fig. 1a–c). Inflammation had decreased by day 28 and there was no significant difference in the EAU in homozygote or heterozygote at this stage (Fig. 1b,c).
Figure 1.

Effect of lack of CX3CR1 on experimental autoimmune uveitis (EAU). (a) Haematoxylin-stained cryostat sections from a CX3CR1GFP/+ mouse (left) and a CX3CR1GFP/GFP mouse (middle) both day 23 postimmunization (p.i.) and a CX3CR1GFP/GFP mouse non-immunized (right) (V, vitreous; GL, ganglion layer; INL, inner nuclear layer; ONL, outer nuclear layer; ROS, rod outer segments; arrows indicate examples of infiltration of inflammatory cells; original magnification × 25). (b) Disease graded by inflammatory infiltrate and (c) structural damage score in CX3CR1GFP/+ and CX3CR1GFP/GFP mice at days 23 and 28 p.i. Hatched bars, CX3CR1GFP/+ mice. Dotted bars, CX3CR1GFP/GFP mice. Error bars indicate SEM; n = 8 mice per group.
When the infiltrate was examined after immunohistochemical staining for the macrophage marker F4/80, the percentage of macrophages in the infiltrate was not significantly changed when CX3CR1GFP/GFP mice were compared with CX3CR1GFP/+ mice at day 23 p.i. or day 28 p.i. (Fig. 2a). In addition, there was no significant difference in the percentage of activated macrophages present in the infiltrate at either day 23 or 28 when the genotypes were compared by staining for iNOS (Fig. 2b). The percentage of NK cells present in the infiltrate, as determined by staining with the pan-NK antibody D X 5, was low and did not change significantly when CX3CR1GFP/GFP and CX3CR1GFP/+ mice were compared either at day 23 or day 28 p.i. (Fig. 2c).
Figure 2.

Effect of lack of CX3CR1 on cellular composition of inflammatory infiltrate in experimental autoimmune uveitis (EAU). (a) Percentage of cells in the inflammatory infiltrate stained positively for F4/80. (b) Percentage of cells in the inflammatory infiltrate stained positively for inducible nitric oxide synthase (iNOS). (c) Percentage of cells in the inflammatory infiltrate stained positively for DX5. Hatched bars, CX3CR1GFP/+ mice. Dotted bars, CX3CR1GFP/GFP mice. Error bars indicate SEM; n = 8 mice per group.
Examination of apoptosis in the inner and outer nuclear layers of the retina by TUNEL staining of sections from mice on day 23 p.i. showed patchy staining but counts of the apoptosing cells throughout the outer and inner nuclear layers revealed no difference between CX3CR1GFP/GFP and CX3CR1GFP/+ mice in the outer nuclear layer, which comprises photoreceptor cell nuclei. However, apoptosis in the inner nuclear layer, which comprises nuclei of other neuronal cell types, was significantly (P= 0·02) increased in CX3CR1GFP/GFP mice compared with CX3CR1GFP/+ mice (Fig. 3a,b). Apoptosing cells were predominantly neuronal as determined by morphology and there was little inflammatory cell infiltrate in this region (Fig. 3a).
Figure 3.
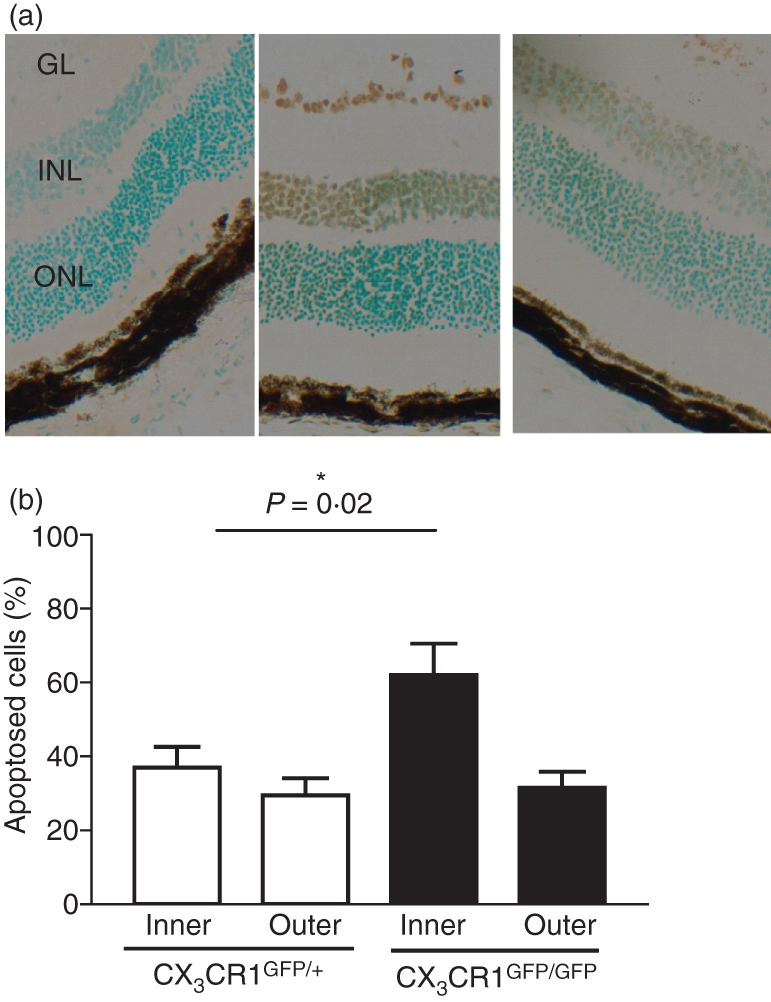
Effect of lack of CX3CR1 on apoptosis in nuclear layers of the retina in experimental autoimmune uveitis (EAU). (a) Cryostat sections from CX3CR1GFP/GFP, assay control (left section), from CX3CR1GFP/GFP TUNEL stained (middle section), and from CX3CR1GFP/+ TUNEL stained (right section), all day 23 postimmunization (p.i.) with apoptosis (brown stain) detected by TUNEL staining using a Klenow FragEL™ DNA fragmentation detection kit (GL, ganglion layer; INL, inner nuclear layer; ONL, outer nuclear layer; original magnification × 50). (b) Percentage of apoptosing cells in the inner and outer nuclear layer of the retina in CX3CR1GFP/+ (open bars) and CX3CR1GFP/GFP mice (filled bars) day 23 p.i. Error bars indicate SEM; n= 8 mice per group.
Changes in resident macrophage/microglial activation during EAU development
Microglia change morphologically from cells with dendrites to rounded cells with minor processes as they become activated.35 We have therefore used dendrite length as a measure of microglial activation. Using images obtained by confocal microscopy of GFP-labelled cells in retinal whole-mounts, dendrite length was measured on 40 randomly chosen cells throughout the retina in six retinae for each genotype at each time-point. For each cell, mean dendrite length for all visible dendrites (at least three dendrites per cell) was calculated. Measurements showed a clear and highly significant (P< 0·0001) decrease in CX3CR1GFP/GFP and CX3CR1GFP/+ dendrite length in immunized compared to non-immunized mice as early as 10 days p.i. (Fig. 4a). These cells could be assumed to be microglia rather than infiltrating macrophages at this stage of the disease because there was no infiltration at day 10 p.i. when EAU is graded. There was, however no difference in dendrite length between CX3CR1GFP/GFP and CX3CR1GFP/+ cells at the same time-point p.i. (Fig. 4a).
Figure 4.

Activation of microglia during early experimental autoimmune uveitis (EAU) development to day 10 postimmunization (p.i.) in CX3CR1GFP/+ and CX3CR1GFP/GFP mice. (a) Average length of microglial dendrites in retinal whole-mounts. All visible dendrites (three or four) were measured per cell. (b) Mean fluorescence intensity (MFI) of individual microglia in retinal whole-mounts. Triangles, CX3CR1GFP/+ mice; circles, CX3CR1GFP/GFP mice. Error bars indicate SEM; n= 40 cells per group.
The MFI of single microglial cells from CX3CR1GFP/GFP and CX3CR1GFP/+ non-immunized mice was the same, showing that the expression of the reporter gene in individual cells was similar in CX3CR1GFP/GFP and CX3CR1GFP/+ as previously reported.26 As dendrites condense and the cells become more compact but with a larger cell body, their fluorescence intensity increases. The MFI of individual cells was therefore used as a second measure of morphological change and was determined for CX3CR1GFP/GFP and CX3CR1GFP/+ cells in the retina as EAU developed. The MFI increased significantly (P< 0·0001) as the disease developed in both genotypes to day 10 p.i. (Fig. 4b). There was, however, no difference in the MFI of individual cells when CX3CR1GFP/GFP and CX3CR1GFP/+ cells in the retina were compared at the same time-point p.i. (Fig. 4b).
Effect of CX3CR1 deletion on location of GFP-positive cells in retinal whole-mounts during EAU development
As EAU developed, GFP-labelled cells were seen to cluster around veins and venules in retinal whole-mounts. As a result of the number of cells clustering, individual cells could not be determined within the clusters, which made counting impossible so the degree of clustering was quantified by measuring the MFI across the vessels using confocal microscopy. The MFI was measured across each vessel at three set distances from the optic nerve, 250, 500 and 750 μm. For CX3CR1GFP/+ mice there was little increase in the clustering of labelled cells around veins and venules as EAU developed at day 15 and day 23 p.i. (Fig. 5a,b). This was the same at each distance from the optic nerve measured. However, in CX3CR1GFP/GFP mice, clustering was increased significantly (P< 0·05) both between day 0 and day 15 p.i., and between day 15 and day 23 p.i. (Fig. 5c,d). Examination of Z stacks using animation showed that these cells surrounded and lined the vessel rather than being located within it.
Figure 5.

Effect of lack of CX3CR1 on distribution of green fluorescent protein (GFP)-positive cells around inflamed veins and venules. Mean fluorescence intensity (MFI) measured perpendicular to the length of the vessel at 250, 500 and 750 μm from the optic disc in retinal whole-mounts from (a) CX3CR1GFP/+ mice and (c) CX3CR1GFP/GFP mice. Open bars, no induction of experimental autoimmune uveitis (EAU); dotted bars, day 15 postimmunization (p.i.); hatched bars, day 23 p.i. Example of confocal image for CX3CR1GFP/+ and CX3CR1GFP/GFP mice day 23 p.i., (b) and (d), respectively, with white bar 180 μm indicating a measurement. Individual GFP-expressing cells counted between 100 and 500 μm away from the centre of the vessel at days 0, 10, 15 and day 23 p.i. (e) Hatched bars, CX3CR1GFP/+ mice. Dotted bars, CX3CR1GFP/GFP mice; n= 3 mice per group. Error bars indicate SEM.
Individual GFP-expressing cells between 100 and 500 μm away from the centre of a vessel were also counted in retinal whole-mounts at days 0 and 10 p.i. when these cells could be assumed to be predominantly microglia rather than infiltrating macrophages and at day 15 and 23 p.i. when they could be infiltrating macrophages. There was no significant increase in the density of GFP-positive cells when CX3CR1GFP/GFP were compared with CX3CR1GFP/+ mice (Fig. 5e). Similarly, there was no decrease in the number of GFP-labelled cells to indicate that microglia were moving from these sites to the affected vessels.
Discussion
Lack of CX3CR1 increased the severity of EAU but did not inhibit resolution. The lack of change in the percentage of monocytes/macrophages in the infiltrate when CX3CR1GFP/GFP and CX3CR1GFP/+ were compared indicated that increased EAU severity at day 23 was not the result of a replacement of macrophages in the infiltrate by neutrophils, as is seen in some situations when monocyte recruitment is reduced,36 but that the increase in inflammatory infiltrate affected all cell types including monocytes. In addition, alteration in the activation of these macrophages did not account for the increase in disease severity.
Lack of requirement for CX3CR1 for monocyte passage across the BRB into the inflamed retina implies the use of alternative chemokine receptors such as CCR2 and CCR5 which have both been implicated in monocyte trafficking into an inflammatory site.11 We and others have shown that ligands for both CCR2 (CCL2) and CCR5 (CCL3, CCL4 and CCL5) are present in the retina and increase as EAU develops.37,38 CCR5 has been reported to be important for T-cell trafficking in EAU39 with CCL3 present on retinal vessels and antibody to CCL3 inhibiting leucocyte recruitment to the retina and the development of EAU.40
It has been suggested from both in vitro and in vivo evidence that CX3CL1 may act as a neuroprotectant preventing the activation of microglia and their neurotoxic effects.19–22 Activation of microglia was markedly increased as EAU developed, occurring as early as day 10 p.i. although infiltration is only detected to a minor extent in some mice at day 15 p.i. and leakage from vessels and other retinal abnormalities is not evident until the peak of the disease on day 23 p.i. Early microglial activation has also been reported in a model of EAU in rats.41 However, there was no evidence to suggest that the response to CX3CL1 could prevent or dampen this activation. In the retina CX3CL1 does not down-regulate the activation of microglia and this may be site-specific or stimulus-specific. Normal microglial activation also occurred in CX3CR1-deficient mice after facial nerve axotomy27 and laser-induced injury.19,28
Increased activation of microglia in CX3CR1-deficient mice cannot therefore account for the increased disease severity or the increase in apoptosis of neuronal cells in the inner nuclear layer of these mice. Although in the past it has been suggested that neuronal expression of CX3CR1 might be important for neuronal survival22 it has recently been shown that CX3CR1 is not expressed on neurons.19,26 The key to these increases is therefore likely to lie with macrophage behaviour. The percentage of macrophages in the increased infiltrate and their activation state in the retina were not altered in CX3CR1-deficient mice. However, the increased infiltration was not reflected in increased migration away from vessels in the retina. In retinal whole-mounts, GFP-positive cells clustered around veins and venules at day 15 p.i. and day 23 p.i. in CX3CR1GFP/GFP mice but only to a very minor extent in mice with CX3CR1. These clustering cells could potentially be resident microglia or infiltrating monocytes differentiating into macrophages. They are less likely to be microglia because GFP-positive cells in the retina away from vessels are not depleted concomitant with the increase in GFP-positive cells at the blood vessels. Although microglia have been reported to be able to move rapidly and substantially in response to injury in the brain and CNS35,42 this may not be the case when injury is more subtle and localized.43 In the retina microglia movement may be highly regulated26,41 and there may be little recruitment to sites of BRB breakdown.
Lack of CX3CR1 therefore appears to result in the accumulation of macrophages around vessels once they have entered the retina. There is substantial constitutive expression of CX3CL1 in the eye38 and this is thought to be by several cell types including retinal neurons and endothelial cells.44 We have confirmed that CX3CL1 expression in the retina is not up-regulated in EAU (data not shown), indicating that CX3CL1 does not have a primary role in monocyte recruitment in EAU. However, it is possible that without CX3CR1 the macrophages, unable to respond to constitutive CX3CL1 in the retina, move away from vessels less efficiently leading to an accumulation of these cells around the inflamed vessels. This would lead to an inflammatory focus in which cytokines and chemokines are produced and cells can be stimulated more efficiently leading to a cascade effect increasing recruitment of inflammatory cells including macrophages and enhancing disease in mice lacking CX3CR1. In this context it is of interest that in brain, adoptively transferred activated microglia from CX3CR1−/− mice showed reduced migration away from the injection site compared with those from CX3CR1+/−.19 Another possible explanation is that in the absence of CX3CR1 and constitutive recruitment of monocytes, the monocyte population in the blood is changed in terms of chemokine receptor expression, resulting in altered recruitment in an inflammatory situation.
Increased apoptosis in the inner rather than the outer nuclear layer is consistent with increased accumulation of macrophages around the vessels of the retinal vasculature. The inner nuclear layer is supplied from central retinal vessels leading to capillary networks and post-capillary venules whereas the outer nuclear layer is supplied by the choriocapillaris at the back of the eye. Accumulating macrophages near the inner nuclear layer would be likely to initiate apoptosis of neuronal cells via Fas ligand or secretion of tumour necrosis factor-α.
It is possible that the increased severity of EAU and increased neuronal apoptosis in mice lacking CX3CR1 is the result of ineffective recruitment of other cells expressing CX3CR1. T cells are not thought to express CX3CR1 in mice27 but NK cells do.27 Interestingly an increase in disease has also been shown in experimental autoimmune encephalomyelitis in mice lacking CX3CR1.45 Evidence suggested that this was the result of deficient recruitment of NK cells, of which a subset are CX3CR1 positive.46 It has been reported that NK cells are able to influence autoimmunity in a variety of ways both preventing and enabling disease but this may depend on different sites, different types of autoimmunity and possibly even different stages of the same disease.45 Deficient recruitment of NK cells is not likely to play a role in the increase in disease seen when EAU is induced in CX3CR1-deficient mice because the percentage of NK cells in the inflammatory infiltrate in these mice was not altered. This is not unexpected because mice depleted of NK cells have diminished EAU.47
We have therefore shown that CX3CR1 does not have a primary role in the recruitment of monocytes to the inflammatory site across the BRB and nor does it prevent microglial activation in the retina during EAU. However, lack of CX3CR1 leads to the accumulation of macrophages at the sites of leucocyte extravasation and increased EAU severity and neuronal apoptosis. These findings contribute to the current debate over the likely value of targeting CX3CR1 in atherosclerosis as it has been suggested that this might be inadvisable if CX3CL1 has an important role as a neuroprotectant.
Acknowledgments
We are very grateful to Dr Steffen Jung, Weizmann Institute of Science, Israel, for his help in providing the CX3CR1-deleted mice and to Dr Delyth Reid, Division of Applied Medicine, University of Aberdeen, for advice on NK cell detection. This work was supported by grants from the National Eye Research Centre, Bristol, UK and NHS Grampian Endowment Trust Fund.
Glossary
Abbreviations:
- BRB
blood–retina barrier
- CNS
central nervous system
- EAU
experimental autoimmune uveitis
- GFP
green fluorescent protein
- IgG2b
immunoglobulin G2b
- iNOS
inducible nitric oxide synthase
- IRBP
human interphotoreceptor retinoid binding protein
- MFI
mean fluorescence intensity
- NK
natural killer
- p.i.
postimmunization
- ROS
rod outer segments
- TUNEL
TdT-mediated biotin–dUTP nick end labelling
Disclosures
None.
References
- 1.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–13. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 2.Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 3.Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–7. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 4.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 5.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 6.Boring L, Gosling J, Chensue SW, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C–C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552–61. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. Journal Exp Med. 2000;192:1075–80. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters W, Scott HM, Chambers HF, Flynn JL, Charo IF, Ernst JD. Chemokine receptor 2 serves an early and essential role in resistance to Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2001;98:7958–63. doi: 10.1073/pnas.131207398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–7. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 10.Tsou CL, Peters W, Si Y, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–9. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–94. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinones MP, Ahuja SK, Jimenez F, et al. Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. J Clin Invest. 2004;113:856–66. doi: 10.1172/JCI20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–40. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Combadiere C, Potteaux S, Gao JL, et al. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–16. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 15.Feng LL, Chen SZ, Garcia GE, et al. Prevention of crescentic glomerulonephritis by immunoneutralization of the fractalkine receptor CX3CR1. Kidney Int. 1999;56:612–20. doi: 10.1046/j.1523-1755.1999.00604.x. [DOI] [PubMed] [Google Scholar]
- 16.Soriano SG, Amaravadi LS, Wang YF, et al. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- 17.Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–70. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 18.Harrison JK, Jiang Y, Chen SZ, et al. Role for neuronally-derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95:10896–901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–24. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 20.Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- 21.Zujovic V, Benavides J, Vige X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–15. [PubMed] [Google Scholar]
- 22.Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX(3)CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A. 2000;97:8075–80. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maciejewski-Lenoir D, Chen S, Feng L, Maki R, Bacon KB. Characterization of fractalkine in rat brain cells: migratory and activation signals for CX3CR-1-expressing microglia. J Immunol. 1999;163:1628–35. [PubMed] [Google Scholar]
- 24.Cross AK, Woodroofe MN. Chemokine modulation of matrix metalloproteinase and TIMP production in adult rat brain microglia and a human microglial cell line in vitro. Glia. 1999;28:183–9. [PubMed] [Google Scholar]
- 25.Boehme SA, Lio FM, Maciejewski-Lenoir D, Bacon KB, Conlon PJ. The chemokine fractalkine inhibits Fas-mediated cell death of brain microglia. J Immunol. 2000;165:397–403. doi: 10.4049/jimmunol.165.1.397. [DOI] [PubMed] [Google Scholar]
- 26.Combadiere C, Feumi C, Raoul W, et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–8. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–14. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 29.Forrester JV, Huitinga I, Lumsden L, Dijkstra CD. Marrow-derived activated macrophages are required during the effector phase of experimental autoimmune uveoretinitis in rats. Curr Eye Res. 1998;17:426–37. doi: 10.1080/02713689808951224. [DOI] [PubMed] [Google Scholar]
- 30.Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick J, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol. 2002;161:1669–77. doi: 10.1016/S0002-9440(10)64444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calder CJ, Nicholson LB, Dick AD. Mechanisms for inducing nasal mucosal tolerance in experimental autoimmune uveoretinitis. Methods. 2006;38:69–76. doi: 10.1016/j.ymeth.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Jiang H-R, Lumsden L, Forrester JV. Macrophages and dendritic cells in IRBP-induced experimental autoimmune uveoretinitis in B10RIII mice. Invest Ophthalmol Vis Sci. 1999;40:3177–85. [PubMed] [Google Scholar]
- 33.Chan-Ling T. Glial, vascular, and neuronal cytogenesis in whole-mounted cat retina. Microsc Res Tech. 1997;36:1–16. doi: 10.1002/(SICI)1097-0029(19970101)36:1<1::AID-JEMT1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 34.Abràmoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 35.Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–66. [PubMed] [Google Scholar]
- 36.Quinones MP, Estrada CA, Kalkonde Y, et al. The complex role of the chemokine receptor CCR2 in collagen-induced arthritis: implications for therapeutic targeting of CCR2 in rheumatoid arthritis. J Mol Med. 2005;83:672–81. doi: 10.1007/s00109-005-0637-5. [DOI] [PubMed] [Google Scholar]
- 37.Crane IJ, McKillop-Smith S, Wallace CA, Lamont GR, Forrester JV. Expression of the chemokines MIP-1α, MCP-1 and RANTES in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2001;42:1547–52. [PubMed] [Google Scholar]
- 38.Foxman EF, Zhang MF, Hurst SD, et al. Inflammatory mediators in uveitis: differential induction of cytokines and chemokines in Th1- versus Th2-mediated ocular inflammation. J Immunol. 2002;168:2483–92. doi: 10.4049/jimmunol.168.5.2483. [DOI] [PubMed] [Google Scholar]
- 39.Crane IJ, Xu H, Wallace C, et al. Involvement of CCR5 in the passage of Th1-type cells across the blood–retina barrier in experimental autoimmune uveitis. J Leukoc Biol. 2006;79:435–43. doi: 10.1189/jlb.0305130. [DOI] [PubMed] [Google Scholar]
- 40.Crane IJ, Xu H, Manivannan A, et al. Effect of anti-macrophage inflammatory protein-1α on leukocyte trafficking and disease progression in experimental autoimmune uveoretinitis. Eur J Immunol. 2003;33:402–10. doi: 10.1002/immu.200310014. [DOI] [PubMed] [Google Scholar]
- 41.Rao NA, Kimoto T, Zamir E, et al. Pathogenic role of retinal microglia in experimental uveoretinitis. Invest Ophthalmol Vis Sci. 2003;44:22–31. doi: 10.1167/iovs.02-0199. [DOI] [PubMed] [Google Scholar]
- 42.Brockhaus J, Moller T, Kettenmann H. Phagocytozing ameboid microglial cells studied in a mouse corpus callosum slice preparation. Glia. 1996;16:81–90. doi: 10.1002/(SICI)1098-1136(199601)16:1<81::AID-GLIA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 43.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 44.Silverman MD, Zamora DO, Pan YZ, et al. Constitutive and inflammatory mediator-regulated fractalkine expression in human ocular tissues and cultured cells. Invest Ophthalmol Vis Sci. 2003;44:1608–15. doi: 10.1167/iovs.02-0233. [DOI] [PubMed] [Google Scholar]
- 45.Huang D, Shi FD, Jung S, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- 46.Campbell JJ, Qin SX, Unutmaz D, et al. Unique subpopulations of CD56(+) NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol. 2001;166:6477–82. doi: 10.4049/jimmunol.166.11.6477. [DOI] [PubMed] [Google Scholar]
- 47.Kitaichi N, Kotake S, Morohashi T, Onoe K, Ohno S, Taylor AW. Diminution of experimental autoimmune uveoretinitis (EAU) in mice depleted of NK cells. J Leukoc Biol. 2002;72:1117–21. [PubMed] [Google Scholar]