

Abstract
To develop into committed T helper type 1 (Th1) cells, naive CD4+ T cells not only need to acquire the capacity to produce interferon-γ (IFN-γ), but they also need to gain the ability to silence their interleukin-4 (IL-4) -producing potential. How Th1 cells silence their Th2 cytokine-producing potential is an important yet unresolved issue in Th1 immunity. We found that a lack of IL-4 stimulation was not sufficient to silence the IL-4-producing potential in activated CD4+ T cells and that Th1-promoting factor was required. Although it has been shown that T-bet is a crucial factor in suppressing Il4 gene expression, it is unclear whether a continuous presence of T-bet is required to silence the Il4 gene in Th1 cells. To address this problem, we used an inducible form of T-bet – a T-bet-oestrogen receptor fusion molecule (T-bet-ER). We found that the activation of T-bet during primary or secondary culture was sufficient to silence IL-4-producing potential. On the other hand, the inactivation of T-bet after naïve CD4+ T cells had differentiated into Th1 cells resulted in derepression of Il4 gene transcription. Additionally, we found that T-bet is required to maintain Ifng expression. Our data demonstrate that the continuous expression of T-bet is required for Th1 cells to silence their IL-4-producing potential.
Keywords: cytokine, gene regulation, signal transduction, T helper type 1/2
Introduction
T helper type 1 (Th1) immunity is of critical importance in protecting against bacterial and viral infections and in causing tissue damage in autoimmune diseases.1–3 To develop the most effective Th1 immunity, naïve CD4+ T cells, after encountering an antigen, must acquire the capacity to produce interferon-γ (IFN-γ) and the ability to silence Th2 cytokine-producing potential. Indeed, failure to silence the interleukin-4 (IL-4) -producing potential in Th1 cells has been shown to result in ineffective protection against Leishmania major infection.4
Although much has been learned about how Th1 and Th2 cells transcribe their cytokine genes, relatively little is known about how Th1 cells silence their potential to differentiate into Th2 cells. Developing Th1 cells can be induced to become Th2 cells, whereas committed Th1 cells lose this ability.5–7 This indicates that the IL-4-producing potential in naïve CD4+ T cells is gradually lost as naïve CD4+ T cells differentiate into committed Th1 cells. Committed Th1 cells express high levels of T-bet, a Th1 master transcription factor, but not GATA3, a Th2 master transcription factor.8–14 Both the IFN-γ–signal transducer and activator of transcription 1 (STAT1) and the IL-12–STAT4 pathways are important in inducing IFN-γ-producing capacity in Th1 cells.15–19 We reported that Th1 cells, which lacked IFN-γ signalling, failed to silence their Il4 gene activity.20 We further showed that IFN-γ suppressed the Il4 gene in Th1 cells, in part, by suppressing the recruitment of STAT6 to the IL-4 receptor-signalling complex.21
Recently, an Il4 silencer region in the mouse Il4 gene has been uncovered.22,23 It is reported that T-bet and Runx3 are recruited into this silencer region to silence the Il4 gene in developing Th1 cells.24,25 However, despite these findings, whether or not the continued presence of T-bet is required to silence the Il4 gene in Th1 cells remains unclear. In this study, we demonstrated that continuous T-bet expression was required for silencing the IL-4-producing potential of Th1 cells.
Materials and methods
Animals and cell cultures
C57BL/6 mice and BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME). T-bet-deficient (Tbx21−/−) mice on a C57BL/6 background9 were purchased from The Jackson Laboratory and maintained in the animal facility of the National Jewish Medical and Research Center (Denver, CO). Naïve CD4+ T cells were purified from the spleen and lymph nodes of various strains of mice using the CD4+ CD62L+ T-cell isolation kit (Miltenyi Biotec Inc., Auburn, CA). Purified naïve CD4+ T cells (0·2 × 106) were stimulated with irradiated T-cell-depleted antigen-presenting cells (0·8 × 106) in 2 ml complete RPMI-1640 medium supplemented with IL-2 (30 U/ml) and anti-CD3 (2C11, 3 μg/ml) and anti-CD28 antibody (3 μg/ml) for 3 days and then expanded with IL-2 (30 U/ml) for a further 8 days. Typically, for Th1 cell priming, IL-12 (10 ng/ml; BD-PharMingen, San Diego, CA) and anti-IL-4 antibody (11B11, 10 μg/ml) were added; for Th2 cell priming, IL-4 (5 ng/ml; BD-PharMingen), anti-IL-12 (10 μg/ml), and anti-IFN-γ (10 μg/ml) antibodies were added (referred to as the Th2-inducing conditions). In the control group, anti-IL-4 (10 μg/ml), anti-IL-12 (10 μg/ml) and anti-IFN-γ (10 μg/ml) were added (referred to the neutralized conditions). On day 11, the primed cells were washed and reprimed under the same conditions or switched to Th2-inducing conditions for an additional 5 days. All animal protocols were approved by the Institutional Animal Care and Users Committees of Loyola University Chicago and the National Jewish Medical and Research Center.
Intracellular staining
Primed cells were stimulated with phorbol 12-myristate 13-acetate (PMA; 50 ng/ml) and ionomycin (1 μm) in the presence of 2 μm monensin (Calbiochem, La Jolla, CA), as described previously.20 After 6 hr of stimulation, cells were fixed with 4% paraformaldehyde and stained with allophycocyanin-labelled anti-CD4, fluorescein isothiocyanate (FITC)-labelled anti-IFN-γ and phycoerythrin-labelled anti-IL-4 (11B11) monoclonal antibodies (BD-PharMingen), as described previously.15 Samples were collected using FACScan or FACScalibur (Becton-Dickinson, Franklin Lakes, NJ) and analysed using flowjo software (Tree Star Inc., Ashland, OR). All fluorescence-activated cell sorting (FACS) data were analysed using a CD4+ gate and dead cells were excluded from FACS data using a low forward scatter.
Enzyme-linked immunosorbent assay
At the end of cultures, T cells were washed and stimulated with PMA (50 ng/ml) and ionomycin (1 μm) at a concentration of 106 cells/1 ml of complete medium overnight. The IL-4 protein concentration in the supernatants was measured by using commercial enzyme-linked immunosorbent assay detection kits (BD-PharMingen).
Real-time polymerase chain reaction
Total RNA was isolated with STAT60 (Tel-test) and complementary DNA was synthesized by reverse transcription. The amounts of T-bet messenger RNA (mRNA) were quantified by real-time polymerase chain reaction (PCR) using specific primers and SYBR® Green PCR master mix (Applied Biosystems, University Park, IL). The amount of mRNA was expressed as an amount relative to that of a house keeping gene, Hprt1. Relative expression = 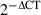 , where
, where 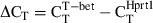 . The primers used were as follows: T-bet forward: 5′-CCTGTTGTGGTCCAAGTT-3′, T-bet reverse, 5′-TTTCCACACTGCACCCACTT-3′; Hprt1 forward, 5′-CTCATGGACTGATTATGGACAGGAC-3′, and Hprt1 reverse, 5′-GCAGGTCAGCAAAGA ACTTATAGCC-3′.
. The primers used were as follows: T-bet forward: 5′-CCTGTTGTGGTCCAAGTT-3′, T-bet reverse, 5′-TTTCCACACTGCACCCACTT-3′; Hprt1 forward, 5′-CTCATGGACTGATTATGGACAGGAC-3′, and Hprt1 reverse, 5′-GCAGGTCAGCAAAGA ACTTATAGCC-3′.
Retroviral infection
The murine stem cell virus-based retroviral T-bet construct was provided by Dr Laurie H. Glimcher of Harvard Medical School (Boston, MA). The T-bet-ER construct was made by Dr Hagman’s laboratory and characterized by Dr Gapin’s laboratory at the National Jewish Medical and Research Center (Denver, CO).26 Retroviral plasmids were cotransfected into Phoenix cells (G. Nolan, University of Stanford) together with the pCL-Eco retroviral packaging vector using LipofectAMINE 2000 (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. The viral supernatants were harvested 2 days after transfection and filtered through a 0·45-μm filter. CD4+ T cells were stimulated with anti-CD3 (3 μg/ml), anti-CD28 (3 μg/ml), IL-2 (30 U/ml), and irradiated antigen-presenting cells in the presence of anti-IL-4 (10 μg/ml), anti-IL-12 (10 μg/ml) and anti-IFN-γ (10 μg/ml) (the neutralized conditions) for 24 hr. The activated cells (1 × 106) were centrifuged at 6000 g for 90 min at 26° in the presence of 1 ml viral supernatant and polybrene (8 μg/ml; Millipore, Billerica, MA). The infected cells were cultured under the neutralized conditions for the indicated time period before they were subjected to Th2-inducing conditions. For T-bet-ER infection, the infected cells were expanded with IL-2 for 48 hr. Two days after the initial infection, the infected cells were cultured in complete RPMI-1640 medium (without phenol red) containing different dosages of 4-hydroxytamoxifen (4HT; Calbiochem) for nine more days. The resultant cells were washed and restimulated for an additional 5 days under either the same conditions or Th2-inducing conditions in the presence or absence of 4HT.26
Statistical analysis
All of the error bars in this report represent the standard deviation. The difference between two samples was analysed using Student’s t-test.
Results
A lack of IL-4 stimulation is not sufficient to silence the IL-4-producing potential in activated CD4+ T cells
There were two possibilities that might have led to the silencing of the IL-4-producing potential of Th1 cells. Initially, we addressed the possibility that the lack of IL-4 signalling and the absence of Th1-inducing factors could be sufficient to silence the IL-4-producing potential. We found that when the Th1- and Th2-inducing factors were neutralized, CD4+ T cells retained the potential to fully differentiate into IL-4-producing cells (comparable to naïve CD4+ T cells), whereas CD4+ T cells primed with Th1-inducing conditions lost such potential (Fig. 1a,b). These data demonstrate that a simple lack of IL-4 signalling is not sufficient to shut down the IL-4-producing potential and Th1-promoting factor is required to silence the Il4 gene.
Figure 1.

A lack of interleukin-4 (IL-4) stimulation is not sufficient to silence the Il4 gene in activated CD4+ T cells. (a) Naive CD4+ T cells from C57BL/6 mice were stimulated with anti-CD3/anti-CD28 and antigen presenting cells under the neutralized conditions or T helper type 1 (Th1)-inducing conditions as indicated under the primary conditions. On day 11, after the initial priming, the primed cells were washed and reprimed under the same conditions or switched to Th2-inducing conditions (indicated as secondary conditions) for an additional 5 days. At the end of culture, the cells were stimulated with phorbol 12-myristate 13-acetate and ionomycin (P&I) for 6 hr and analysed by intracellular staining using allophycocyanin-labelled anti-CD4, phycoerythrin-labelled anti-IL-4 and fluorescein isothiocyanate-labelled anti-interferon-γ (IFN-γ) antibodies. The numbers inside the FACS plots indicate the positive percentage within gated CD4+ T cells. (b) The resultant cells were also stimulated with P&I overnight at a concentration of 106 cells in 1 ml complete medium. The IL-4 protein concentration in the supernatants was measured by enzyme-linked immunosorbent assay. The error bars represent SD derived from triplicate measurements. These data are representative of three independent experiments with similar results. Neu–Neu means that cells were treated under neutralized conditions both in the primary and secondary cultures; Th1–Th1 means that cells were treated under Th1-inducing conditions both in the primary and secondary cultures; Neu–Th2 means that cells were treated under neutralized conditions in the primary culture and under Th2-inducing conditions in the secondary culture; and Th1–Th2 means that cells were treated under Th1-inducing conditions in the primary and under Th2-inducing conditions in the secondary cultures.
T-bet is sufficient to silence the IL-4-producing potential in Th1 cells
To assess the role of T-bet in silencing the IL-4-producing potential of Th1 cells, we primed naïve CD4+ T cells from wild-type (WT) and Tbx21−/− mice with the neutralized conditions or Th1 priming conditions for 11 days and then tested the commitment status of the primed cells by providing them with Th2-inducing conditions. We found that in the absence of T-bet, the IL-12-primed CD4+ T cells fully retained their potential to develop into IL-4-producing cells, whereas the WT control cells lost the potential to differentiate into IL-4-producing cells as measured by intracellular staining (Fig. 2a) and by enzyme-linked immunosorbent assay (Fig. 2b). Similar results were obtained when the resultant cells were stimulated with anti-CD3 and anti-CD28 antibodies plus irradiated antigen-producing cells or by immobilized anti-CD3 and anti-CD28 antibodies (data not shown). On the other hand, T-bet expression was maintained in WT Th1 cells even after they were switched to Th2-inducing conditions (Fig. 2c). These results demonstrate that T-bet is required for Th1 cells and suggest that T-bet might be required for the ‘switched’ Th1 cells to silence their IL-4-producing potential.
Figure 2.

T-bet is required for T helper type 1 (Th1) cells to silence their interleukin-4 (IL-4)-producing potential. (a) Naive CD4+ T cells from wild-type (WT) control and Tbx21−/− mice were stimulated under the neutralized conditions, or Th1 conditions before they were reprimed under the same conditions or switched to Th2-inducing conditions. IL-4 and interferon-γ (IFN-γ) proteins were measured by either intracellular staining or by enzyme-linked immunosorbent assay (b). The error bars represent SD derived from triplicate measurements. (c) Total RNA was isolated from the resultant cells and used for complementary DNA synthesis. The amount of T-bet messenger RNA was quantified by real-time polymerase chain reaction and expressed as the amount relative to that of Hprt1. The error bars indicate the standard deviation of duplicate measurements. These data are representative of three independent experiments with similar results.
Previous work that has focused on investigating the role of T-bet in suppressing the Il4 gene in Th2 cells has produced conflicting results. Glimcher and colleagues reported that T-bet overexpression achieved a 70% suppression of IL-4 expression,8,9 whereas Murphy and colleagues failed to observe any suppression.27 Recently, Rao and colleagues demonstrated that T-bet is not sufficient to suppress the Il4 gene in the presence of Th2-inducing factors; rather, it co-operates with Runx3 to silence the Il4 gene.24 On the other hand, Naoe et al.25 reported that Cbfβ, but not Runx3, is required for silencing the Il4 gene. To determine whether or not T-bet is sufficient to silence the IL-4-producing potential in Th1 cells, we overexpressed T-bet or green fluorescent protein (GFP) in WT and Tbx21−/− CD4+ T cells and found that the overexpression of T-bet, but not GFP, completely suppressed the IL-4-producing potential (Fig. 3). These data together demonstrate that T-bet is necessary and sufficient to silence the IL-4-producing potential of Th1 cells.
Figure 3.

T-bet over-expression suppresses the interleukin-4(IL-4)-producing potential. Naive CD4+ T cells from wild-type (WT) or Tbx21−/− mice were stimulated under the neutralized conditions and infected with green fluorescent protein (GFP) or T-bet retrovirus (RV). After infection, cells were cultured for 9 days before they were reprimed under T helper type 2 (Th2)-inducing conditions. IL-4 expression was measured by intracellular staining and infected cells (GFP+) are shown. The percentages in the fluorescence-activated cell sorting plots were calculated by dividing the number of positive events by the number of GFP+ cells analysed. These data are representative of four independent experiments with similar results.
The continuous expression of T-bet is required to silence the IL-4-producing potential of Th1 cells
To determine whether the continuous expression of T-bet is necessary for maintaining the silenced state of the Il4 gene, we used an inducible form of T-bet – the T-bet-oestrogen receptor fusion molecule (T-bet-ER) that can translocate inside the nucleus and become activated when it binds to tamoxifen26– to examine this issue. Wild-type CD4+ T cells expressing T-bet-ER were treated with 4HT for 9 days. The resultant cells were then washed and recultured in the presence or absence of 4HT for an additional 5 days. In the presence of 4HT, IL-4 protein expression in WT CD4+ T cells was no longer induced by Th2-inducing conditions, whereas in the absence of 4HT, IL-4 protein expression was induced by Th2-inducing conditions (Fig. 4a). Similar results were obtained for WT CD4+ expressing T-bet-ER T cells that were treated with 4HT for 5 or 11 days before they were exposed to Th2-inducing conditions (data not shown). The ability of T-bet-ER-expressing WT CD4+ T cells to express IL-4 after they were exposed to Th2-inducing conditions was inversely correlated with the dosage of 4HT added during the priming phase (Fig. 4b).
Figure 4.

Continuous T-bet expression is required to silence the interleukin-4 (IL-4)-producing potential in T helper type 1 (Th1) cells. (a) Naïve CD4+ T cells of wild-type (WT) mice were stimulated under the neutralized conditions and infected with green fluorescent protein (GFP), T-bet or a T-bet-oestrogen receptor fusion molecule (T-bet-ER) retrovirus. The GFP- and T-bet-infected cells were cultured for 9 days before they underwent Th2-inducing conditions for a period of 5 days. The GFP- and T-bet-ER-infected cells were cultured in the presence of 50 nm 4-hydroxytamoxifen (4HT) for 9 days before they were reprimed under Th2 conditions in the absence or presence of 50 nm of 4HT. Interferon-γ (IFN-γ) and IL-4 expression were measured by intracellular staining and GFP+ cells are shown. (b) Naïve CD4+ T cells were used and the priming and repriming conditions were the same as those described in (a) except for the doses of 4HT. The percentages of IL-4-positive cells in the infected cells (GFP+ cells) are shown. (c,d) Naïve CD4+ T cells from Tbx21−/− mice were stimulated and infected with GFP, T-bet or T-bet-ER retrovirus. The T-bet-ER-infected cells were then cultured in the absence or presence of different doses of 4HT as indicated for 9 days. The resultant cells were then reprimed under Th2 conditions. The 4HT treatments in the secondary priming conditions are indicated. The percentages of IL-4-positive cells in the infected cells (GFP+ cells) are shown. The error bars represent the standard deviation derived from triplicate measurements. The presented data are representative of three independent experiments.
Although WT CD4+ T cells express low levels of T-bet, we cannot exclude the possibility that the endogenous T-bet expression could be up-regulated by T-bet-ER, which could then suppress the Il4 gene. Additionally, the endogenous T-bet, after the inducible T-bet was removed, might also interfere with the accuracy of the measure of the T-bet dosage effect. To address these potential problems, we expressed T-bet-ER in Tbx21−/− CD4+ T cells and showed that induced T-bet expression under the secondary priming conditions (e.g. no T-bet expression in the primary priming conditions) suppressed the IL-4-producing potential of CD4+ T cells (Fig. 4c,d), indicating that the silencing function mediated by T-bet is not restricted to an initial developmental window.
To determine whether T-bet expression induced during the primary culture can induce an irreversible silencing effect, we treated the T-bet-ER-expressing Tbx21−/− CD4+ T cells with 4HT during the primary priming phase and then reprimed the treated cells in the presence or absence of 4HT. In the presence of 4HT, the T-bet-ER-expressing Tbx21−/− CD4+ T cells failed to develop into IL-4-producing cells. In contrast, in the absence of 4HT, the T-bet-ER-expressing Tbx21−/− CD4+ T cells fully regained their ability to differentiate into IL-4-producing cells and showed no residual suppression effect as the result of the T-bet expression induced during the primary priming phase (even with doses of 4HT as high as 250 nm) (Fig. 4c,d). Additionally, we found that T-bet was sufficient to induce IFN-γ. We also observed that the removal of 4HT in the secondary conditions resulted in increased IL-4 expression and greatly reduced IFN-γ expression (Fig. 4c). These data demonstrate that T-bet is required to maintain both IL-4 silencing and IFN-γ expression. STAT5-ER was used as a control and we did not find that the ER fusion protein silenced the IL-4-producing potential of CD4+ T cell (data not shown). Together, these data demonstrate that T-bet silences the Il4 gene and that continuous T-bet expression is essential for maintaining the silenced state of the Il4 gene.
Discussion
How Th1 cells silence their Th2 cytokine-producing potential is an important but not yet resolved issue in Th1 immunity. Here we define the IL-4-producing potential in Th1 cells as the ability to differentiate into Th2 cells when Th2-inducing conditions are provided. Our experiments were conducted in conditions different from those of published works. Our investigation focused on the role of T-bet in silencing/suppressing the IL-4-producing potential in Th1 cells (e.g. Th1 cell commitment). Under Th1 conditions, T-bet should not have to deal with an ‘opened’Il4 locus or to face competition from Th2-specific transcription factors, such as GATA3. The difference in culture conditions might explain why T-bet over-expression alone is largely sufficient to silence the IL-4-producing potential in WT or Tbx21−/− Th1 cells.
Our data show that continuous T-bet expression is required for maintaining the silenced state of the Il4 gene and support a mechanism of action, in which T-bet directly binds to the regulatory regions of the Il4 gene; this binding might then form a repressive complex. In the absence of T-bet the repressive complex would fall apart. This model is consistent with a recent report, in which authors show that T-bet binds to the ‘silencer’ region of the Il4 gene and recruits Runx3 and Cbfβ.24,25 Determining whether the Il4 silencer region is the only region to which T-bet binds requires further investigation. Alternatively, T-bet might silence the IL-4-producing potential by suppressing GATA3 expression. In fact, it has been shown that T-bet suppresses GATA3 mRNA expression.28 We previously reported that committed Th1 cells failed to up-regulate their GATA3 mRNA even when they were treated under Th2-inducing conditions.20 The present study shows that T-bet expression was maintained in WT Th1 cells even after they were switched to Th2-inducing conditions. Together, these data suggest that suppressing GATA3 expression might be one mechanism by which T-bet silences the IL-4-producing potential.
If the proposed model were true, then Th1 cells must find a way to constantly maintain high levels of T-bet expression. Previously, we have shown that in the absence of IFN-γ signalling, Th1 cells failed to maintain an inheritable pattern of Ifng and Il4 gene expression.15 Together with the current findings, we formally demonstrate that committed Th1 cells might maintain their inheritable patterns of cytokine gene expression by a constant, automatic feedback mechanism, in which IFN-γ, produced by Th1 cells, maintains high levels of T-bet expression, which silences the Il4 gene.
A repressive chromatin-remodelling enzyme that participates in silencing the Il4 gene remains unidentified. Bix and colleagues showed that a repressive modification of histone 3 molecules (H3 trimethylation at Lysine 27) was detected in Th1 cells that had gone through two rounds of differentiation under Th1-inducing conditions.29,30 This delayed modification might not reflect the initial T-bet-mediated formation of the repressive chromatin-remodelling complex. They also reported that they did not detect another repressive modification (trimethylation modification of H3 at Lysine 9) at the Il4 locus.29 It would be of great future interest to investigate which repressive chromatin-remodelling enzyme participates in silencing the Il4 gene and how T-bet recruits the putative repressive enzyme into the Il4 gene.
Additionally, our data demonstrate that T-bet is required to maintain IFN-γ expression. However, we attempted to culture T-bet-ER-expressing CD4+ T cells in the presence of 4HT for longer than 11 days before they were subjected to Th2-inducing conditions and failed to obtain sufficient numbers of cells for the subsequent analyses because of antigen-induced cell death. Consequently, our data do not rule out the possibility that Th1 cells, after undergoing repeated activation and cytokine stimulation, might not require T-bet for maintaining IFN-γ expression.31 Our work provides knowledge on how external silencing stimuli connect to silencing actions that occur inside the nucleolus and contributes to a fuller understanding of the development of Th1 immunity. This provides useful information for designing immune interventions to reverse autoimmunity mediated by a dominant Th1 immune response.
Acknowledgments
This work was supported by the National Institutes of Health (RO1 AI48568 and RO1 AI068083) and National Jewish Health. We would like to thank Drs Laurent Gapin, Jennifer Matsuda and James Hagman for providing us with the T-bet-ER construct and for their help in setting up experiments using this construct and we thank Dr Laurie H. Glimcher of Harvard Medical School for providing us with the retroviral T-bet construct. We would also like to thank Drs Laurent Gapin, Jennifer Matsuda, Binfeng Lu and Hongwei Chu for critically reading our manuscript, Leah Cho and J.D. Williams for assistance in manuscript preparation, and Dr Manuel Diaz’s support for Zan Huang’s graduate study. We are grateful to Pat Simms and Josh Looms for their assistance with the FACS analysis and to Tom Startz for technical assistance. The authors have no conflicting financial interests.
Glossary
Abbreviations:
- 4HT
4-hydroxytamoxifen
- FACS
fluorescence-activated cell sorting
- IFN-γ
interferon-γ
- IL-4
interleukin-4
- mRNA
messenger RNA
- Neu
neutralized conditions
- STAT
signal transducer and activator of transcription
- T-bet
T box transcription factor (Tbx)
- T-bet-ER
a T-bet-oestrogen receptor fusion molecule
- Th1
T helper type 1
- WT
wild-type
Disclosure
The authors have no conflicting financial interests.
References
- 1.Del Prete G, Romagnani S. The role of TH1 and TH2 subsets in human infectious diseases. Trends Microbiol. 1994;2:4–6. doi: 10.1016/0966-842x(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 2.Liblau RS, Singer SM, McDevitt HO. Th1 and Th2 CD4+ T cells in the pathogenesis of organ-specific autoimmune diseases. Immunol Today. 1995;16:34–8. doi: 10.1016/0167-5699(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 3.Moss RB, Moll T, El-Kalay M, Kohne C, Soo Hoo W, Encinas J, Carlo DJ. Th1/Th2 cells in inflammatory disease states: therapeutic implications. Expert Opin Biol Ther. 2004;4:1887–96. doi: 10.1517/14712598.4.12.1887. [DOI] [PubMed] [Google Scholar]
- 4.Ansel KM, Greenwald RJ, Agarwal S, et al. Deletion of a conserved Il4 silencer impairs T helper type 1-mediated immunity. Nat Immunol. 2004;5:1251–9. doi: 10.1038/ni1135. [DOI] [PubMed] [Google Scholar]
- 5.Huang H, Paul WE. Impaired interleukin 4 signaling in T helper type 1 cells. J Exp Med. 1998;187:1305–13. doi: 10.1084/jem.187.8.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy E, Shibuya K, Hosken N, Openshaw P, Maino V, Davis K, Murphy K, O’Garra A. Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J Exp Med. 1996;183:901–13. doi: 10.1084/jem.183.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–56. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 8.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 9.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–42. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 10.Mullen AC, High FA, Hutchins AS, et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 2001;292:1907–10. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 11.Grogan JL, Locksley RM. T helper cell differentiation: on again, off again. Curr Opin Immunol. 2002;14:366–72. doi: 10.1016/s0952-7915(02)00340-0. [DOI] [PubMed] [Google Scholar]
- 12.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–58. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 13.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–96. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 14.Zhu J, Min B, Hu-Li J, et al. Conditional deletion of Gata3 shows its essential function in T(H)1–T(H)2 responses. Nat Immunol. 2004;5:1157–65. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–9. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 16.Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–73. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–7. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 18.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 19.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Apilado R, Coleman J, Ben-Sasson S, Tsang S, Hu-Li J, Paul WE, Huang H. Interferon gamma stabilizes the T helper cell type 1 phenotype. J Exp Med. 2001;194:165–72. doi: 10.1084/jem.194.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Z, Xin J, Coleman J, Huang H. IFN-gamma suppresses STAT6 phosphorylation by inhibiting its recruitment to the IL-4 receptor. J Immunol. 2005;174:1332–7. doi: 10.4049/jimmunol.174.3.1332. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal S, Rao A. Modulation of chromatin structure regulates cytokine gene expression during T cell differentiation. Immunity. 1998;9:765–75. doi: 10.1016/s1074-7613(00)80642-1. [DOI] [PubMed] [Google Scholar]
- 23.Lee GR, Fields PE, Flavell RA. Regulation of IL-4 gene expression by distal regulatory elements and GATA-3 at the chromatin level. Immunity. 2001;14:447–59. doi: 10.1016/s1074-7613(01)00125-x. [DOI] [PubMed] [Google Scholar]
- 24.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–53. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 25.Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F, Littman DR, Taniuchi I. Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med. 2007;204:1749–55. doi: 10.1084/jem.20062456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuda JL, George TC, Hagman J, Gapin L. Temporal dissection of T-bet functions. J Immunol. 2007;178:3457–65. doi: 10.4049/jimmunol.178.6.3457. [DOI] [PubMed] [Google Scholar]
- 27.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–57. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 28.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O’Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–66. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koyanagi M, Baguet A, Martens J, Margueron R, Jenuwein T, Bix M. EZH2 and histone 3 trimethyl lysine 27 associated with Il4 and Il13 gene silencing in Th1 cells. J Biol Chem. 2005;280:31470–7. doi: 10.1074/jbc.M504766200. [DOI] [PubMed] [Google Scholar]
- 30.Baguet A, Bix M. Chromatin landscape dynamics of the Il4-Il13 locus during T helper 1 and 2 development. Proc Natl Acad Sci USA. 2004;101:11410–5. doi: 10.1073/pnas.0403334101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martins GA, Hutchins AS, Reiner SL. Transcriptional activators of helper T cell fate are required for establishment but not maintenance of signature cytokine expression. J Immunol. 2005;175:5981–5. doi: 10.4049/jimmunol.175.9.5981. [DOI] [PubMed] [Google Scholar]