

Abstract
Granulocyte colony-stimulating factor (G-CSF) is used commonly in an attempt to reduce the duration of neutropenia and hospitalization in patients undergoing chemotherapy and to obtain hematopoietic stem cells (HSC) for transplantation applications. Despite the relative safety of administration of G-CSF in most individuals, including subjects with sickle cell trait, severe and life-threatening complications have been reported when used in individuals with sickle cell disease (SCD), including those who were asymptomatic and undiagnosed prior. The administration of G-CSF has now been reported in a total of 11 individuals with SCD. Seven developed severe adverse events including vaso-occlusive episodes, acute chest syndrome, multi-organ system failure, and death. Precautions including minimizing the peak white blood cell count, dividing or reducing the G-CSF dose, and red blood cell transfusions to reduce HbS levels have been employed with no consistent benefit. These reported data indicate that administration of G-CSF in individuals with SCD should be undertaken only in the absence of alternatives, and after full disclosure of the risks involved. Unless further data demonstrate safety, routine usage of G-CSF in individuals with SCD should be avoided.
Keywords: SCD, G-CSF, hematopoietic stem cell mobilization, complications, multi-organ failure, pain crisis
Introduction
Sickle cell disease (SCD) is caused by a single point mutation at codon 6 of the β-globin gene, alone or in combination with other genetic abnormalities, leading to abnormal hemoglobin polymerization, cell sickling in areas of low oxygen tension, chronic hemolysis, and recurrent vaso-occlusive crises. G-CSF, cytokine essential for myeloid differentiation and proliferation, is routinely used for chemotherapy-induced neutropenia (CIN) and for hematopoietic stem cell (HSC) mobilization for transplantation applications. Though generally safe when used in cancer patients and healthy donors, unique complications have been described in individuals with SCD that include severe vaso-occlusive crises (VOC), multiorgan failure, and death. We summarize the hitherto reported experience of G-CSF usage in 11individuals with SCD and further caution against its routine usage in such individuals.
Case reports of G-CSF use in SCD
After an attempt to collect autologous HSCs for gene therapy studies from an individual with SCD through G-CSF mobilization, Abboud and colleagues described severe pain crisis and concluded that this approach was not safe [1]. In a second case report, an individual with hemoglobin SC disease was described who served as an allogeneic donor for a sibling undergoing transplantation for a malignant disease [2]. After extensive deliberations with the Institutional Review Board (IRB), this subject was mobilized with G-CSF and in the process succumbed of multi-organ system failure, despite disease so mild it was not known prior to the donor screening process [2]. Now, ten years after the first report, there are 11 total individuals with SCD who have received G-CSF and have been reported in the literature (summarized in Table 1). Age ranged from 18 to 58 years. The indications for G-CSF administration were prophylaxis for chemotherapy-induced neutropenia (CIN) in two individuals, procurement of allogeneic HSCs in one individual and autologous HSCs in nine individuals (one individual received G-CSF for two separate indications). G-CSF was administered subcutaneously, with doses ranging from 2.5 mcg/kg per day to 16 mcg/kg per day, and the duration ranged from 3 to 20 days. The dosing schedule was twice daily in five individuals and once daily in the remainder.
Table I.
Details of individuals with sickle cell disease who received G-CSF.
| Reference, Year |
SCD Type/ Age |
Indication | Peak WBC 109/L |
% HbS/ % HbC |
G-CSF dose; Duration; Time between initiation to symptoms |
Precautions | Complications | Hospitalized? |
|---|---|---|---|---|---|---|---|---|
| Abboud et al., 1998 [1] | HbSS/34 years | Gene therapy study | 63.4 | NR | 2.5mcg/kg/day; 3 days; 3 days | None | VOC, ACS | Yes |
| Wei & Grigg, 2001[3] | HbS-β+thal/58 years | Stage II invasive ductal breast cancer | 6.4 | NR | 480mcg/day; 4 days; 4 days | None | Multi-organ failure, survived | Yes |
| Adler et al., 2001[2] | HbSC/47 years | Donor for sibling with CML | 70.9 | NR | 400mcg/m2/dayb; 4 days; 4 days | None | Multi-organ failure and death | Yes |
| Onitilo et al., 2003 [6] | HbSS/40 years | Diffuse large B cell lymphoma | NR | NR | 10mcg/kg/day; 20 days; N/A | Hyper-transfusionc | None | No |
| Kamble et al.a, 2006[4] | HbSC/22 years | Stage IV large B cell lymphoma | NR | NR | NR | None | VOC | Yes |
| 18.7 | 6 | 5mcg/kg BID; 5 days; N/A | Exchange transfusion | None | No | |||
| Tormey et al., 2008 [7] | HbSC/45 years | Multiple myeloma | 69.6 | 10.2/9.4 | 16mcg/kg/day; 4 days; N/A | Erythrocytapheresis, normal saline | None | No |
| Rosenbaum et al., 2008 [5] | HbSC/56 years | Backup collection for allo-BMT | 53 | 46/47 | 5mcg/kg BID; 3 days; N/A | None | None | No |
| HbSS/40 years | Backup collection for allo-BMT | 41 | NR | 5mcg/kg BID; 3 days; 3 days | None | HTN, VOC | Yes | |
| HbSS/18 years | Backup collection for allo-BMT | 44.1 | 75 | 5mcg/kg BID; 3 days; N/A | None | None | No | |
| HbSS/23 years | Backup collection for allo-BMT | 105 | 25 | 5mcg/kg BID; NR; NR | None | VOC, HA | Yes | |
| HbSS/30 years | Recurrent Hodgkin’s disease | NR | 14.4 | 5mcg/kg daily; 3 days; 3 days | Transfusiond | Back/extremity pain | Yes |
Individual was given G-CSF to prevent chemotherapy-induced neutropenia and developed vaso-occlusive crises requiring hospitalization. Many months later, G-CSF was used to mobilize HSCs after he was prophylactically transfused with red cells and then tolerated G-CSF without complications.
Individual also received dexamethasone 10mg/m2/d starting 3 days after initiation of G-CSF
Individual was transfused to maintain Hct >40%
Individual was transfused to maintain HbS <30%
Abbreviations: ACS, acute chest syndrome; BMT, bone marrow transplant; CML, chronic myelogenous leukemia; HA, headaches; HbSC, compound heterozygous for hemoglobin S and C; HbSS, homozygous for hemoglobin S; HTN, hypertension; N/A, not applicable; NR, not reported; VOC, vaso-occlusive crises;
Of these reported individuals, two developed multi-organ failure resulting in death in one, and several months of hospitalization in the other [2, 3]. Five individuals experienced pain severe enough to require hospitalization and narcotic therapy [1, 4, 5]. One individual received G-CSF on two occasions; he experienced a VOC requiring hospitalization with prophylactic doses of G-CSF to prevent CIN, but tolerated subsequent G-CSF for autologous HSC mobilization without complications [4]. In total, 7 of the 11 individuals required hospitalization [1–5], while 4 others experienced no complications [5–7].
Plausible determinants of SCD-related G-CSF complications
SCD has been described as a chronic inflammatory state, with elevated leukocyte counts, increased serum levels of C-reactive protein and cytokines [8, 9], and a propensity for erythrocyte and platelet activation [10], all processes that might be exacerbated by G-CSF usage. A number of prospective clinical trials have demonstrated a correlation between leukocytosis and sickle-related complications including increased mortality [11–13]. Further, the beneficial effects of hydroxyurea have been attributed in part to lowering leukocyte counts [14–16]. G-CSF was detectable in five out of six samples of bronchial fluid from children with ACS, suggesting that endogenous G-CSF may facilitate the development of ACS [17]. Neutrophils from SCD patients display longer survival [18], increased activation and erythrocyte adhesion [19–22], and elevated production of reactive oxygen species [19]. Lastly, significant serum phosphate depletion associated with rapid leukocyte proliferation can lead to a reduction of red cell 2,3 diphosphoglycerate (DPG), resulting in tissue hypoxia and HbS polymerization within the red cell [23].
Attempted strategies to reduce SCD-related G-CSF complications in SCD
Reducing HbS concentrations to <30% in SCD patients has been demonstrated as effective secondary stroke prophylaxis [24, 25] and primary stroke prophylaxis in patients with elevated transcranial Doppler velocity [26]. This modality has been performed by some as a precautionary measure to prevent G-CSF associated complications in individuals with SCD. However, transfusing to a low HbS level in SCD patients has been incorrectly extrapolated as being equivalent to the clinical situation in individuals with sickle cell trait [5] when in fact there is a marked clinical difference between these 2 scenarios (Figure 1). Assays to quantitate HbS measure total protein levels after red blood cell lysis, and thus give only an average percentage of HbS among all cells. Individuals with sickle cell trait have 35–40% HbS in each red blood cell and have been mobilized with G-CSF without complications [27]. The presence of 35–40% HbS within each red cell of sickle cell trait subjects presents virtually no risk for sickling. In contrast, individuals with SCD have levels approaching 100% HbS within each red cell. Targeting overall levels similar to that seen in sickle cell trait by exchange red cell transfusion in individuals with SCD results in 35–40% of cells still containing close to 100% HbS, thus retaining their propensity to sickle. This paradigm is supported not only by our controlled clinical trial in which sickle cell trait subjects received G-CSF without complications [27], but also in vitro experiments demonstrating that the presence of HbA or HbF within sickle red cells improves HbS solubility and delays HbS polymerization dramatically, by a factor of 104 and 10 7, respectively [28].
Figure 1.
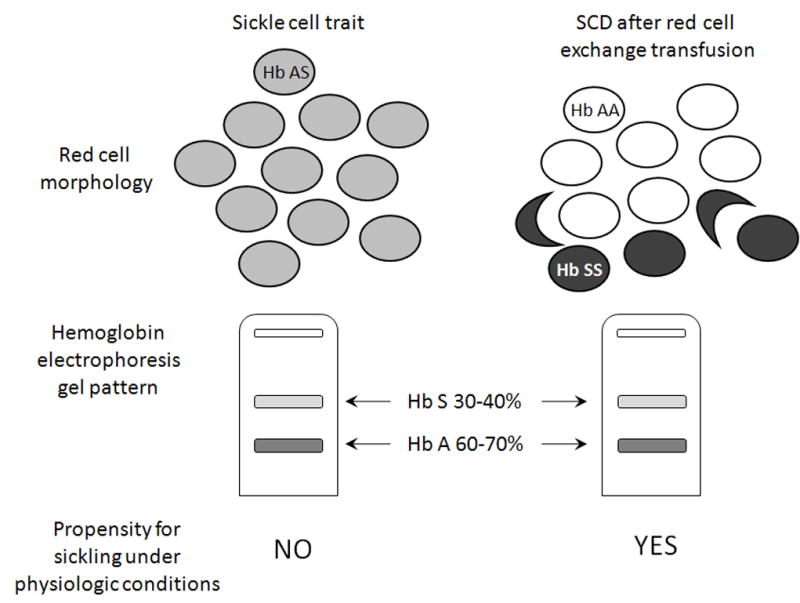
Depicted in the upper panel is the red blood cell morphology of an individual with sickle cell trait, and an individual with SCD after red cell exchange transfusion. The percentage of HbS is shown in grey-scale within each red cell from 0% in normal transfused red cells (Hb AA) to 30–40% in sickle cell trait red cells (Hb AS) to nearly 100% in SCD (Hb SS). Bottom panel, a hemoglobin electrophoresis is depicted demonstrating that in both scenarios, the overall percentage of HbS is identical, yet the propensity for sickling at physiologic conditions exists only among cells containing nearly 100% HbS.
Two patients with SCD underwent red cell exchange transfusion therapy to reduce the sickle hemoglobin (HbS) level to <30% and did not experience any complications (Table I). Two others received simple red cell transfusion therapy, and one had back and extremity pain similar to prior VOC. In total, 3 of 4 patients in the red cell transfusion group [4–7], and 2 of 8 patients in the no transfusion group [1–5] were free of adverse events (again, one patient received G-CSF on two occasions). However, there was no threshold HbS under which VOC was prevented, as a patient with a HbS as low as 14.4% experienced VOC requiring hospitalization, and another patient with a HbS level of 75% did not have complications (Table I).
Controlling peak leukocyte counts by divided or reduced dosing has also been proposed as a means to prevent SCD related complications with G-CSF usage. Peak leukocyte counts following G-CSF in these individuals ranged from 6.4 to 105 × 109/L. As one patient that developed multi-organ failure had a peak leukocyte count of only 6.4 × 109/L, and a G-CSF dose as low as 2.5 mcg/kg/day, and another developed symptoms when the WBC reached 60 × 109/L with twice daily dosing yet these did not recur when G-CSF was held and then restarted daily with a peak of 105 × 109/L, there is no apparent threshold leukocyte count or daily G-CSF dose which prevented the development of SCD related complications. Others have suggested the use of hydroxyurea to mitigate the risks of growth factor use in SCD [29]; however, no data are yet available in the literature to support this strategy.
Assessing the risks and benefits of G-CSF administration in SCD
When determining whether any intervention should be undertaken, careful consideration of the risks and benefits should be performed. Current approved indications for G-CSF usage include decreasing the incidence of infection in cancer patients receiving myelosuppressive chemotherapy, reducing the time to neutrophil recovery in patients receiving induction or consolidation chemotherapy for acute myeloid leukemia, reducing the duration of neutropenia in cancer patients receiving bone marrow transplantation, reducing the incidence and duration of sequelae of neutropenia in patients with severe chronic neutropenia, and collection of peripheral blood progenitor cells by apheresis [30–37]. Though hospitalization duration is reduced with G-CSF usage in patients undergoing chemotherapy, a reduction in overall mortality has not been demonstrated. In a recent meta-analysis of 17 randomized controlled trials, including 3,493 patients with solid tumors and lymphomas [38], G-CSF was demonstrated to decrease the incidence of febrile neutropenia from 39.5% to 22.4% (RR 0.54, p<0.0001) and early mortality from 5.7% to 3.4% (RR 0.6, p=0.002). However, decreased overall and event-free survival were not demonstrated. Another meta-analysis evaluated eight randomized controlled trials involving 1221 patients with malignancies who received antibiotics alone versus antibiotics plus G-CSF for treatment of established febrile neutropenia [39]. Length of hospitalization decreased in patients that received G-CSF (HR 0.63, p=0.0006), but there was again no difference in overall mortality between the two groups. Thus, in the setting of chemotherapy, the benefit of G-CSF is shorter hospitalization duration. The risks of G-CSF use in SCD patients include severe adverse reactions in a significant number of cases (7 out of 11); these complications led to a significantly longer overall hospitalization due to VOC and even death. Since these risks clearly outweigh the benefit of G-CSF use, the routine use of G-CSF in SCD should be avoided. This assessment is supported by the package insert of G-CSF, which states that “severe sickle cell crises, in some cases resulting in death, have been associated with the use of Neupogen® (G-CSF) in patients with sickle cell disorders. Only physicians qualified by specialized training or experience in the treatment of patients with sickle cell disorders should prescribe Neupogen® for such patients, and only after careful consideration of the potential risks and benefits” [40]. Further, the National Marrow Donor Program continues to maintain a policy against the collection of HSCs through G-CSF mobilization from subjects with not only SCD, but also sickle cell trait (personal communication Dr. Willis Navarro, Medical Director, NMDP).
Regarding the usage of G-CSF for peripheral blood progenitor cell collection, a risk/benefit assessment for individuals with SCD is not even necessary. The existence of a safe and viable alternative to G-CSF mobilization proves that its use in SCD, and the unnecessary exposure to the ensuing risks, are not justified. Indeed, multiple studies have compared outcomes between recipients of G-CSF-mobilized peripheral blood and harvested bone marrow as the allogeneic HSC source. While most studies have found no difference in the rates of graft versus host disease (GVHD), overall survival, and disease-free survival [41–43], in some settings, such as pediatric patients with severe aplastic anemia, bone marrow was found to be superior as a stem cell source due to an increased risk of GVHD and mortality with G-CSF-mobilized peripheral blood HSCs [44–46]. Autologous transplantation studies also demonstrate similar long-term outcomes with bone marrow as the HSC source [47]. With respect to gene therapy applications, we have previously demonstrated equivalent engraftment of genetically modified HSCs derived from bone marrow when compared to that from G-CSF mobilized peripheral blood in non-human primates [48]. Though often used in bone marrow harvesting, the safety of general anesthesia in SCD patients undergoing surgical procedures has been optimized over the last 3 decades, and the morbidity and mortality has been significantly reduced [49, 50]. Furthermore, bone marrow harvesting can be safely performed under conscious sedation and local anesthesia [51, 52] or spinal anesthesia [53]. Thus, bone marrow harvesting should be considered in any scenario when the risk of G-CSF mobilization in the donor is significant.
Another potential alternative to G-CSF mobilization is the CXCR4 antagonist, AMD-3100, which blocks the interaction between the CXCR4 receptor of HSC and the SDF-1 ligand on the bone marrow stroma, and releases HSCs into circulation allowing for collection by apheresis. However, AMD3100 also increases leukocyte counts, albeit to a lesser degree than G-CSF [54, 55], suggesting that similar related side effects might also be encountered.
Final thoughts and recommendations
Of the 11 reported individuals spanning the last 10 years, one had asymptomatic SCD that developed multi-organ failure and died within 36 hours of receiving 4 doses of G-CSF; another asymptomatic individual developed multi-organ failure after 4 doses that required prolonged hospitalization for months. Of the remaining 9 reported individuals, 5 individuals developed VOC severe enough to require hospitalization. Though reporting bias may account in part for the high complication rate reported thus far, the physiology of G-CSF administration in SCD patients, including leukocytosis, the propensity of sickle red cells for vaso-occlusion, and the chronic inflammatory state of patients with SCD all implicate a potentially hazardous role for G-CSF administration in patients with SCD. The reported patients and their clinical outcomes were heterogeneous, with no evidence of a “threshold” leukocyte count, G-CSF dose or administration schedule effect, or HbS percentage for which sickle-related complications could be anticipated or reliably prevented.
Unless further data demonstrate safety, we argue that the routine usage of G-CSF (i.e. for chemotherapy-induced neutropenia and for HSC mobilization) in individuals with SCD is not justified by a risk benefit assessment and therefore should be avoided. In the rare event in which an individual risk benefit assessment favors the use of G-CSF in SCD, one should only proceed after an informed consent process, which includes disclosure of the risk of vaso-occlusive crisis requiring hospitalization and a lesser risk of multi-organ failure and even death, along with standard precautions similar to those employed for major surgery in such individuals.
Acknowledgments
This work was supported by the intramural research program of the National Institute of Diabetes, Digestive and Kidney Diseases, the National Heart, Lung and Blood Institute, and the Clinical Center Department of Bioethics at the National Institutes of Health. The opinions expressed here are the authors’ and do not reflect the policies and positions of the National Institutes of Health, the U.S. Public Health Service, or the U.S. Department of Health and Human Services.
References
- 1.Abboud M, Laver J, Blau CA. Granulocytosis causing sickle-cell crisis. Lancet. 1998 Mar 28;351(9107):959. doi: 10.1016/S0140-6736(05)60614-9. [DOI] [PubMed] [Google Scholar]
- 2.Adler BK, Salzman DE, Carabasi MH, Vaughan WP, Reddy VV, Prchal JT. Fatal sickle cell crisis after granulocyte colony-stimulating factor administration. Blood. 2001 May 15;97(10):3313–4. doi: 10.1182/blood.v97.10.3313. [DOI] [PubMed] [Google Scholar]
- 3.Grigg AP. Granulocyte colony-stimulating factor-induced sickle cell crisis and multiorgan dysfunction in a patient with compound heterozygous sickle cell/beta+ thalassemia. Blood. 2001 Jun 15;97(12):3998–9. doi: 10.1182/blood.v97.12.3998. [DOI] [PubMed] [Google Scholar]
- 4.Kamble RT, Tin UC, Carrum G. Successful mobilization and transplantation of filgrastim mobilized hematopoietic stem cells in sickle cell-hemoglobin C disease. Bone Marrow Transplant. 2006 Jun;37(11):1065–6. doi: 10.1038/sj.bmt.1705376. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbaum C, Peace D, Rich E, VanBesien K. Granulocyte colony-stimulating factor-based stem cell mobilization in patients with sickle cell disease. Biol Blood Marrow Transplant. 2008 Jun;14(6):719–23. doi: 10.1016/j.bbmt.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Onitilo AA, Lazarchick J, Brunson CY, Frei-Lahr D, Stuart RK. Autologous bone marrow transplant in a patient with sickle cell disease and diffuse large B-cell lymphoma. Transplant Proc. 2003 Dec;35(8):3089–92. doi: 10.1016/j.transproceed.2003.10.085. [DOI] [PubMed] [Google Scholar]
- 7.Tormey CA, Snyder EL, Cooper DL. Mobilization, collection, and transplantation of peripheral blood hematopoietic progenitor cells in a patient with multiple myeloma and hemoglobin SC disease. Transfusion. 2008 May 29; doi: 10.1111/j.1537-2995.2008.01777.x. [DOI] [PubMed] [Google Scholar]
- 8.Dallalio G, Brunson CY, Means RT., Jr Cytokine concentrations in bone marrow of stable sickle cell anemia patients. J Investig Med. 2007 Mar;55(2):69–74. doi: 10.2310/6650.2007.06029. [DOI] [PubMed] [Google Scholar]
- 9.Akohoue SA, Shankar S, Milne GL, Morrow J, Chen KY, Ajayi WU, et al. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatric research. 2007 Feb;61(2):233–8. doi: 10.1203/pdr.0b013e31802d7754. [DOI] [PubMed] [Google Scholar]
- 10.Lee SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2006 Jul;26(7):1626–31. doi: 10.1161/01.ATV.0000220374.00602.a2. [DOI] [PubMed] [Google Scholar]
- 11.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. The New England journal of medicine. 1994 Jun 9;330(23):1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 12.Miller ST, Sleeper LA, Pegelow CH, Enos LE, Wang WC, Weiner SJ, et al. Prediction of adverse outcomes in children with sickle cell disease. The New England journal of medicine. 2000 Jan 13;342(2):83–9. doi: 10.1056/NEJM200001133420203. [DOI] [PubMed] [Google Scholar]
- 13.Quinn CT, Lee NJ, Shull EP, Ahmad N, Rogers ZR, Buchanan GR. Prediction of adverse outcomes in children with sickle cell anemia: a study of the Dallas Newborn Cohort. Blood. 2008 Jan 15;111(2):544–8. doi: 10.1182/blood-2007-07-100719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. The New England journal of medicine. 1995 May 18;332(20):1317–22. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 15.Saleh AW, Hillen HF, Duits AJ. Levels of endothelial, neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1999;102(1):31–7. doi: 10.1159/000040964. [DOI] [PubMed] [Google Scholar]
- 16.Miller MK, Zimmerman SA, Schultz WH, Ware RE. Hydroxyurea therapy for pediatric patients with hemoglobin SC disease. J Pediatr Hematol Oncol. 2001 Jun-Jul;23(5):306–8. doi: 10.1097/00043426-200106000-00014. [DOI] [PubMed] [Google Scholar]
- 17.Abboud MR, Taylor EC, Habib D, Dantzler-Johnson T, Jackson SM, Xu F, et al. Elevated serum and bronchoalveolar lavage fluid levels of interleukin 8 and granulocyte colony-stimulating factor associated with the acute chest syndrome in patients with sickle cell disease. British journal of haematology. 2000 Nov;111(2):482–90. doi: 10.1046/j.1365-2141.2000.02358.x. [DOI] [PubMed] [Google Scholar]
- 18.Conran N, Almeida CB, Lanaro C, Ferreira RP, Traina F, Saad ST, et al. Inhibition of caspase-dependent spontaneous apoptosis via a cAMP-protein kinase A dependent pathway in neutrophils fromsickle cell disease patients. British journal of haematology. 2007 Oct;139(1):148–58. doi: 10.1111/j.1365-2141.2007.06748.x. [DOI] [PubMed] [Google Scholar]
- 19.Hofstra TC, Kalra VK, Meiselman HJ, Coates TD. Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood. 1996 May 15;87(10):4440–7. [PubMed] [Google Scholar]
- 20.Fadlon E, Vordermeier S, Pearson TC, Mire-Sluis AR, Dumonde DC, Phillips J, et al. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998 Jan 1;91(1):266–74. [PubMed] [Google Scholar]
- 21.Stroncek DF, Jaszcz W, Herr GP, Clay ME, McCullough J. Expression of neutrophil antigens after 10 days of granulocyte-colony-stimulating factor. Transfusion. 1998 Jul;38(7):663–8. doi: 10.1046/j.1537-2995.1998.38798346635.x. [DOI] [PubMed] [Google Scholar]
- 22.Assis A, Conran N, Canalli AA, Lorand-Metze I, Saad ST, Costa FF. Effect of cytokines and chemokines on sickle neutrophil adhesion to fibronectin. Acta Haematol. 2005;113(2):130–6. doi: 10.1159/000083451. [DOI] [PubMed] [Google Scholar]
- 23.Lichtman MA, Miller DR, Cohen J, Waterhouse C. Reduced red cell glycolysis, 2, 3-diphosphoglycerate and adenosine triphosphate concentration, and increased hemoglobin-oxygen affinity caused by hypophosphatemia. Ann Intern Med. 1971 Apr;74(4):562–8. doi: 10.7326/0003-4819-74-4-562. [DOI] [PubMed] [Google Scholar]
- 24.Russell MO, Goldberg HI, Hodson A, Kim HC, Halus J, Reivich M, et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood. 1984 Jan;63(1):162–9. [PubMed] [Google Scholar]
- 25.Pegelow CH, Adams RJ, McKie V, Abboud M, Berman B, Miller ST, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. The Journal of pediatrics. 1995 Jun;126(6):896–9. doi: 10.1016/s0022-3476(95)70204-0. [DOI] [PubMed] [Google Scholar]
- 26.Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. The New England journal of medicine. 1998 Jul 2;339(1):5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 27.Kang EM, Areman EM, David-Ocampo V, Fitzhugh C, Link ME, Read EJ, et al. Mobilization, collection, and processing of peripheral blood stem cells in individuals with sickle cell trait. Blood. 2002 Feb 1;99(3):850–5. doi: 10.1182/blood.v99.3.850. [DOI] [PubMed] [Google Scholar]
- 28.Sunshine HR, Hofrichter J, Eaton WA. Gelation of sickle cell hemoglobin in mixtures with normal adult and fetal hemoglobins. J Mol Biol. 1979 Oct 9;133(4):435–67. doi: 10.1016/0022-2836(79)90402-9. [DOI] [PubMed] [Google Scholar]
- 29.Abboud MR, Dover GJ, Jackson S, Brunson CY, Forcier A, Oette D, et al. The National Sickle Cell Disease Program. New York City: 1994 March, 1994. A pilot study of recombinant human granulocyte-macrophage colony stimulating factor and hydroxyurea in patients with sickle cell disease. abstract. [Google Scholar]
- 30.Bronchud MH, Scarffe JH, Thatcher N, Crowther D, Souza LM, Alton NK, et al. Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. British journal of cancer. 1987 Dec;56(6):809–13. doi: 10.1038/bjc.1987.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabrilove JL, Jakubowski A, Fain K, Grous J, Scher H, Sternberg C, et al. Phase I study of granulocyte colony-stimulating factor in patients with transitional cell carcinoma of the urothelium. The Journal of clinical investigation. 1988 Oct;82(4):1454–61. doi: 10.1172/JCI113751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gabrilove JL, Jakubowski A, Scher H, Sternberg C, Wong G, Grous J, et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional-cell carcinoma of the urothelium. The New England journal of medicine. 1988 Jun 2;318(22):1414–22. doi: 10.1056/NEJM198806023182202. [DOI] [PubMed] [Google Scholar]
- 33.Morstyn G, Campbell L, Souza LM, Alton NK, Keech J, Green M, et al. Effect of granulocyte colony stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet. 1988 Mar 26;1(8587):667–72. doi: 10.1016/s0140-6736(88)91475-4. [DOI] [PubMed] [Google Scholar]
- 34.Bronchud MH, Howell A, Crowther D, Hopwood P, Souza L, Dexter TM. The use of granulocyte colony-stimulating factor to increase the intensity of treatment with doxorubicin in patients with advanced breast and ovarian cancer. British journal of cancer. 1989 Jul;60(1):121–5. doi: 10.1038/bjc.1989.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neidhart J, Mangalik A, Kohler W, Stidley C, Saiki J, Duncan P, et al. Granulocyte colony-stimulating factor stimulates recovery of granulocytes in patients receiving dose-intensive chemotherapy without bone marrow transplantation. J Clin Oncol. 1989 Nov;7(11):1685–92. doi: 10.1200/JCO.1989.7.11.1685. [DOI] [PubMed] [Google Scholar]
- 36.Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993 May 15;81(10):2496–502. [PMC free article] [PubMed] [Google Scholar]
- 37.Heil G, Hoelzer D, Sanz MA, Lechner K, Liu Yin JA, Papa G, et al. A randomized, double-blind, placebo-controlled, phase III study of filgrastim in remission induction and consolidation therapy for adults with de novo acute myeloid leukemia. The International Acute Myeloid Leukemia Study Group. Blood. 1997 Dec 15;90(12):4710–8. [PubMed] [Google Scholar]
- 38.Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol. 2007 Jul 20;25(21):3158–67. doi: 10.1200/JCO.2006.08.8823. [DOI] [PubMed] [Google Scholar]
- 39.Clark OA, Lyman GH, Castro AA, Clark LG, Djulbegovic B. Colony-stimulating factors for chemotherapy-induced febrile neutropenia: a meta-analysis of randomized controlled trials. J Clin Oncol. 2005 Jun 20;23(18):4198–214. doi: 10.1200/JCO.2005.05.645. [DOI] [PubMed] [Google Scholar]
- 40.Amgen I. Neupogen® Prescribing Information. September, 2007 ; cited 2008 September 22]; Available from: http://www.neupogen.com/pi.html.
- 41.Irfan M, Hashmi K, Adil S, Shamsi T, Farzana T, Ansari S, et al. Beta-thalassaemia major: bone marrow versus peripheral blood stem cell transplantation. Jpma. 2008 Mar;58(3):107–10. [PubMed] [Google Scholar]
- 42.Meisel R, Laws HJ, Balzer S, Bernbeck B, Kramm C, Schonberger S, et al. Comparable long-term survival after bone marrow versus peripheral blood progenitor cell transplantation from matched unrelated donors in children with hematologic malignancies. Biol Blood Marrow Transplant. 2007 Nov;13(11):1338–45. doi: 10.1016/j.bbmt.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 43.Remberger M, Ringden O. Similar outcome after unrelated allogeneic peripheral blood stem cell transplantation compared with bone marrow in children and adolescents. Transplantation. 2007 Aug 27;84(4):551–4. doi: 10.1097/01.tp.0000275184.41831.6d. [DOI] [PubMed] [Google Scholar]
- 44.Schmitz N, Eapen M, Horowitz MM, Zhang MJ, Klein JP, Rizzo JD, et al. Long-term outcome of patients given transplants of mobilized blood or bone marrow: A report from the International Bone Marrow Transplant Registry and the European Group for Blood and Marrow Transplantation. Blood. 2006 Dec 15;108(13):4288–90. doi: 10.1182/blood-2006-05-024042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrezenmeier H, Passweg JR, Marsh JC, Bacigalupo A, Bredeson CN, Bullorsky E, et al. Worse outcome and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anemia. Blood. 2007 Aug 15;110(4):1397–400. doi: 10.1182/blood-2007-03-081596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dey BR, Shaffer J, Yee AJ, McAfee S, Caron M, Power K, et al. Comparison of outcomes after transplantation of peripheral blood stem cells versus bone marrow following an identical nonmyeloablative conditioning regimen. Bone marrow transplantation. 2007 Jul;40(1):19–27. doi: 10.1038/sj.bmt.1705688. [DOI] [PubMed] [Google Scholar]
- 47.Benboubker L, Cartron G, Roingeard F, Delain M, Degenne M, Linassier C, et al. Long-term marrow reconstitutive ability of autologous grafts in lymphoma patients using peripheral blood mobilized with granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor compared to bone marrow. Experimental hematology. 2003 Jan;31(1):89–97. doi: 10.1016/s0301-472x(02)01007-x. [DOI] [PubMed] [Google Scholar]
- 48.Hematti P, Tuchman S, Larochelle A, Metzger ME, Donahue RE, Tisdale JF. Comparison of retroviral transduction efficiency in CD34+ cells derived from bone marrowversus G-CSF-mobilized or G-CSF plus stem cell factor-mobilized peripheral blood in nonhuman primates. Stem Cells. 2004;22(6):1062–9. doi: 10.1634/stemcells.22-6-1062. [DOI] [PubMed] [Google Scholar]
- 49.Vichinsky EP, Haberkern CM, Neumayr L, Earles AN, Black D, Koshy M, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. The New England journal of medicine. 1995 Jul 27;333(4):206–13. doi: 10.1056/NEJM199507273330402. [DOI] [PubMed] [Google Scholar]
- 50.Neumayr L, Koshy M, Haberkern C, Earles AN, Bellevue R, Hassell K, et al. Surgery in patients with hemoglobin SC disease. Preoperative Transfusion in Sickle Cell Disease Study Group. Am J Hematol. 1998 Feb;57(2):101–8. doi: 10.1002/(sici)1096-8652(199802)57:2<101::aid-ajh2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 51.de Vries EG, Vriesendorp R, Meinesz AF, Mulder NH, Postmus PE, Sleijfer DT. No narcosis for bone marrow harvest in autologous bone marrow transplantation. Blut. 1984 Nov;49(5):419–21. doi: 10.1007/BF00319890. [DOI] [PubMed] [Google Scholar]
- 52.Dicke KA, Hood DL, Hanks S, Vaughan M, Fulbright L, Dicke JA, et al. A marrow harvest procedure under local anesthesia. Exp Hematol. 1995 Oct;23(11):1229–32. [PubMed] [Google Scholar]
- 53.Karlsson L, Quinlan D, Guo D, Brown C, Selinger S, Klassen J, et al. Mobilized blood cells vs bone marrow harvest: experience compared in 171 donors with particular reference to pain and fatigue. Bone Marrow Transplant. 2004 Apr;33(7):709–13. doi: 10.1038/sj.bmt.1704418. [DOI] [PubMed] [Google Scholar]
- 54.Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004 Oct 1;37(2):1253–62. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- 55.Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003 Oct 15;102(8):2728–30. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]