

Abstract
From a screen of small molecule libraries to identify potential therapeutics for psychostimulant abuse, 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-ones were shown to be isoform-selective PKC-ζ inhibitors.
Protein kinase C (PKC) describes a family of kinases consisting of at least 10 distinct isoforms of lipid-regulated serine/threonine kinases.1 These isoforms can be classified into three groups based on their structure and regulatory/activation requirements: (i) classical (α, β1, β2, γ), (ii) novel (δ, ε, η, θ), and (iii) atypical (ζ, ι). Each of these isoforms contains an amino-terminal regulatory domain (REG) and a carboxy-terminal catalytic kinase domain (CAT). PKC plays pivotal roles in several signal transduction pathways that regulate immune responses, cellular growth, transformation, differentiation, and apoptosis. Given these diverse physiological roles, it is not surprising that dysfunctional PKC-dependent signaling has been implicated in a wide variety of pathological conditions, including a variety of neurological and psychiatric disorders such as Parkinson's disease and drug abuse.2,3
PKC-ζ and PKM-ζ (the constitutively-active form lacking the auto-inhibitory PKC-ζ regulatory domain) are highly expressed in the brain4 and have been implicated in various neurobiological functions, including learning and memory.5 Various neuropsychiatric disorders are now being considered as long-term maladaptive (dysfunctional) memory processes;6 therefore, detailed elucidation of the role of PKC-ζ and PKM-ζ in long-term memory processes could provide new insights into potential mechanisms that underlie the long-term persistence (chronic clinical courses) of neuropsychiatric disorders. Psychostimulant addiction, which continues to present profound socioeconomic, legal, and medical problems worldwide, is an example of such a disorder. Total abstinence from a drug such as methamphetamine or cocaine is difficult to sustain, mainly because, even after abstinence is achieved, abusers remain vulnerable for years to episodes of craving and relapse triggered by stimuli previously associated with drug abuse.7 Recent findings from animal studies suggest that certain PKC isoforms (e.g., PKC-β2, -γ, -δ, -ζ) may play important roles in the neurobiological process of addiction,8,9 and PKC-ζ in particular may be important in the long-term persistence of neurobiological changes associated with the relapse vulnerability in psychostimulant abusers.9
The importance of PKC in the mediation of normal and pathological processes has motivated considerable efforts to identify PKC inhibitors. Although a wealth of inhibitory compounds are available, generation of PKC inhibitors with selectivity toward individual PKC isoforms has proven to be a challenge.10 In particular, isoform-selective PKC-ζ inhibitors are rare.11 Herein, we report initial evidence showing that 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-ones are isoform-selective PKC-ζ inhibitors and have potential for the treatment of psychostimulant abuse.
To identify small molecule inhibitors of PKC-ζ, we have screened 1200 compounds using purified PKC-ζ enzymes and a standard luciferase-based kinase assay. These 1200 compounds represent structurally diverse and druggable 20000-member heterocyclic compound libraries, including benzopyrans, oxazoles, thiazoles, thiadiazoles, and pyrimidinediones.12 Chelerythrine,13 a non-specific PKC-ζ inhibitor, was used in the assay as a positive control.
The primary screen identified several 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-ones (1–4)14 as inhibitors of PKC-ζ enzymatic activity (Fig. 1(a)). Despite their structural similarity to the flavonol scaffold, 5–11 from the library showed no or little PKC-ζ inhibitory activity in the primary screen (Fig. 1(b)). From preliminary structure–activity relationship studies of 1–4, it appears that the 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-one moiety is critical to their PKC-ζ inhibitory activities. Since compounds in which the hydroxyl group at the meta position on the C2-phenyl group is protected with a benzyl group exhibit no activity (see 5–7 in Fig. 1(b)), the hydroxyl group should play an important role as an H-bond donor. Flavonols exhibit a variety of biological activities, including anti-oxidant, anti-tumour, and anti-inflammatory activities.15 Several flavonols such as quercetin and fisetin are known as weak and non-selective PKC inhibitors,16 but none of them is a PKC-ζ inhibitor.
Fig. 1.
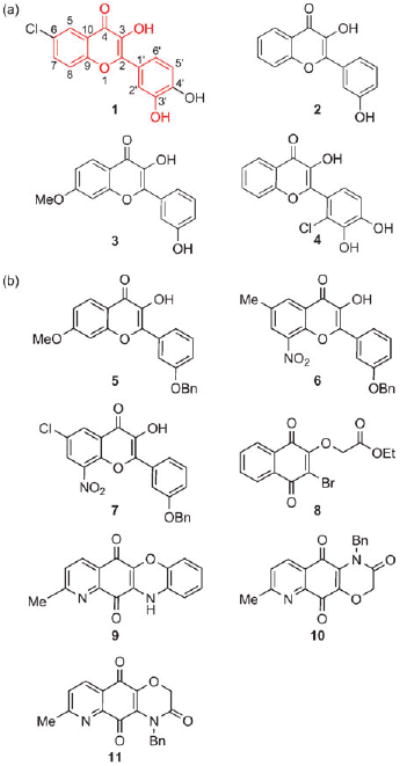
(a) Structure of PKC-ζ inhibitors identified in the primary screen. (b) Structure of compounds with no or little PKC-ζ inhibitory activity.
After the initial hit compounds were identified from the primary screen, we tested 1–4 for their cellular toxicity using Cos-7 cells. We first measured initial dose-response curves (0.01–50 μM) to identify the dose at which 1–4 began to display cytotoxic effects after a 24-h treatment. Results revealed that a subset of compounds began to display cytotoxicity at a concentration of 40 μM, and subsequent temporal toxicity screens were performed below this threshold to further validate the least toxic compound (15 μM). Specifically, we performed a standard MTT assay to accurately determine the toxicity of 1–4 after 24 and 48 h (Fig. 2). Data for each experiment were normalized to % of the untreated group (i.e., no compound or DMSO added) and analyzed using between-subject, two-way analysis of variance (ANOVA; compound × time), followed by individual group comparisons with post-hoc Tukey's test. A two-way ANOVA revealed significant main effects of compound on cell survival (F(4,49) = 2.935, p < 0.03; Fig. 2) without main effects of time (F(1,49) = 1.865, p > 0.17) or a compound × time interaction (F(4,49) = 1.35, p > 1.265). Compound 1 displayed a small, but statistically significant toxicity compared to DMSO or 4 (<15% each, p < 0.05, Tukey's test) at 48 h, but not at the earlier time point. Results were in good agreement with parallel total protein quantification assays, which displayed little or no cellular toxicity over the DMSO controls (data not shown).
Fig. 2.
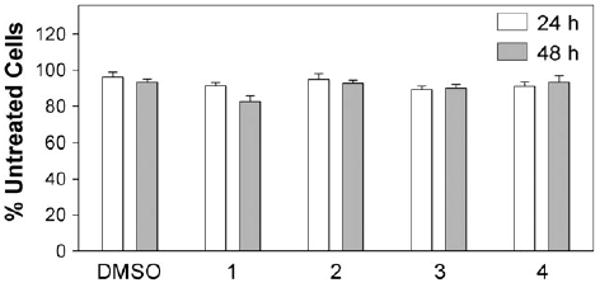
Cellular toxicity of 1–4 (15 μM final concentration).
To determine the biological efficacy of 1–4 and to rule out any false positives from the luciferase-based assay, we used a radioisotope-based PKC-ζ immunoprecipitation (IP) kinase assay to assess inhibition of cellular PKC-ζ activity in Cos-7 cells by 1–4 (Fig. 3). All compounds displayed inhibition with similar or greater efficacy relative to chelerythrine (p < 0.001).
Fig. 3.
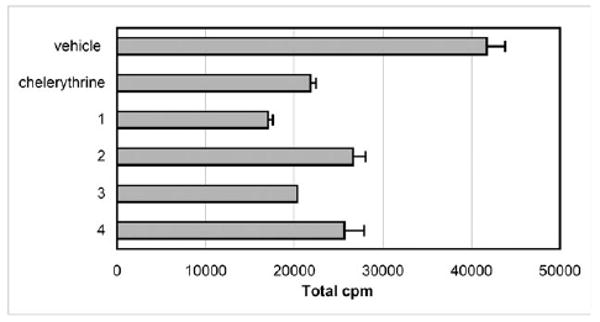
Inhibition of immunoprecipitated PKC-ζ activity by 1–4 (15 μM final concentration).
To further characterize the initial hit compounds, we determined IC50 values of 1–3 with serial dilutions of each inhibitor (Fig. 4). These assays identified 2 and 3 as the most potent inhibitors of PKC-ζ (IC50 values of 1 = 9.8 μM, 2 = 2.1 μM, 3 = 2.2 μM). Together with cellular toxicity data, the present study demonstrates that 2 and 3 are more effective inhibitors of PKC-ζ than the well-characterized chelerythrine, while producing no greater cytotoxic effects than the DMSO control.
Fig. 4.
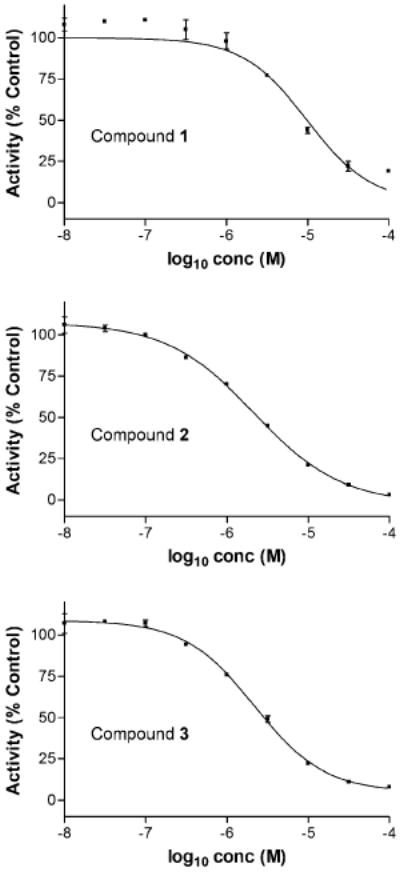
IC50 titration curve of 1–3.
The success of kinase inhibitors in clinical applications relies on their kinase selectivity, and this has been the most challenging and important problem associated with developing kinase inhibitors. To evaluate the isoform-selectivity of 1–3, Millipore KinaseProfiler was performed on a panel of kinases including PKC-β2 (a classical PKC) and PKC-ε (a novel PKC) (Table 1). Compounds 1–3 showed useful levels of selectivity for PKC-ζ in comparison with PKA, PKB, and other PKC isoforms.
Table 1.
Isoform-selectivity of 1–3 (% activity at 10 μM)
| 1 | 2 | 3 | |
|---|---|---|---|
| PKC-ζ | 36 | 33 | 32 |
| PKA | 48 | 115 | 91 |
| PKB-α | 61 | 115 | 99 |
| PKC-β2 | 71 | 71 | 91 |
| PKC-ε | 97 | 99 | 92 |
Computational docking and scoring of small molecules into macromolecule structures is a useful method for identifying and optimizing lead compounds.17 The crystal structure of the catalytic domain of PKC-ζ has not been reported, but the amino acid sequence of PKC-ι, with an available crystal structure,18 shows the greatest homology to PKC-ζ, with 72% identity overall rising to 84% in the catalytic domain. To identify key structural requirements for potency and isoform-selectivity of 1–3, we used two different computational methods, molecular docking simulations using LigandFit19 and QM/MM calculations using CHARMM (see ESI for details‡). As shown in Fig. 5, 1–3 can adopt two remarkably different orientations. As seen from other flavonol kinase inhibitors,20 1–3 are anchored in two orientations in the binding pocket by the formation of a pair of hydrogen bonds between the 3-hydroxyl and 4-keto groups of 1–3 and backbone groups in Glu324 and Val326. In Orientation I generated by LigandFit (the same binding mode as BIM1 bound to the active site of PKC-ι18), 1–3 are bound in an orientation that allows the C2–phenyl group to occupy the hydrophobic pocket formed by Ile323 and Phe297. In this orientation, the 3′-hydroxyl group forms a hydrogen bond with Asp387, Thr386, or Lys274. The C6–Cl in 1 and C7–OMe in 3 are outside of the active site and exposed to water. Adding a substituent at the 2′-position of the 2-(3-hydroxyphenyl) group of 1–3 may create a ligand that is better able to fill this pocket and bind with higher affinity to PKC-ζ. In Orientation II (Fig. 5), the C6–Cl in 1 and C7–OMe in 3 occupy the hydrophobic pocket formed by Ile323 and Phe297, respectively. We have identified two possible orientations using LigandFit for each of 1–3. In comparison, Orientation I utilizes hydrogen bonds with Asp387, Thr386, or Lys274 as well as hydrogen bonds with Glu324 and Val326, while Orientation II best utilizes the hydrophobic pocket formed by Ile323 and Phe297. Orientation I scored higher in the docking simulation19 and was further investigated with a QM/MM method. The compound orientation found in the QM/MM analysis is nearly identical to one in the docking study, supporting Orientation I as the more stable orientation. Sequence alignment of PKC-ζ, PKA, PKB-α, PKC-β2, and PKC-ε by ClustalW221 showed that the ATP-binding site is highly conserved among the kinases, but the hydrophobic pocket formed by Ile323 and Phe297 is unique in PKC-ζ and may be important for the observed isoform-selectivity of 1–3 (see ESI for details‡).11 The other kinases have Met and Leu instead of Ile and Phe, respectively. Thus, designing a ligand that combines the interactions characteristic of these two orientations and strong interactions with all binding sites may provide a more potent and isoform-selective PKC-ζ inhibitor. As such, computational methods may provide insights to further optimize the potency and selectivity of 1–3 and to design new small molecule libraries.
Fig. 5.
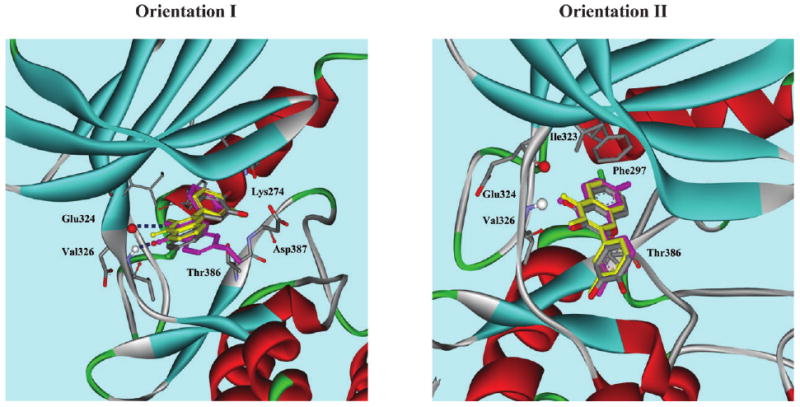
Proposed binding modes (Orientation I and Orientation II) of 1–3 (1 in full color, 2 in yellow, and 3 in magenta) in the active site of PKC-ι.
In summary, we have screened 1200 heterocyclic small molecules and identified 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-ones as isoform-selective inhibitors of PKC-ζ enzymatic activity. Further studies are currently underway to optimize the potency and selectivity of 3-hydroxy-2-(3-hydroxyphenyl)-4H-1-benzopyran-4-ones and address their in vivo efficacy and therapeutic potential. These molecules may serve as useful mechanistic probes of the cellular function of PKC-ζ and the neurobiological mechanisms underlying compulsive chronic psychostimulant abuse.
Supplementary Material
Acknowledgments
This work was supported by grants from Duke University (J.H.), the Center for Biological Modulators of the 21st Century Frontier R&D Program, Ministry of Education, Science and Technology, Korea (CBM32-B1000-01-00-00 to Y.D.G.), National Institutes of Health (DA12768 and NS42124 to T.H.L), National Institutes of Health through a Center of Excellence in Chemical Methodologies and Library Development (P50GM067082 to D.N.B), and TeraDisc (D.N.B).
Footnotes
This article is part of the 2009 Molecular BioSystems ‘Emerging Investigators’ issue: highlighting the work of outstanding young scientists at the chemical- and systems-biology interfaces.
Electronic Supplementary Information (ESI) available: Detailed assay and molecular docking procedures; ClustalW2 multiple sequence alignment; spectroscopic and analytical data for 1–4. See DOI: 10.1039/b903036k/
References
- 1.Newton AC. J Biol Chem. 1995;270:28495. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 2.Lee AM, Messing RO. Ann N Y Acad Sci. 2008;1141:22. doi: 10.1196/annals.1441.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandel SA, Sagi Y, Amit T. Neurochem Res. 2007;32:1694. doi: 10.1007/s11064-007-9351-8. [DOI] [PubMed] [Google Scholar]
- 4.Naik MU, Benedikz E, Hernandez I, Libien J, Hrabe J, Valsamis M, Dow-Edwards D, Osman M, Sacktor TC. J Comp Neurol. 2000;16:243. doi: 10.1002/1096-9861(20001016)426:2<243::aid-cne6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton A, Sacktor TC. Science. 2006;313:1141. doi: 10.1126/science.1128657. [DOI] [PubMed] [Google Scholar]
- 6.Centonze D, Siracusano A, Calabresi P, Bernardi G. Mol Neurobiol. 2005;32:123. doi: 10.1385/MN:32:2:123. [DOI] [PubMed] [Google Scholar]
- 7.Elkashef A, Vocci F, Hanson G, White J, Wickes W, Tiihonen J. Subst Abus. 2008;29:31. doi: 10.1080/08897070802218554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narita M, Akai H, Nagumo Y, Sunagawa N, Hasebe K, Nagase H, Kita T, Hara C, Suzuki T. Neuroscience. 2004;127:941. doi: 10.1016/j.neuroscience.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Lee TH, Wetsel WC, Sun QA, Liu Y, Davidson C, Xiong X, Ellinwood EH, Zhang X. Biochem Biophys Res Commun. 2007;356:733. doi: 10.1016/j.bbrc.2007.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackay HJ, Twelves CJ. Nat Rev Cancer. 2007;7:554. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 11.Trujillo JI, Kiefer JR, Huang W, Thorarensen A, Xing L, Caspers NL, Day JE, Mathis KJ, Kretzmer KK, Reitz BA, Weinberg RA, Stegeman RA, Wrightstone A, Christine L, Compton R, Li X. Bioorg Med Chem Lett. 2009;19:908. doi: 10.1016/j.bmcl.2008.11.105. [DOI] [PubMed] [Google Scholar]
- 12.Hwang JY, Choi HS, Seo JS, La HJ, Yoo SE, Gong YD. J Comb Chem. 2006;8:897. doi: 10.1021/cc0600526. [DOI] [PubMed] [Google Scholar]; Hwang JY, Gong YD. J Comb Chem. 2006;8:297. doi: 10.1021/cc050149c. [DOI] [PubMed] [Google Scholar]; Hwang JY, Choi HS, Lee DH, Yoo SE, Gong YD. J Comb Chem. 2005;7:136. doi: 10.1021/cc049931n. [DOI] [PubMed] [Google Scholar]; Hwang JY, Choi HS, Lee DH, Gong YD. J Comb Chem. 2005;7:816. doi: 10.1021/cc0500957. [DOI] [PubMed] [Google Scholar]; Lee IY, Kim SY, Lee JY, Yu CM, Lee DH, Gong YD. Tetrahedron Lett. 2004;45:9319. [Google Scholar]
- 13.Herbert JM, Augereau JM, Gleye J, Maffrand JP. Biochem Biophys Res Commun. 1990;15:993. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- 14.Fougerousse A, Gonzalez E, Brouillard R. J Org Chem. 2000;65:583. doi: 10.1021/jo990735q. [DOI] [PubMed] [Google Scholar]; Daskiewicz JB, Depeint F, Viornery L, Bayet C, Comte-Sarrazin G, Comte G, Gee GM, Johnson IT, Ndjoko K, Hostettmann K, Barron D. J Med Chem. 2005;48:2790. doi: 10.1021/jm040770b. [DOI] [PubMed] [Google Scholar]
- 15.Brahmachari G, Gorai D. Curr Org Chem. 2006;10:873. [Google Scholar]
- 16.Ferriola PC, Cody V, Middleton E., Jr Biochem Pharmacol. 1989;38:1617. doi: 10.1016/0006-2952(89)90309-2. [DOI] [PubMed] [Google Scholar]; Gamet-Payrastre L, Manenti S, Gratacap MP, Tulliez J, Chap H, Payrastre B. Gen Pharmacol. 1999;32:279. doi: 10.1016/s0306-3623(98)00220-1. [DOI] [PubMed] [Google Scholar]
- 17.Joseph-McCarthy D, Baber JC, Feyfant E, Thompson DC, Humblet C. Curr Opin Drug Discovery Dev. 2007;10:264. [PubMed] [Google Scholar]
- 18.Messerschmidt A, Macieira S, Velarde MB, Benda C, Jestel A, Brandstetter TN, Blaesse M. J Mol Biol. 2005;352:918. doi: 10.1016/j.jmb.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 19.Venkatachalam CM, Jiang X, Waldman OM. J Mol Graphics Modell. 2003;21:289. doi: 10.1016/s1093-3263(02)00164-x. [DOI] [PubMed] [Google Scholar]
- 20.Lu H, Chang DJ, Baratte B, Meijer L, Schulze-Gahmen U. J Med Chem. 2005;48:737. doi: 10.1021/jm049353p. [DOI] [PubMed] [Google Scholar]; Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Mol Cell. 2000;6:909. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 21.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Bioinformatics. 2007;23:2947. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.