

Summary
Bacterial pathogenesis involves social behavior including biofilm formation and swarming, processes that are regulated by the bacterially unique second messenger cyclic di-GMP (c-di-GMP). Diguanylate cyclases containing GGDEF and phosphodiesterases containing EAL domains have been identified as the enzymes controlling cellular c-di-GMP levels, yet less is known regarding signal transmission and the targets of c-di-GMP. FimX, a protein from Pseudomonas aeruginosa that governs twitching motility, belongs to a large subfamily containing both GGDEF and EAL domains. Biochemical and structural analyses reveals its function as a high-affinity receptor for c-di-GMP. A model for full-length FimX was generated combining solution scattering data and crystal structures of the degenerate GGDEF and EAL domains. While FimX forms a dimer in solution via the N-terminal domains, a crystallographic EAL domain dimer suggests modes for the regulation of FimX by c-di-GMP binding. The results provide the structural basis for c-di-GMP sensing via degenerate phosphodiesterases.
Introduction
Bis-(3′-5′)-cyclic dimeric guanosine monophosphate (cyclic di-GMP or c-di-GMP) emerged as a central second messenger in eubacteria that controls community behavior, secretion, adhesion, and motility, ultimately contributing to the virulence of pathogens (D'Argenio and Miller, 2004; Romling et al., 2005; Ross et al., 1987). Diguanylate cyclases and phosphodiesterases have been identified in large numbers in bacterial genomes, and GGDEF and EAL domains, respectively, and have been shown to be responsible for the production and degradation of c-di-GMP (Christen et al., 2005; Galperin et al., 2001; Hickman et al., 2005; Paul et al., 2004; Ryjenkov et al., 2005; Schmidt et al., 2005; Simm et al., 2004; Tal et al., 1998; Tamayo et al., 2005). GGDEF and EAL represent conserved amino acid motifs at the active site within the domains. These proteins cluster into three classes which are often encoded in the same genome: GGDEF domain-containing proteins, EAL domain-containing proteins, and proteins that contain a tandem GGDEF-EAL domain. In many cases, the signature motif is degenerate and the domains function as allosteric regulators of adjacent domains (Christen et al., 2005; Newell et al., 2009). In the case of GGDEF domains, a second c-di-GMP binding site is characterized by an RxxD motif, called the inhibitory or I-site, and has been shown to restrict the signaling potential of active diguanylate cyclases via a negative feedback loop (Chan et al., 2004; Christen et al., 2006; De et al., 2008; Wassmann et al., 2007).
Lagging behind the structural and functional characterization of diguanylate cyclases and phosphodiesterases, the targets and signaling mechanisms for c-di-GMP are largely unknown, especially considering the high abundance and apparent signaling specificity of cyclases and phosphodiesterases encoded in bacterial genomes (Beyhan et al., 2008; Kader et al., 2006; Kulasakara et al., 2006; Sommerfeldt et al., 2009). In addition, it is believed that the majority of the cellular c-di-GMP pool, typically 100-200 molecules per bacterial cell, is sequestered by proteins, calling for receptors with high apparent affinity for the nucleotide (Simm et al., 2009; Weinhouse et al., 1997). Yet, the affinities reported for c-di-GMP receptors identified thus far span multiple orders of magnitude, with many of the proteins showing rather weak binding in vitro (Hengge, 2009).
Initial predictions, subsequently corroborated experimentally, identified PilZ domains as c-di-GMP-binding modules (Amikam and Galperin, 2006; Ryjenkov et al., 2006). Other studies revealed FleQ and PelD from P. aeruginosa, proteins involved in exopolysaccharide synthesis, as functional, PilZ-unrelated c-di-GMP receptors (Hickman and Harwood, 2008; Lee et al., 2007). While both proteins affect the same pathway, they are structurally unrelated. FleQ belongs to the AAA superfamily and interacts with the alternative RNA polymerase sigma factor ⌠54 at enhancer sites to reciprocally regulate flagella biosynthesis and secretion on a transcriptional level (Hickman and Harwood, 2008). In contrast, PelD is a transmembrane protein that contains an RxxD motif that mimics the I-site found on GGDEF domains (Lee et al., 2007). In addition, a prevalent riboswitch class in messenger RNA of many bacteria possesses high affinity for c-di-GMP and impacts gene expression (Sudarsan et al., 2008), demonstrating the remarkable diversity in c-di-GMP targets and signaling mechanisms.
In genome-wide studies of P. aeruginosa and E. coli, a fraction of the GGDEF and/or EAL domain-containing proteins lacked activity despite being expressed (Kulasakara et al., 2006; Sommerfeldt et al., 2009). While this could suggest that these proteins may become active upon receiving a signal, many display a degenerate diguanylate cyclase and phosphodiesterase domain, and it has been proposed that inactive enzymes could serve as c-di-GMP receptors. Such function has been attributed to the GGDEF domain of PopA from Caulobacter crescentus and the EAL domain of the GGDEF-EAL domain-containing transmembrane protein LapD from P. fluorescence (Duerig et al., 2009; Newell et al., 2009).
Here, we identify FimX (PA4959), a dual GGDEF-EAL domain-containing protein involved in swarming behavior of P. aeruginosa, as a high-affinity c-di-GMP receptor (Figure 1A) (Huang et al., 2003). FimX regulates twitching motility and biofilm formation (Huang et al., 2003; Kazmierczak et al., 2006). While overexpression of FimX had no affect on pellicle formation and cytotoxicity, transposon insertions and chromosomal deletions at the FimX locus abolished biofilm formation (Huang et al., 2003; Kulasakara et al., 2006). Although FimX was initially described as an active phosphodiesterase (Kazmierczak et al., 2006), degeneration of its EAL domain and lack of phosphodiesterase activity has been predicted based on homology modeling and was corroborated in enzyme assays (Rao et al., 2008), suggesting an alternative mechanism for FimX function.
Figure 1. Structure of the FimXEAL.
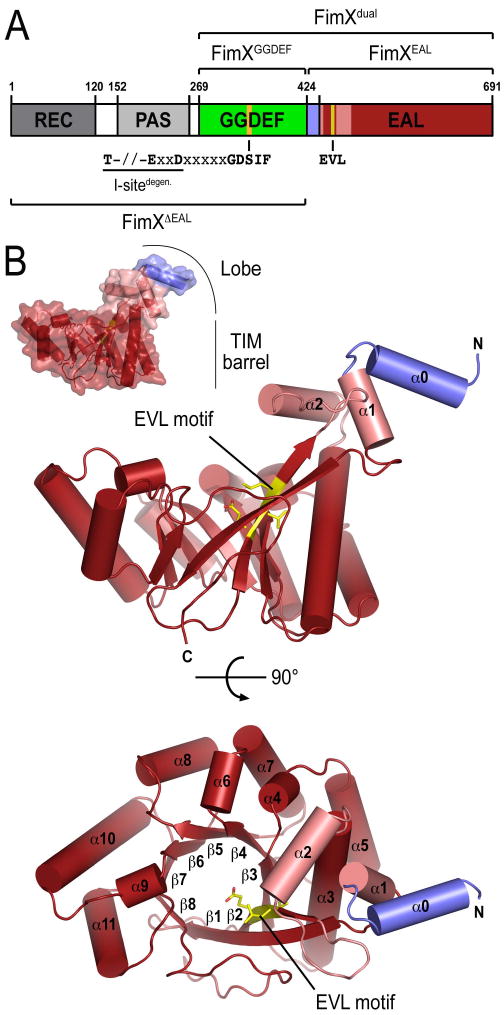
(A) Domain organization of FimX. Protein constructs used in this study are indicated. The sequence motifs of the putative inhibitory site (I-site), diguanylate active site, and phosphodiesterase active site are shown.
(B) Crystal structure of apo-FimXEAL. The structure of the isolated EAL domain of FimX from P. aeruginosa is shown as ribbon presentation in two orthogonal views, with the molecular surface shown as inset. The EAL motif (E475VL in FimX) is colored in yellow. The linker between the GGDEF and EAL domains of FimX forms a helix in this structure and is colored in blue.
We present the crystal structure of the EAL domain of FimX in its c-di-GMP-bound and apo-state, and in the context of the GGDEF-EAL dual-domain module. In addition, we determined the structure of the degenerate GGDEF domain, presenting atomic models for the large family of degenerate GGDEF and EAL domains. While the GGDEF domain may not accommodate nucleotides, the EAL domain binds c-di-GMP with high affinity in solution. Combining small-angle X-ray scattering-based modeling with available crystal structures of the isolated domains, a low-resolution envelope for dimeric full-length FimX was derived. Based on the structural data, we discuss potential molecular mechanisms that may explain how c-di-GMP binding to the EAL domain of FimX could affect biofilm formation.
Results and Discussion
Crystal structures of the EAL domain of FimX
Studies described here were carried out with the full-length protein (FimXfull-length) as well as four truncation constructs comprising the N-terminal response receiver, PAS and GGDEF domains (residues 1-431, FimXΔEAL), the GGDEF-EAL dual-domain module (residues 260-691, FimXdual), and the isolated GGDEF and EAL domains (residues 260-431/FimXGGDEF and 429-691/FimXEAL, respectively) (Figure 1A). Proteins were over-expressed in E.coli and purified to homogeneity using standard affinity and size exclusion chromatography (see Material and Methods for details). From all the protein constructs, FimXEAL, FimXGGDEF and FimXdual yielded suitable crystals for structure determination (Table 1). The structure of FimXdual was solved by single-wavelength anomalous dispersion (SAD) phasing in the space group P41212 with two molecules in the asymmetric unit. The structure will be discussed later. FimXEAL crystallized in both, the apo- and c-di-GMP-bound state in the space group P6422 and P41212, respectively, and the structures were solved at 2.3 Å and 1.5 Å resolution by molecular replacement using the EAL domain model determined from crystals of FimXdual as the search model.
Table 1.
X-ray data collection and refinement statistics.
| FimXEAL + c-di-GMP | FimXEAL | FimXdual | FimXGGDEF | |
|---|---|---|---|---|
| Data collection | ||||
| X-ray source | CHESS, F1 | NSLS, X29 | CHESS, A1 | CHESS, A1 |
| Wavelength (Å) | 0.9180 | 1.0809 | 0.9771 | 0.9771 |
| Space group | P41212 | P6422 | P41212 | P21 |
| Unit cell parameters | ||||
| a, b, c (Å) | 93.9, 93.9, 68.4 | 107.9, 107.9, 131.4 | 106.2, 106.2, 193.4 | 33.2, 94.1, 45.5 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 | 90.0, 90.6, 90.0 |
| Resolution range (Å) | 42.1 – 1.5 (1.56 – 1.5)a | 46.7 – 2.3 (2.43 – 2.3) | 48.9 – 3.0 (3.18 – 3.0) | 47.0 – 2.0 (2.09 – 2.0) |
| No. of reflections | ||||
| Total | 696341 (63645) | 243699 (38570) | 317432 (47627) | 63637 (6265) |
| Unique | 54448 (8121) | 20719 (3245) | 42159 (6702) | 17415 (2270) |
| Completeness (%) | 98.4 (92.3) | 99.8 (99.5) | 99.5 (97.9) | 93.6 (76.1) |
| Redundancy | 12.8 (7.8) | 11.8 (11.8) | 7.5 (7.1) | 3.6 (2.8) |
| I/σ(I) | 25.6 (3.5) | 31.7 (5.2) | 25.7 (4.4) | 18.7 (6.4) |
| Rmeas (%) | 5.3 (61.9) | 6.2 (62.3) | 6.2 (49.4) | 5.8 (17.4) |
| Refinement | ||||
| Rwork / Rfree (%) | 19.1 / 21.2 | 19.5 / 23.7 | 20.2 / 25.2 | 21.3 / 24.6 |
| r.m.s. deviations | ||||
| Bond length (Å) | 0.007 | 0.008 | 0.010 | 0.003 |
| Bond angles (°) | 1.171 | 1.106 | 1.362 | 0.723 |
| No. of atoms | ||||
| Protein | 1977 | 2059 | 3916 | 2666 |
| Water | 318 | 117 | 0 | 143 |
| Average B-factors (Å2) | ||||
| Protein | 32.2 | 51.1 | 89.8 | 26.3 |
| Water | 44.2 | 51.2 | -- | 31.1 |
| Ramachandram (%)b | ||||
| Favored | 94.6 | 93.6 | 85.7 | 92.8 |
| Allowed | 5.4 | 6.4 | 14.1 | 7.2 |
| Generous | 0.0 | 0.0 | 0.2 | 0.0 |
| Disallowed | 0.0 | 0.0 | 0.0 | 0.0 |
Values for the highest resolution bin
Calculated in PROCHECK (Laskowski et al., 1993)
The overall fold of EAL domain of FimX is very similar to that of the putative, EAL domain-containing phosphodiesterases tdEAL from Thiobacillus dentrificans (PDB code 2r6o) and YkuI from Bacilus subtilis (PDB code 2bas), both determined recently by the Midwest Center for Structural Genomics (unpublished; Minasov et al., 2009). Despite low sequence identity (25% and 21% compared to tdEAL and YkuI, respectively), the superimposed Cα atoms yield an r.m.s. deviation of 1.5 Å (over 215 Cα-positions) and 1.8 Å (over 194 Cα-positions), respectively. The EAL domain of FimX possesses 11 α-helices and 8 β-strands, and its fold resembles a TIM-like barrel. However, in contrast to the classical (β/α)8 TIM barrel fold, the succession of secondary structure elements in the EAL domain barrel is αβ(β/α)6β, where the first β-strand is anti-parallel with respect to the other 7 strands forming the barrel core (Banner et al., 1975) (Figure 1B). In addition, the EAL domain of FimX lacks two of the helices that surround the internal β-strands in canonical TIM barrels. An EAL domain-specific feature, a lobe formed by two N-terminal helices (α0 and α1) and a helical insertion between strand β2 and helix α3 in the TIM barrel (α2), protrudes perpendicularly from the barrel plane (Figure 1B). Helix α0 of the lobe in FimX is part of the linker region that connects the GGDEF and EAL domains (Figure 1).
Soaking in a crystallization solution containing c-di-GMP dissolved the crystals of FimXEAL initially but the nucleotide-bound protein re-crystallized in an alternative space group (see above; Table 1). Similar to several other TIM-barrel-containing enzymes and the EAL domain of YkuI (Hocker et al., 2001; Minasov et al., 2009), the c-di-GMP binding site of FimXEAL, including the degenerate EAL motif (E475VL in FimX), is located at the C-terminal end of the β-strands composing the barrel, and a c-di-GMP molecule was modeled with high confidence into the density at that site (Figures 1B and 2A). Unlike the stacked c-di-GMP dimers bound at the active site and I-site of GGDEF domains (Chan et al., 2004; De et al., 2008), the nucleotide bound to FimXEAL assumed an extended conformation that is perpendicular to the axis defined by the strands of the TIM barrel (Figure 2A), and similar to the conformation observed in the structure of the EAL domain from YkuI (Figure 2B) (Minasov et al., 2009).
Figure 2. Structural comparison between the c-di-GMP-bound form and the apo-state.
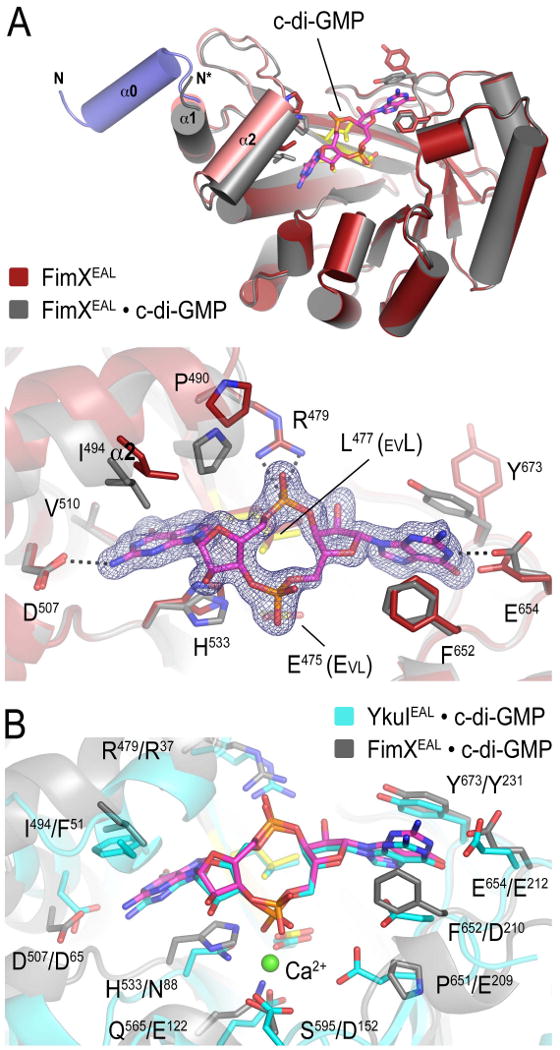
(A) Crystal structure of FimXEAL•c-di-GMP. The c-di-GMP-bound structure of FimXEAL, (colored in grey) was superimposed onto the nucleotide-free structure (colored in red) shown in Figure 1. Although present in the construct, the linker helix (blue) is disordered in FimXEAL•c-di-GMP. The bottom panel shows a close-up view of the nucleotide binding pocket, with residues involved in c-di-GMP binding shown as sticks. The (|Fo|-|Fc|) electron density map shown as calculated from a model prior to inclusion of nucleotide and is contoured at 3σ.
(B) Comparison of the degenerate EAL domain of FimX and the EAL domain of YkuI. The c-di-GMP bound state of FimXEAL (grey) and YkuIEAL (PDB code 2w27; cyan) were superimposed. Nucleotide and magnesium coordinating residues are shown as sticks. Residue type and sequence position are indicated (left: FimXEAL, right: YkuIEAL). YkuI was crystallized in the presence of calcium (green sphere).
Cyclic di-GMP binding had very little impact on the conformation of the TIM barrel, exemplified by an overall r.m.s. deviation of 0.32 Å (over all atoms) between the two structures. The most notable difference was a bilateral clamping down of the protein onto the c-di-GMP molecule. While this involved aromatic π-stacking interactions between one guanosine base and Y673, adopting an alternative rotamer conformation compared to the apo-state, and F652 of FimX on one side, the second guanosine base of the nucleotide occupied a somewhat hydrophobic cavity formed at the interface of the lobe (helix α2) and the barrel core (Figure 2A). Interactions of helix α2 with the second guanosine base of c-di-GMP induce a rigid body shift of the lobe by 3.7 Å, concomitant with a melting of the linker helix α0 (Figure 2A, top panel).
Several highly conserved residues in EAL domains have been identified based on homology modeling, and were shown to be essential for phosphodiesterase activity (Rao et al., 2008). Structural characterization of the c-di-GMP-bound EAL domain of YkuI corroborated these findings (Minasov et al., 2009). Residues crucial for activity included a conserved general base catalyst (E352 in RocR/SadR, E239 in tdEAL/2R60, or E209 in YkuI) and the Mg2+-coordinating residues N88, E122, D152 and E523, the glutamic acid residue from the EAL motif (YkuI numbering). In FimX, the general base-catalyst is substituted by a proline (P651). In addition, three of the Mg2+-coordinating side chains have been replaced non-conservatively in FimX (H533, Q565 and S595 corresponding to N88, E122 and D152 of YkuI, respectively) (Figure 2B). Indeed, no metal ion could be identified in the respective binding site of FimX. However, several other residues that potentially play an important role in c-di-GMP binding (Minasov et al., 2009; Rao et al., 2008), such as Y231, E212, D65, R37 and L35 of YkuI, are conserved in FimX and mediate numerous specific polar and hydrophobic interactions between the protein and the nucleotide (Y673, E654, D507, R479 and L477 in FimX; Figure 2B). One of the phosphates of c-di-GMP participates in a charge-charge interaction with the conserved R479, while residue L477 of the EVL motif serves as a platform to support the phosphate ring of the cyclic nucleotide.
We determined the affinity and stoichimetry of c-di-GMP binding by using isothermal titration calorimetry. The isolated EAL domain of FimX bound c-di-GMP with high affinity (Kd=104.2 nM) and stoichiometry of 1:1.11 (Figure 3A). A similar affinity and stoichiometry (Kd=125.0 nM; N=0.93) were found for full-length FimX (Figure 3B). The titration with FimXfull-length showed an initial slope that may indicate a cooperative or sequential binding of c-di-GMP, and fits using such models were marginally better. Since the molecular basis for such a phenomenon is currently unknown, we report values for a simple one-site binding model. The affinity of FimX for c-di-GMP was at the higher end of the spectrum reported for other EAL domains and c-di-GMP receptor proteins, which spans three orders of magnitude (60 nM to low μM) (Hengge, 2009).
Figure 3. Isothermal titration calorimetry data for c-di-GMP binding to FimX.
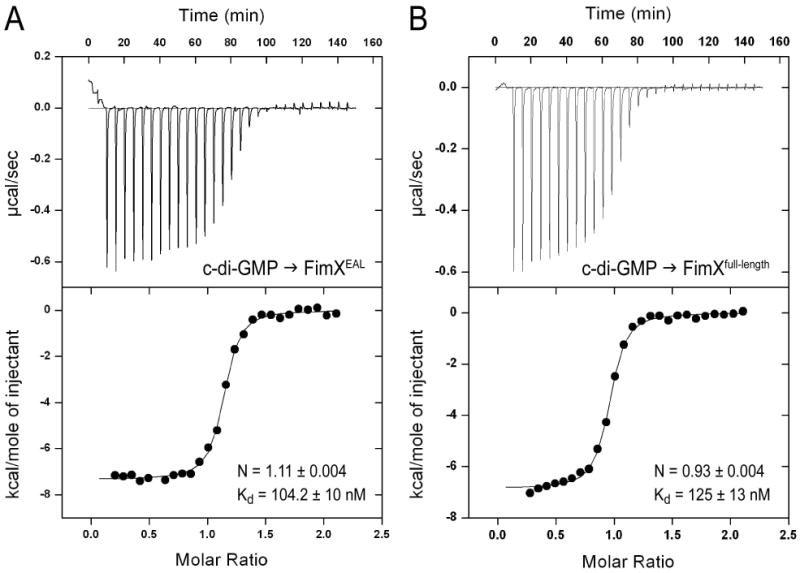
(A) Binding of c-di-GMP to FimXEAL. Calorimetric titration for c-di-GMP (250 μM) titrated into FimXEAL (25 μM) is shown. Derived values for Kd and stoichiometry (N) are shown.
(B) Binding of c-di-GMP to full-length FimX. Calorimetric titration for c-di-GMP (250 μM) titrated into full-length FimX (25μM) is shown. Derived values for Kd and stoichiometry (N) are shown.
Taken together, the structural analysis revealed the molecular basis for c-di-GMP binding to a degenerate EAL domain. The results suggest that FimX functions as a high-affinity c-di-GMP-binding protein that lacks enzymatic activity, consistent with previous studies and our biochemical characterization (Kulasakara et al., 2006; Rao et al., 2008) (Supplementary Figure 1).
Structure of the degenerate GGDEF domain of FimX
Crystals of FimXGGDEF diffracted X-rays to 2.0 Å resolution, and the structure was solved by molecular replacement in space group P21 with the GGDEF domain of PleD as the search model (PDB code 1w25; Chan et al., 2004). The asymmetric unit contained two GGDEF domain molecules. Although GGDEF domain dimerization plays an important role in the catalytic activity and regulation of active cyclases (Chan et al., 2004; De et al., 2008; Wassmann et al., 2007), packing contacts in FimXGGDEF crystals differ from the ones observed previously and are likely to be biologically irrelevant, considering the small dimensions of interfacial areas (614 Å2 which corresponds to 7.5% of the total GGDEF domain surface) (Krissinel and Henrick, 2007).
Similar to the GGDEF domains of PleD and WspR, the overall fold of the FimX diguanylate cyclase domain consists of a central 5-stranded β-sheet surrounded by four α-helices (Figure 4A) (Chan et al., 2004; De et al., 2008). With the exception of some flexible loops, the GGDEF domain of FimX superimposed very well on the cyclase domains from PleD (r.m.s. deviation of 1.2 Å for 150 Cα) and WspR (r.m.s. deviation of 1.3 Å for 152 Cα). The characteristic GGEEF motif (G346DSIF in FimX) formed a loop between β2 and β3. However, several residues important for catalysis and nucleotide binding underwent non-conservative changes in FimX, yielding a degenerate GGDEF domain that is inactive (Supplementary Figure 1). The GGDEF domain of FimX lacks residues that coordinate the γ-phosphate moiety of GTP (K442 and R446 in PleD, substituted by A418 and G422 in FimX), as well as residues involved in Mg2+ coordination (E370 of the GGEEF motif and D327 in PleD, substituted by S348 and H305 in FimX; closed arrows in Figure 4D; Figure 4B) (Wassmann et al., 2007). Some of the residues that contribute to a binding pocket for the guanosine base, in particular D322 and Y309, are conserved or similar to active cyclases, suggesting that FimX may still be able to bind GTP (asterisks in Figure 4D). In the conformation observed in the crystals, D347 of the GDSIF motif and Y309 occupy the nucleotide binding cleft, and GTP binding would require a remodeling of this site. CC3396 from Caulobacter crescentus, a dual-domain phosphodiesterase that becomes activated allosterically by GTP binding to its degenerate GGDEF domain, contains a glutamate substitution instead of the second glycine at its signature motif (GEDEF), but unlike FimX displays residues that can interact with the phosphate moieties of GTP (open arrow in Figure 4D) (Christen et al., 2005). Considering the already rather low affinity of CC3396 for GTP (∼4 μM), nucleotide binding to the GGDEF domain of FimX is likely to be even weaker, consistent with a lack of GTP binding in vitro (data not shown).
Figure 4. Structure of the degenerate GGDEF domain of FimX.
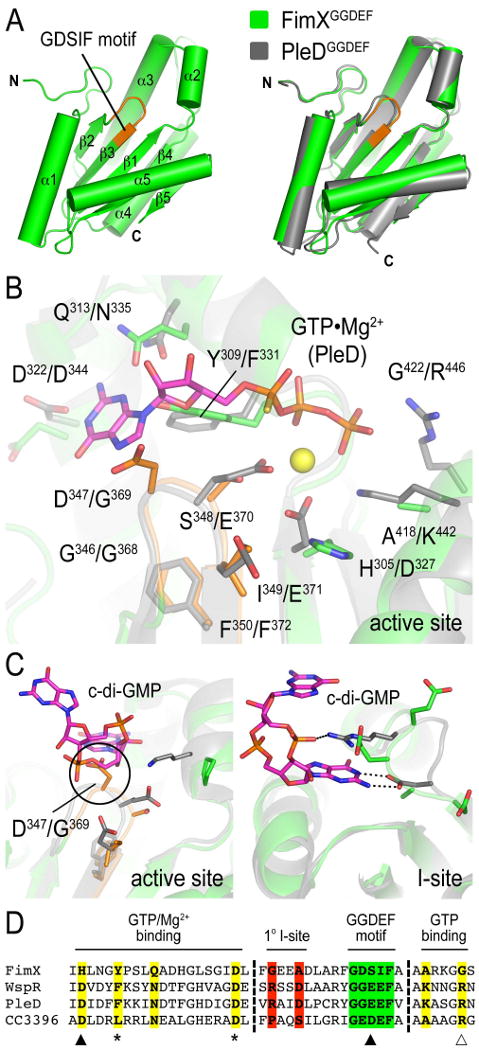
(A) Crystal structure of FimXGGDEF. The GGDEF domain of FimX is shown in green. The degenerate GGDEF motif (G346DSIF in FimX) at the active site is colored in orange. The right panel shows a superposition with the GGDEF domain of the active diguanylate cyclase PleD from Caulobacter crescentus (PDB code 1w25; grey).
(B) Close-up view of the degenerate nucleotide binding site of FimXGGDEF. The structures of nucleotide-free FimXGGDEF (green) and the GGDEF domain of PleD bound to a nonhydrolyzable GTP analog (PDB code 2v0n; grey) were superimposed. The degenerate GDSIF motif of FimX is colored in organge. Nucleotide and magnesium (yellow sphere) coordinating residues are shown as sticks. Residue type and sequence position are indicated (left: FimXGGDEF, right: PleD).
(C) Cyclic di-GMP-incompatible active and I-sites of FimXGGDEF. The structures of nucleotide-free FimXGGDEF (green) and the GGDEF domain of PleD bound to c-di-GMP at the active (left panel) and I-site (right panel) (PDB code 2w25; grey) were superimposed. Cyclic di-GMP coordinating residues of PleD and corresponding residues of FimX are shown as sticks.
(D) Sequence alignment of nucleotide binding motifs in GGDEF domain-containing proteins. GGDEF domain sequences from FimX, WspR, PleD and CC3396 were aligned using ClustalW. Sequence blocks spanning the nucleotide binding sites (colored residues) are shown. Residues involved in magnesium binding (filled arrows), GTP binding (open arrow and asterisks) are highlighted. The GGDEF motif and the I-site are colored in green and red, respectively.
Superimposing the nucleotide-free GGDEF domain of FimX with that of c-di-GMP-bound PleD revealed the incompatibility of product binding to FimX at the active site. FimX lacks side chains important for c-di-GMP binding, and D347 of the GDSIF motif, located at the position of the second conserved glycine residue in active GGDEF domains, introduced a major steric clash with the nucleotide (Figure 4C, left panel) (Chan et al., 2004). In addition to the GGDEF and EAL domain active sites, a third c-di-GMP binding site has been identified in diguanylate cyclase structures, and has been shown to function as an inhibitory site (I-site) (Chan et al., 2004; Christen et al., 2006; De et al., 2008; Wassmann et al., 2007). The I-site plays an important role in the regulation of cyclase activity and its architecture is distinct compared to the active site. It is formed by a characteristic primary site motif with the consensus RxxD (R359xxD and R242xxD in PleD and WspR, respectively). From sequence and structural alignments, the GGDEF domain of FimX lacks this motif, ruling out c-di-GMP binding via an I-site (Figure 4C, right panel; Figure 4D). Nucleotide binding studies in solution confirmed the EAL domain as sole c-di-GMP binding module of FimX (Supplementary Figure 2).
EAL domain dimerization in the structure of FimXdual
Next, we determined the crystal structure of FimXdual. Although the protein contained both the GGDEF and EAL domains (residues 260-691 of FimX), density for the first 177 N-terminal residues covering the entire GGDEF domain was absent after refinement of the EAL domain. Disordered domains have been reported before in crystal structures such as in the case of some flavoproteins (Gruez et al., 2000; Leys et al., 2003). To rule out the possibility of limited protein degradation during crystallization, crystals were washed and analyzed by SDS-PAGE. The silver-stained gels showed a single band corresponding to the intact dual-domain protein that was used as the input material (Supplementary Figure 3A). Inspection of the crystal lattice revealed large cavities that could accommodate the GGDEF domain from both molecules of FimXdual in the asymmetric unit (Supplementary Figure 3B). Furthermore, the crystal solvent content (VM) estimated based on the protein molecular weight and crystal lattice dimensions indicated the dual-domain module as protomer rather than with the final structural model, which comprised only the EAL domains (residues 437-687), considering average solvent contents observed for protein crystals (VMdual=58.5% vs VMfinal model=74.8%). Given the relatively low Rfree of 25.2% of the refined model at a resolution of 3.0 Å, the GGDEF domain is presumably disordered in the crystals with insignificant contribution to the diffraction of X-rays.
The EAL domain of FimXdual crystallized in a dimeric state that was formed by the two molecules in the asymmetric unit. In such an assembly, the lobes of the protomers engage in mutual interactions with the TIM barrels of the adjacent chains (Figure 5A). Helix α0 melted and part of the loop structure contributes to dimerization. Although pair-wise contacts in the interface involve mainly backbone atoms and lack ionic bonds, there is a remarkable shape and physicochemical complementarity across ∼3300 Å2 of buried surface area (Figure 5A). Computational analysis of the protein interface suggested that the interface is of biological relevance with an estimated solvation free energy (ΔiG) of -11.9 kcal/mol, according to the PISA server (Krissinel and Henrick, 2007).
Figure 5. Structure of an EAL domain dimer observed in FimXdual crystals.
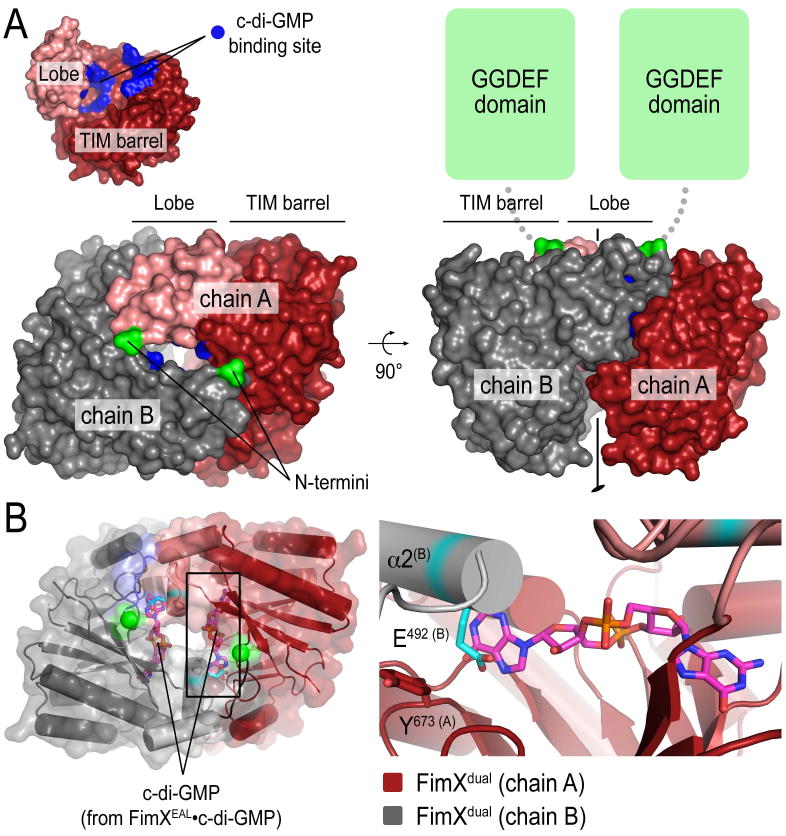
(A) Crystal structure of a homodimeric EAL domain of FimX. In the structure of FimXdual, the two EAL domains in the asymmetric unit (colored in red and grey) form a dimeric assembly. The inset (top left panel) shows a surface presentation of an EAL domain monomer. The nucleotide binding site is colored in blue. Two orthogonal views are shown. The N-termini of the EAL domain are colored in green
(B) Blocked c-di-GMP binding in the dimer. The structures of FimXEAL•c-di-GMP and FimXdual were superimposed. Only the nucleotide of the c-di-GMP-bound structure is shown as sticks. The close-up view (right panel) shows clashes of c-di-GMP with the dimeric EAL domain.
The EAL domain dimer forms a cage burying the nucleotide binding sites in an inner cavity inaccessible for nucleotide binding (Figure 5A). In addition, placing a c-di-GMP molecule into the binding site based on a structural alignment with FimXEAL•c-di-GMP revealed that the conformation of FimX in the dimer is incompatible with c-di-GMP binding (Figure 5B). Major clashes between the nucleotide and the protein involved the backbone of helix α2 of the dimerization partner and the side chain of E492 in that helix (Figure 5B, right panel). By accommodating helix α2 of an adjacent protomer, the side chain of Y673, one of the two residues that clamps the guanosine base via aromatic stacking interactions, faces away from the nucleotide-binding site (Figure 5B, right panel). We predict that nucleotide binding would disrupt such dimer conformation.
Interestingly, apo-FimXEAL formed a similar dimer in the crystal lattice with a symmetry-related molecule (Figure 6A). Despite the different construct boundaries and crystallographic space groups (P41212 for FimXdual and P6422 for FimXEAL), the relative domain orientation and core interfacial surfaces in both dimers were comparable (Figure 6). While the interactions between the lobes of adjacent FimXEAL molecules were preserved preventing concomitant c-di-GMP binding, the cage formed by dimerization was less closed than that observed in the crystals grown with FimXdual. Differences originated from a rigid body rotation of one EAL domain by ∼11° with respect to the adjacent EAL domain, which resulted in a reduction in buried surface area of ∼1000 Å2 (Figure 6). While the N-terminal linker participates in dimerization of FimXdual placing the N-termini at the center of the assembly, this motif forms helix α0 in apo-FimXEAL. The engagement of the helix in the dimer interface is less extensive and the N-termini are more separated in this alternative dimer conformation, pointing away from the core (Figure 6A, right panel). Energetic estimations predicted that the weaker interface retained its biological relevance, suggesting that structural changes in the linker helix α0 that connects the GGDEF and EAL domains could be functionally coupled to the opening of the dimer interface or vice versa.
Figure 6. Dimer interfaces in EAL domain-containing structures of FimX.
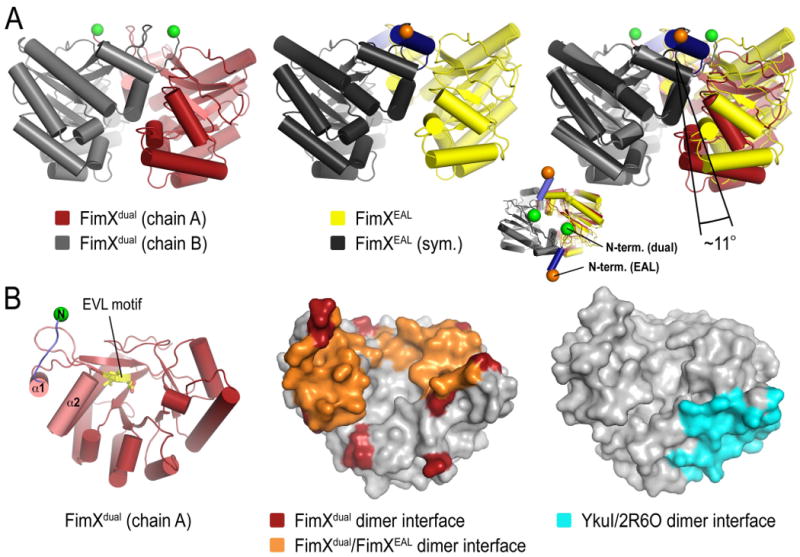
(A) Conformational variation in FimXEAL and FimXdual domain dimers. A comparison of the EAL domain dimer observed in the crystal structure of FimXdual (left panel) and the FimX dimer in apo-FimXEAL (middle panel) crystals is shown. The structure of FimXdual is colored as in Figure 4. The protomers of the FimXEAL structure are colored in yellow and grey, with their N-termini shown as orange spheres. The right panel shows a superposition of the two dimers using a single EAL domain as reference. The inset is an orthogonal view highlighting the position of the N-termini in both dimers.
(B) Mapping of dimer interfaces onto the surface of the EAL domain. The structure of FimXEAL shown as ribbons (left panel) depicts the orientation of the surface presentations (middle and right panel). In the middle panel, interfacial residues of the apo-FimXEAL dimer are colored orange. Additional residues contributing to dimerization in FimXdual are colored in red. In the right panel, a dimer interface observed in structures of the EAL domain-containing protein tdEAL and YkuI is colored in cyan on the surface of the crystal structure of YkuI (PDB code 2w27).
The potential biological significance of the dimer interface presented here is unclear. As we describe below, the EAL domain is monomeric in solution and unlikely to form this interaction in the context of the full-length FimX dimer conformation observed in solution. However, it is tempting to speculate that similar interactions could mediate higher-order oligomerization or complex formation with other proteins (see discussion below).
Mapping of sequence conservation considering all 16 GGDEF-EAL domain-containing proteins from P. aeruginosa onto the accessible surface of FimXEAL indicated that this dimer interface may not be a general feature in EAL domains (Supplementary Figure 4). In addition, dimerization of the EAL domains of FimX was not observed in solutions of purified proteins (see below). The interface is distinct from and non-overlapping with those observed in structures of tdEAL and YkuI, two unrelated, presumably active EAL domains (Figure 6B) (Minasov et al., 2009). Despite low overall sequence similarity, both tdEAL and YkuI use a common, conserved interface for dimerization located on the TIM barrel distal to the active site and the lobe. Although the interface is moderately conserved in FimX, dimerization via this surface is prohibited due to backbone clashes in the current conformation of the FimX EAL domain (data not shown). Interestingly, this dimerization motif that corresponds to a loop region in the EAL domain has recently been identified as being functionally linked to catalysis in active phosphodiesterases (Rao et al., 2009).
Structural characterization of FimX in solution
In size exclusion chromatography, full-length FimX and the truncated forms eluted in single sharp peaks, indicating that the proteins were monodispersed with regard to their oligomeric state in solution (Figure 7A). Calibration using globular standard proteins indicated that FimXdual and constructs that contained the N-terminal response receiver and PAS domains, FimXΔEAL and FimXfull-length, eluted at volumes more consistent with higher molecular weight forms than predicted from the protein sequence, or with proteins of non-globular shape, while FimXEAL appeared monomeric in solution (Figure 7A; Table 2).
Figure 7. Oligomerization state of FimX in solution.
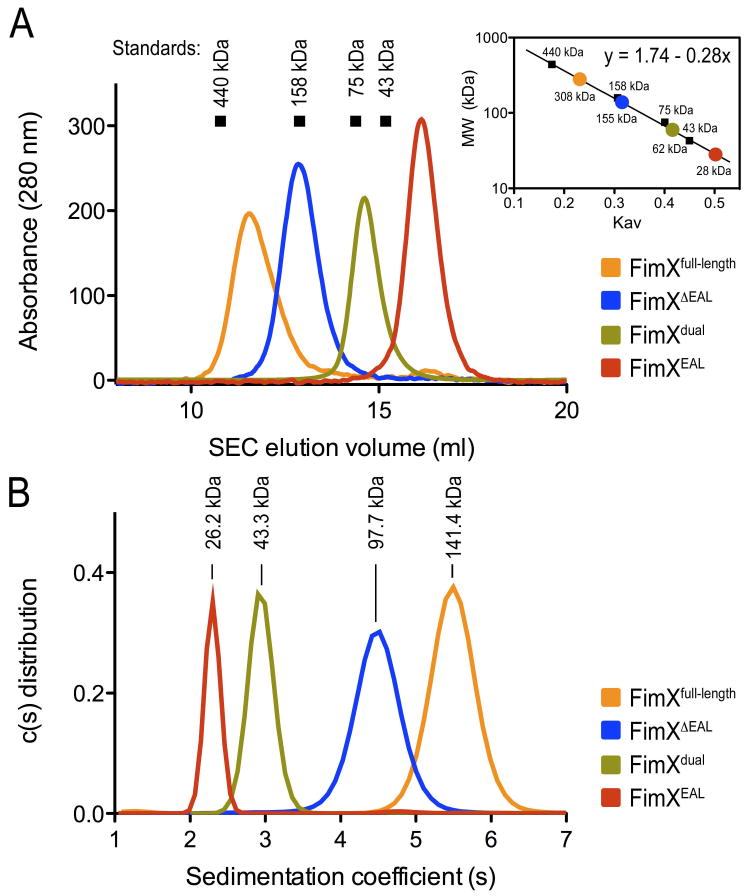
(A) Size exclusion chromatography. Standard proteins (arrows), FimX full-length and truncations were analyzed by size exclusion chromatography in gel filtration buffer. Standardization using elution times, molecular weights of the standard proteins, and column specifics yielded a standard curve for molecular weight estimations for FimX.
(B) Analytical ultracentrifugation. FimX full-length and truncations were analyzed by sedimentation velocity analytical ultracentrifugation in gel filtration buffer. Molecular weights were analyzed by using the program SedFit.
Table 2.
Molecular weight of FimX in solution.
| Molecular mass (kDa) | Sedimentation coefficient (S) | Frictional ratio | ||||
|---|---|---|---|---|---|---|
| Sequencea | SECb | SAXSc | AUCd | |||
| FimXfull-length | 76.0 | 308.0 | 137.6 | 141.4 | 5.69 | 1.63 |
| FimXΔEAL | 47.4 | 155.0 | 101.1 | 97.7 | 4.65 | 1.67 |
| FimXdual | 47.3 | 61.6 | 44.4 | 43.3 | 2.96 | 1.50 |
| FimXEAL | 28.9 | 28.2 | 26.2 | 26.2 | 2.38 | 1.31 |
Molecular mass calculated from the primary sequence.
Molecular mass calculated from size exclusion chromatography (SEC).
Molecular mass calculated from small-angle X-ray scattering data (see Supplementary Table 2).
Molecular mass calculated from sedimentation velocity analytical ultracentrifugation.
The accurate molecular weight and oligomeric state of FimX was elucidated by sedimentation velocity analytical ultracentrifugation (Figure 7B). Based on their sedimentation coefficients and frictional ratios, FimXEAL and FimXdual were monomeric (Table 2). The larger frictional ratio for FimXdual (and the elution volume in gel filtration experiments) indicated that the protein is in an extended conformation. For FimXΔEAL and FimXfull-length, the sedimentation coefficients and frictional ratios described dimeric proteins with non-globular shape in solution, which explained the higher apparent molecular masses estimated from size exclusion chromatography data (Table 2). These results elucidated that FimX is an elongated protein in solution, which dimerizes through its N-terminal domains.
To determine the overall architecture and solution conformation of FimX and its nucleotide-bound states, we turned to small-angle X-ray scattering. This method yields molecular geometry parameters such as the radius of gyration (Rg) and maximum diameter of a molecule or assembly (Dmax), as well as the distance distribution or P(r) function, describing the shape of a molecule in solution. We collected and analyzed data for FimXfull-length, FimXΔEAL, FimXdual and FimXEAL, and for the c-di-GMP-bound states of the EAL domain-containing constructs (Figure 8; Supplementary Table 1). The apo-FimXEAL and its c-di-GMP-bound state showed very similar dimensions and shapes that were in agreement with the parameters calculated from the crystal structures (Figure 8D; Supplementary Table 1). Similarly, Rg, Dmax, and P(r) functions determined for FimXfull-length and FimXdual were insensitive to c-di-GMP binding, ruling out major conformational rearrangements upon nucleotide binding (Figures 8A and 8C; Supplementary Table 1). The P(r) functions calculated for the full-length protein showed two main peaks and a shoulder at larger distances, characteristic of an elongated conformation comprising multiple folded domains. The P(r) function for FimXΔEAL revealed a similar pattern with two main peaks but a smaller shoulder and overall shorter Dmax (Figure 8B). The analysis of the solution scattering data suggested that the EAL domains are located at the distal tips of the elongated dimer, contributing to a larger diameter of the full-length assembly.
Figure 8. Solution scattering analysis of FimX.
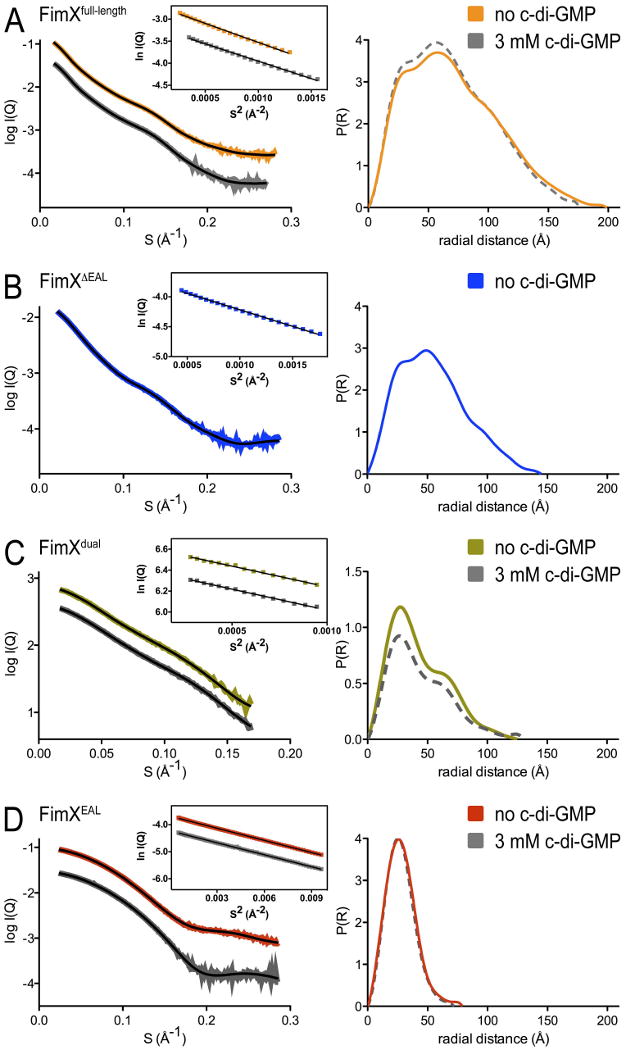
Small-angle X-ray scattering curves and distance distribution functions of FimXfull-length (A), FimXΔEAL (B), FimXdual (C), and FimXEAL (D) are shown after averaging and subtraction of solvent background scattering. Theoretical scattering profiles calculated from the ab initio models with the lowest χ values are shown (black line; see Figure 8). The inset shows Guinier plots (including linear fits) at the low angle region (Smax*Rg<1.3). For constructs containing the EAL domain, experiments were also performed in the presence of c-di-GMP (dashed grey lines).
Modeling of the SAXS data provided low-resolution envelopes corroborating the results described above. Positions of spherical scattering units representing residues in a protein were adjusted by a simulated annealing procedure to optimally match the scattering data (Svergun et al., 2001). FimXfull-length and FimXΔEAL were shown previously to be dimeric in solution by analytical ultracentrifugation, and 2-fold symmetry was imposed during the modeling of these proteins (trials in the absence of symmetry yielded comparable results; data not shown). For each protein, at least twenty independent simulated annealing runs were carried out, and the accuracy of each model was indicated by its goodness of fit to the experimental scattering data (Svergun, 1999). Superposition, averaging over all models, and filtering of the most reproducibly modeled regions generated a consensus or filtered envelope (Figure 9, colored envelopes) (Volkov and Svergun, 2003). The envelopes colored in grey represent the sum of all models prior to filtering.
Figure 9. SAXS-based shape reconstruction of FimX.
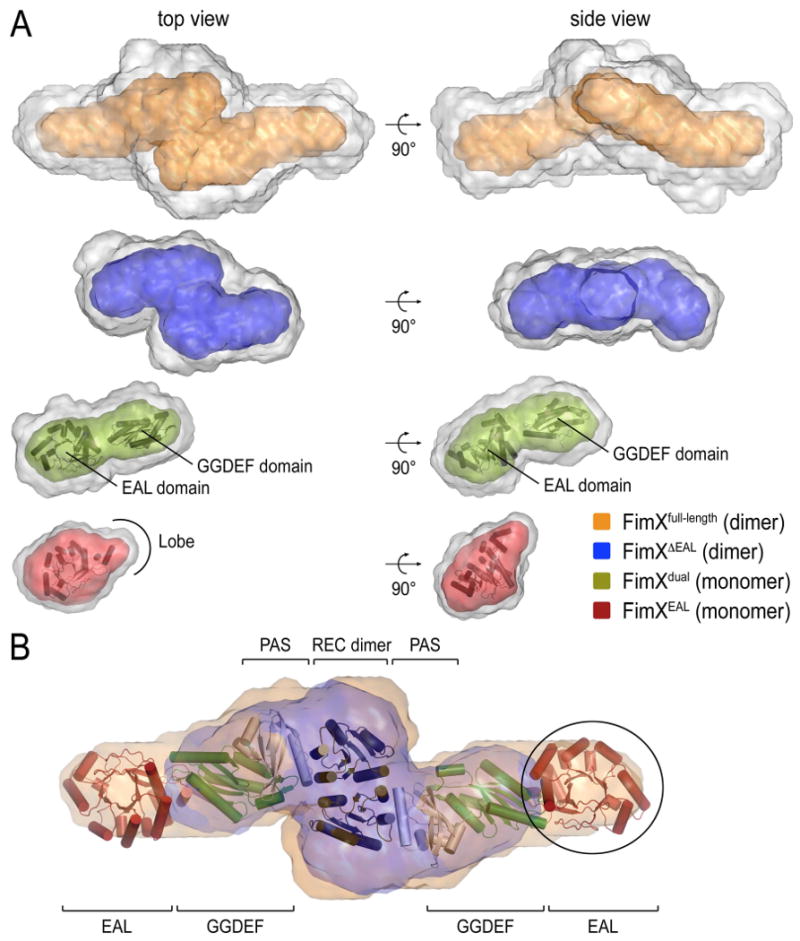
(A) SAXS-based shape reconstruction of FimXEAL (red), FimXdual (green), FimXΔEAL (blue), and FimXfull-length (orange) are shown after averaging of twenty independent models (grey envelopes) and filtering (colored enveloped). High-resolution crystal structures of the EAL and GGDEF domains were docked into the appropriate envelopes and are shown in cartoon presentation.
(B) Model for full-length FimX. High-resolution crystal structures of the EAL and GGDEF domains, as well as models for the dimeric response receiver and PAS domains (PDB codes 3BRE and 1DRM, respectively; De et al., 2008; Gong et al., 1998) were docked into the envelopes modeled from the solution scattering of full-length FimX, and are shown in cartoon presentation. A superposition of solution envelopes for FimXΔEAL (blue) and FimXfull-length is shown. The circle highlights a region in the FimXfull-length envelope that is missing in the FimXΔEAL envelope.
As expected from the Gaussian-shaped P(r) function, models for the isolated EAL domain of FimX described a globular domain into which the crystal structure could be docked (Figure 9A, bottom panel). A short extension observed in the solution envelopes accommodated the lobe of the EAL domain, highlighting the quality and information content of the ab initio modeling. Models for FimXdual described a monomeric, bean-shaped module, and the high-resolution structures of the isolated EAL and GGDEF domains could be placed into the envelope with high confidence (Figure 9A). The refined envelope covering both domains is in contrast to the lack of density for the GGDEF domain in FimXdual crystals, and suggests that the GGDEF domains were not coordinated by crystal packing contacts and might undergo conformational sampling as rigid bodies.
Envelopes calculated for FimXΔEAL were consistent with an elongated assembly containing a central dimerization hub (Figure 9A). Models of dimeric FimXfull-length had a similar serpentine shape but were extended in the longest dimension (Figure 9A). Superposition of the FimXΔEAL and full-length envelopes allowed us to map the position of EAL domains in the context of the entire protein (Figure 9B). EAL domains were located at the far ends of the protein, separated by the N-terminal and GGDEF domains. Given the domain organization, the response receiver domains are likely to mediate dimerization while the GGDEF domains form the protrusions observed in the FimXΔEAL models. Manual docking of crystal structures and the tendency of response receiver domains to dimerize support such a model (Figure 9B).
Taken together, the solution scattering-based analysis revealed an extended conformation of full-length FimX, in which the N-terminal domains separate the EAL domains. In this conformation, the EAL domains are unpaired, in contrast to the dimeric assemblies observed in the crystal structures of apo-FimXEAL and FimXdual. The monomeric state of the EAL domains in solution suggests that dimerization via this interface is kinetically unfavorable, in agreement with the lack of long-range electrostatic interactions (Elcock et al., 1999).
Putative models for FimX function in c-di-GMP signaling
Functional studies elucidated a role for FimX in twitching motility with the phenotypes of gene disruptions suggesting that FimX resembles either a c-di-GMP receptor or negative regulator of phosphodiesterases, rather than an active phosphodiesterase (Huang et al., 2003; Kulasakara et al., 2006). Interestingly, proper localization of FimX to a single pole in cells relies on intact G346DSIF and E475VL motifs in FimX, suggesting that both the degenerate GGDEF and EAL domains are important for molecular interactions (Kazmierczak et al., 2006). Our structural and biochemical analyses revealed that the EAL domain of FimX is still capable of c-di-GMP binding but lacks important residues for catalysis, consistent with previous reports (Kulasakara et al., 2006; Rao et al., 2008). Two of the EAL domain-containing crystal structures showed an extensive dimerization interface that is incompatible with c-di-GMP binding. In contrast, solution scattering-based shape reconstructions indicated monomeric EAL domains in the context of full-length FimX, which forms a dimer via its N-terminal domains.
Integrating our data and previous studies, two plausible molecular mechanisms for the cellular function of FimX can be proposed. In a canonical receptor model for FimX function, c-di-GMP would bind to the EAL domain with high-affinity. Complex formation with yet unidentified binding partners may be regulated by c-di-GMP binding to the EAL domain. Complex formation could be facilitated by c-di-GMP binding to FimX (as shown in Supplementary Figure 5A), but it is equally feasible that the monomeric, nucleotide-free EAL domain is engaged in interactions that become destabilized upon nucleotide binding. Interestingly, the degenerate EAL domain of a blue light sensor from E. coli has recently been identified as a module for controlled protein interactions (Tschowri et al., 2009). Binding and release of the partner protein is regulated by the light-sensing domain located adjacent to the EAL domain, but is independent of c-di-GMP degradation or binding. In another recent case, a catalytically inactive GGDEF-EAL domain-containing protein has been shown to transmit signals across the cell membrane via c-di-GMP binding to its degenerate EAL domain (Newell et al., 2009). The two studies reveal the usage of EAL domains as switchable protein or nucleotide binding modules, and FimX may combine both features in controlling swarming behavior.
An extension of this model could involve c-di-GMP-triggered dissociation of FimX EAL domain dimers (Supplementary Figure 5A). Disrupting EAL domain dimers or oligomers may already provide a switch, either in conformation or localization. While we cannot exclude conformational changes within the full-length protein that would bring the EAL domains into close proximity for dimerization, an alternative model could involve the formation of higher-order oligomers at regions of high local protein concentration such as the pole of the bacterial cell, where FimX accumulates (Huang et al., 2003; Kazmierczak et al., 2006).
In an alternative model, other EAL or dual GGDEF-EAL domain-containing proteins could form specific complexes with FimX via interactions similar to the ones observed in FimX EAL domain homodimers. Such assemblies would sequester the active site of the partner EAL domain (and possibly the GGDEF domain in the case of dual domain proteins), rendering them unavailable for catalysis (Supplementary Figure 5B). The FimXEAL and FimXdual dimerization is in part facilitated by shape complementarity and backbone interactions, which are likely to be conserved features in EAL domains. This model would predict that c-di-GMP binding to the EAL domain of FimX would liberate the partner EAL domain, revealing its phosphodiesterase active site. Such a mechanism would be also consistent with the mutational studies (Kulasakara et al., 2006). An attractive feature of the phosphodiesterase inhibitor model is the prediction that the activation of partner EAL domains would be self-limiting since the freed activity would eventually degrade the c-di-GMP molecule that is bound to FimX, reversing the process.
Degenerate domains with alternative functionality may account in part for the large number of GGDEF and EAL domains encoded in eubacterial genomes and for the complexitity in c-di-GMP-mediated signaling. Little is known about the binding partners of GGDEF and EAL domain-containing proteins and the targets of c-di-GMP, and how the signal is transmitted. Genetic and phenotypic analyses provided invaluable information regarding proteins that affect similar pathways and clearly demonstrated signaling specificity and hierarchy (Beyhan et al., 2008; Kader et al., 2006; Kulasakara et al., 2006; Sommerfeldt et al., 2009). Deciphering the interactions of FimX and other factors involved in c-di-GMP signaling on a molecular level will be instrumental in determining the mechanism underlying FimX function, and signal transmission in general.
Material and Methods
Protein expression and purification
The coding regions corresponding to full-length FimX from P. aeruginosa PAO1 (Huang et al., 2003) was amplified by standard PCR using genomic DNA as a template. Coding regions corresponding to FimXΔEAL (residues 1-431), FimXdual (residues 260-691), FimXEAL (residues 429-691), and FimXGGDEF (residues 260-431) were PCR-amplified from an expression plasmid encoding FimXfull-length. DNA inserts were cloned into a modified pProExHTb (Invitrogen) expression plasmid yielding N-terminally hexahistidine-tagged proteins (FimXfull-length, FimXdual, and FimXGGDEF), or into a modified pET28a expression plasmid (Novagen) yielding N-terminally hexahistidine-tagged SUMO fusion proteins (FimXΔEAL and FimXEAL). The hexahistidine tag and the hexahistidine-tagged SUMO-moiety were cleavable by using Prescission protease or the protease Ulp-1 from S. cerevisiae, respectively. Proteins were produced following standard liquid chromatography techniques as described previously (De et al., 2008). A detailed protocol is provided as Supplementary Information.
Crystallization, X-ray data collection, and structure solution
All crystals were obtained by hanging drop vapor diffusion by mixing equal volumes of protein (5-30 mg/ml) and reservoir solution followed by incubation at 20°C. For FimXEAL and FimXEAL•c-di-GMP the reservoir solution comprised 0.1 M Hepes, pH 7.1, 1.1 M ammonium sulfate, 0.5% PEG8000. Cyclic di-GMP bound crystals were obtained by soaking nucleotide-free crystals in reservoir solution supplemented with 1 mM c-di-GMP. Crystals of FimXGGDEF were obtained by incubation over a reservoir solution comprising 0.1 M BisTris, pH 6.5, 29-33% Peg MME 5000. Crystals of FimXdual were obtained by incubation over a reservoir solution comprising 0.1 M NaOAc, pH 5.0, 0.5-0.9 M Na-formate, and 5 mM GTP/MgCl2. For cryoprotection, single crystals were soaked in reservoir solution supplemented with 10% xylitol or 20% glycerol. All crystals were flash-frozen in liquid nitrogen and kept at 100K during data collection.
Data sets were collected using synchrotron radiation at the Cornell High Energy Synchrotron Source (CHESS, Ithaca, beamlines A1 and F1) and at the National Synchrotron Light Source (NSLS, Brookhaven, beamline X29A). Data reduction was carried out with the software XDS (Kabsch, 1993). Structures were solved by molecular-replacement or single anomalous dispersion using the software package Phenix (Adams et al., 2002). The models were built and refined manually using Coot (Emsley and Cowtan, 2004), followed by iterative minimization cycles in Phenix (Adams et al., 2002). Crystallographic statistics for data collections and refinements are shown in Table 1.
Structures were analyzed by using and the protein interfaces, surfaces and assemblies service PISA at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) (Krissinel and Henrick, 2007). Structure superimpositions were performed with LSQMAN (Kleywegt, 1996) and illustrations were made in Pymol (DeLano Scientific).
Nucleotide loading state and phosphodiesterase assays
Reverse-phase HPLC was used to separate nucleotides as described previously (De et al., 2008). A detailed protocol is provided as Supplementary Information.
Isothermal titration calorimetry
Apparent dissociation constants (Kd) and stoichiometry of interactions were measured by isothermal titration calorimetry (ITC) using a VP calorimeter (Microcal, Amherst, MA). Calorimetric titrations of c-di-GMP (250 μM in the syringe; 10 μl injections) and FimX (25 μM in the cuvette) were carried out at 20°C in assay buffer (25 mM Tris-Cl, pH 5.5, 100 mM NaCl) with a spacing of 300 seconds between injections. ITC data were analyzed by integrating heat effects normalized to the amount of injected protein and curve-fitting based on a single-site binding model using the Origin software package (Microcal, Northampton, MA). The dissociation constant was derived from the data by using standard procedures.
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out using an XL-I analytical ultracentrifuge (Beckman Coulter) equipped with an AN-60 Ti rotor. Proteins (0.5-2 mg/ml) were diluted in assay buffer and were analyzed at a centrifugation speed of 130,000 × g. Data collection was carried out at 280 nm, followed by data analysis using the program SedFit (version 11.0).
Small angle X-ray scattering (SAXS) and SAXS-based shape reconstruction
SAXS data were collected at the Cornell High Energy Synchrotron Source (CHESS, Ithaca, beamline G1) at an electron energy of 8KeV or at the Advanced Photon Source (APS, BESSRC-CAT, beamline 12-IDC, Argonne, IL) at the electron energy of 12 KeV. Data was collected at 4°C on homogeneous samples. Data reduction, analysis and free-atom modeling of the SAXS data was carried out by using the program package ATSAS (31). A detailed description is provided as Supplementary Information.
Supplementary Material
Acknowledgments
We are grateful to Bill Horne for providing access to isothermal titration calorimetry and to Cynthia Kinsland for technical assistance with analytical ultracentrifugation. We thank the scientists and staff at the Cornell High Energy Synchrotron Source (CHESS), the Advanced Photon Source (APS), and the National Synchrotron Light Source (NSLS) for assistance with synchrotron data collection. This work is based upon research conducted at CHESS/MacCHESS, which is supported by the National Science Foundation (award DMR-0225180) and the National Institutes of Health (NIH)/National Institute of General Medical Sciences (award RR-01646). NSLS is supported by the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the NIH. This work was supported by the NIH under award 1R01GM081373 (H.S.) and a PEW Scholar award in Biomedical Sciences (H.S.).
Footnotes
Accession Numbers: Atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank under ID code 3HV8, 3HV9, 3HVA, and 3HVB.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams P, Grosse-Kunstleve R, Hung L, Ioerger T, McCoy A, Moriarty N, Read R, Sacchettini J, Sauter N, Terwilliger T. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Amikam D, Galperin MY. PilZ domain is part of the bacterial c-di-GMP binding protein. Bioinformatics. 2006;22:3–6. doi: 10.1093/bioinformatics/bti739. [DOI] [PubMed] [Google Scholar]
- Banner DW, Bloomer AC, Petsko GA, Phillips DC, Pogson CI, Wilson IA, Corran PH, Furth AJ, Milman JD, Offord RE, Priddle JD, Waley SG. Structure of chicken muscle triose phosphate isomerase determined crystallographically at 2.5 angstrom resolution using amino acid sequence data. Nature. 1975;255:609–614. doi: 10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- Beyhan S, Odell LS, Yildiz FH. Identification and characterization of cyclic diguanylate signaling systems controlling rugosity in Vibrio cholerae. J Bacteriol. 2008;190:7392–7405. doi: 10.1128/JB.00564-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Paul R, Samoray D, Amiot N, Giese B, Jenal U, Schirmer T. Structural basis of activity and allosteric control of diguanylate cyclase. Proc Natl Acad Sci USA. 2004;101:17084–17089. doi: 10.1073/pnas.0406134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen B, Christen M, Paul R, Schmid F, Folcher M, Jenoe P, Meuwly M, Jenal U. Allosteric control of cyclic di-GMP signaling. J Biol Chem. 2006;281:32015–32024. doi: 10.1074/jbc.M603589200. [DOI] [PubMed] [Google Scholar]
- Christen M, Christen B, Folcher M, Schauerte A, Jenal U. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J Biol Chem. 2005;280:30829–30837. doi: 10.1074/jbc.M504429200. [DOI] [PubMed] [Google Scholar]
- D'Argenio DA, Miller SI. Cyclic di-GMP as a bacterial second messenger. Microbiology. 2004;150:2497–2502. doi: 10.1099/mic.0.27099-0. [DOI] [PubMed] [Google Scholar]
- De N, Pirruccello M, Krasteva PV, Bae N, Raghavan RV, Sondermann H. Phosphorylation-independent regulation of the diguanylate cyclase WspR. PLoS Biol. 2008;6:e67. doi: 10.1371/journal.pbio.0060067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerig A, Abel S, Folcher M, Nicollier M, Schwede T, Amiot N, Giese B, Jenal U. Second messenger-mediated spatiotemporal control of protein degradation regulates bacterial cell cycle progression. Genes Dev. 2009;23:93–104. doi: 10.1101/gad.502409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock AH, Gabdoulline RR, Wade RC, McCammon JA. Computer simulation of protein-protein association kinetics: acetylcholinesterase-fasciculin. J Mol Biol. 1999;291:149–162. doi: 10.1006/jmbi.1999.2919. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Nikolskaya AN, Koonin EV. Novel domains of the prokaryotic two-component signal transduction systems. FEMS Microbiol Lett. 2001;203:11–21. doi: 10.1111/j.1574-6968.2001.tb10814.x. [DOI] [PubMed] [Google Scholar]
- Gong W, Hao B, Mansy SS, Gonzalez G, Gilles-Gonzalez MA, Chan MK. Structure of a biological oxygen sensor: a new mechanism for heme-driven signal transduction. Proc Natl Acad Sci USA. 1998;95:15177–15182. doi: 10.1073/pnas.95.26.15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruez A, Pignol D, Zeghouf M, Coves J, Fontecave M, Ferrer JL, Fontecilla-Camps JC. Four crystal structures of the 60 kDa flavoprotein monomer of the sulfite reductase indicate a disordered flavodoxin-like module. J Mol Biol. 2000;299:199–212. doi: 10.1006/jmbi.2000.3748. [DOI] [PubMed] [Google Scholar]
- Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci USA. 2005;102:14422–14427. doi: 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman JW, Harwood CS. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol. 2008;69:376–389. doi: 10.1111/j.1365-2958.2008.06281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocker B, Jurgens C, Wilmanns M, Sterner R. Stability, catalytic versatility and evolution of the (beta alpha)(8)-barrel fold. Curr Opin Biotechnol. 2001;12:376–381. doi: 10.1016/s0958-1669(00)00230-5. [DOI] [PubMed] [Google Scholar]
- Huang B, Whitchurch CB, Mattick JS. FimX, a multidomain protein connecting environmental signals to twitching motility in Pseudomonas aeruginosa. J Bacteriol. 2003;185:7068–7076. doi: 10.1128/JB.185.24.7068-7076.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- Kader A, Simm R, Gerstel U, Morr M, Römling U. Hierarchical involvement of various GGDEF domain proteins in rdar morphotype development of Salmonella enterica serovar Typhimurium. Mol Microbiol. 2006;60:602–616. doi: 10.1111/j.1365-2958.2006.05123.x. [DOI] [PubMed] [Google Scholar]
- Kazmierczak BI, Lebron MB, Murray TS. Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol Microbiol. 2006;60:1026–1043. doi: 10.1111/j.1365-2958.2006.05156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr D Biol Crystallogr. 1996;52:842–857. doi: 10.1107/S0907444995016477. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, Lee D, Neely A, Hyodo M, Hayakawa Y, Ausubel F, Lory S. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′-5′)-cyclic-GMP in virulence. Proc Natl Acad Sci USA. 2006;103:2839–2844. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, Lory S. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol. 2007;65:1474–1484. doi: 10.1111/j.1365-2958.2007.05879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys D, Basran J, Talfournier F, Sutcliffe MJ, Scrutton NS. Extensive conformational sampling in a ternary electron transfer complex. Nat Struct Biol. 2003;10:219–225. doi: 10.1038/nsb894. [DOI] [PubMed] [Google Scholar]
- Minasov G, Padavattan S, Shuvalova L, Brunzelle JS, Miller DJ, Basle A, Massa C, Collart FR, Schirmer T, Anderson WF. Crystal structures of YkuI and its complex with second messenger c-di-GMP suggests catalytic mechanism of phosphodiester bond cleavage by EAL domains. J Biol Chem. 2009 doi: 10.1074/jbc.M808221200. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell PD, Monds RD, O'Toole GA. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc Natl Acad Sci USA. 2009;106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Weiser S, Amiot N, Chan C, Schirmer T, Giese B, Jenal U. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 2004;18:715–727. doi: 10.1101/gad.289504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1-towards automated and web-supported small-angle scattering data analysis. J Appl Cryst. 2007;40:S223–S228. [Google Scholar]
- Rao F, Yang Y, Qi Y, Liang ZX. Catalytic mechanism of cyclic di-GMP-specific phosphodiesterase: a study of the EAL domain-containing RocR from Pseudomonas aeruginosa. J Bacteriol. 2008;190:3622–3631. doi: 10.1128/JB.00165-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao F, Qi Y, Chong HS, Kotaka M, Li B, Li J, Lescar J, Tang K, Liang ZX. The functional role of a conserved loop in EAL domain-based c-di-GMP specific phosphodiesterase. J Bacteriol. 2009 doi: 10.1128/JB.00327-09. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romling U, Gomelsky M, Galperin MY. C-di-GMP: the dawning of a novel bacterial signalling system. Mol Microbiol. 2005;57:629–639. doi: 10.1111/j.1365-2958.2005.04697.x. [DOI] [PubMed] [Google Scholar]
- Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Mayer R, Braun S, de Vroom E, van der Marel GA, van Boom JH, Benziman M. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature. 1987;325:279–281. doi: 10.1038/325279a0. [DOI] [PubMed] [Google Scholar]
- Ryjenkov DA, Simm R, Romling U, Gomelsky M. The PilZ domain is a receptor for the second messenger c-di-GMP: the PilZ domain protein YcgR controls motility in enterobacteria. J Biol Chem. 2006;281:30310–30314. doi: 10.1074/jbc.C600179200. [DOI] [PubMed] [Google Scholar]
- Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol. 2005;187:1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AJ, Ryjenkov DA, Gomelsky M. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol. 2005;187:4774–4781. doi: 10.1128/JB.187.14.4774-4781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simm R, Morr M, Kader A, Nimtz M, Römling U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol. 2004;53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- Simm R, Morr M, Remminghorst U, Andersson M, Romling U. Quantitative determination of cyclic diguanosine monophosphate concentrations in nucleotide extracts of bacteria by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Anal Biochem. 2009;386:53–58. doi: 10.1016/j.ab.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Sommerfeldt N, Possling A, Becker G, Pesavento C, Tschowri N, Hengge R. Gene expression patterns and differential input into curli fimbriae regulation of all GGDEF/EAL domain proteins in Escherichia coli. Microbiology. 2009;155:1318–1331. doi: 10.1099/mic.0.024257-0. [DOI] [PubMed] [Google Scholar]
- Sudarsan N, Lee ER, Weinberg Z, Moy RH, Kim JN, Link KH, Breaker RR. Riboswitches in eubacteria sense the second messenger cyclic di-GMP. Science. 2008;321:411–413. doi: 10.1126/science.1159519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal R, Wong HC, Calhoon R, Gelfand D, Fear AL, Volman G, Mayer R, Ross P, Amikam D, Weinhouse H, Cohen A, Sapir S, Ohana P, Benziman M. Three cdg operons control cellular turnover of cyclic di-GMP in Acetobacter xylinum: genetic organization and occurrence of conserved domains in isoenzymes. J Bacteriol. 1998;180:4416–4425. doi: 10.1128/jb.180.17.4416-4425.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo R, Tischler A, Camilli A. The EAL domain protein VieA is a cyclic diguanylate phosphodiesterase. J Biol Chem. 2005;280:33324–33330. doi: 10.1074/jbc.M506500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschowri N, Busse S, Hengge R. The BLUF-EAL protein YcgF acts as a direct anti-repressor in a blue-light response of Escherichia coli. Genes Dev. 2009;23:522–534. doi: 10.1101/gad.499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann P, Chan C, Paul R, Beck A, Heerklotz H, Jenal U, Schirmer T. Structure of BeF3--modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure. 2007;15:915–927. doi: 10.1016/j.str.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Weinhouse H, Sapir S, Amikam D, Shilo Y, Volman G, Ohana P, Benziman M. c-di-GMP-binding protein, a new factor regulating cellulose synthesis in Acetobacter xylinum. FEBS Lett. 1997;416:207–211. doi: 10.1016/s0014-5793(97)01202-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.