

Abstract
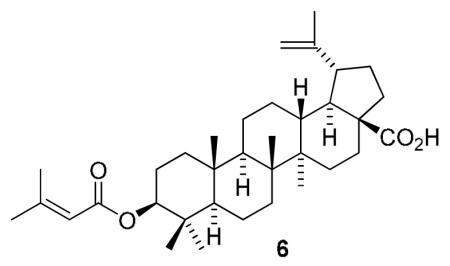
C-3 Esterifications of betulinic acid (BA, 1) and its A-ring homolog, ceanothic acid (CA, 2), were carried out to provide sixteen terpenoids, 4-19, including nine new compounds (4-12). All synthesized compounds were evaluated in an in vitro antitumor-promoting assay using the Epstein-Barr virus early antigen (EBV-EA) activation induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) in Raji cells. Among them, compounds 4-6, 11-14, 16, and 17 displayed remarkable inhibitory effects of EBV-EA activation. BA analog 6, which contains a prenyl-like group, showed the most potent inhibitory effect (100, 76, 37, and 11% inhibition of EBA activation at 1000, 500, 100 and 10 mol ratio/TPA, respectively, with IC50 value of 285 mol ratio/32pmol TPA). Compound 6 merits further development as a cancer preventive agent.
Keywords: Betulinic acid, Ceanothic acid, Antitumor-promoter, Cancer preventive agents, Epstein-Barr virus
Betulinic acid (BA, 1), a pentacyclic lupane-type triterpene, is widely distributed in the bark of several plant species, including the white birch (Betula pubescens), ber tree (Ziziphus mauritiana), tropical carnivorous plants (Triphyophyllum peltatum, Ancistrocladus heyneanus, Diospyros leucomelas), a member of the persimmon family (Tetracera boiviniana), the jambul (Syzygium formosanum), flowering quince (Chaenomeles sinensis), and Pulsatilla chinensis.1,2 Compound 1 and its derivatives exhibit a broad range of biological activities, including inhibition of human immunodeficiency virus (HIV)3,4 and cytotoxic activity against various human cancer cell lines,5-9 as well as antibacterial,10,11 antiinflamatory,12 antimalarial,13,14 and anthelmintic effects. Importantly, 1 has low toxicity and a high safety profile. It is safe even at a dose of 500 mg/kg body weight as an antitumor agent.1 In our continuing study of 1-analogs, many compounds have been synthesized and evaluated for biological activity. Among them, we discovered 3′,3′-dimethylsuccinyl betulinic acid (DSB, PA-457 or bevirimat, 3), which shows extremely potent anti-HIV activity with unique inhibition of a viral maturation step.4,15 Compound 3 is now in clinical development and has shown great promise to date through Phase IIb clinical trials.
Inhibitory effects on Epstein-Barr virus early antigen (EBV-EA) activation induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) in Raji cells correlate well with antitumor promoting activity in vivo.16 Some triterpenoids, including 1, possess potent antitumor promoting activity based on results in this assay.17-19 From our previous studies, the presence of a double bond tends to increase the inhibitory activity. Accordingly, various unsaturated fatty acids including ω3 and ω6 essential fatty acids were incorporated as ester side chains at the C3 position of 1. Chiral monomethyl succinyl analogs of 3, as well as analogs of ceanothic acid (CA, 2), were also prepared to determine structure-activity relationship (SAR) correlations. All synthesized compounds were evaluated for in vitro inhibitory activity against EBV-EA activation as mentioned above. Herein, we describe the syntheses of BA analogs and their potency as antitumor promoters.
The syntheses of 1-analogs are outlined in Scheme 1. Crotonic, sorbic, and dimethylacrylic acids were chosen as lower unsaturated fatty acids, and docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), linolenic, arachidoic, linoleic, and oleic acids were selected as higher unsaturated fatty acids. Condensation of 1 with the corresponding fatty acid was carried out by a well-known esterification procedure using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI) in the presence of N,N-4-dimethylaminopyridine (DMAP) in pyridine. The target compounds 4-12 were obtained in moderate yields after reaction overnight at room temperature. Compounds 17-19 were synthesized previously.15,20 To establish SAR for A-ring, CA (2) and 1-decarboxy-CA (16) were also treated with 3,3-dimethylsuccinic anhydride in the presence of catalytic p-TsOH to afford 13 and 14, respectively (Scheme 2). Interestingly, basic conditions using pyridine did not produce the target compounds, even though 3 is prepared by esterification of 1 with succinyl anhydride under basic conditions. The following two-step sequence gave 1-decarboxy 16 in 47% overall yield without using any protecting group. Oxidation of 2 with pyridinium dichromate (PDC) following reflux in the presence of 2N NaOH in benzene provided 15 in 66% yield. The resulting ketone was reduced by sodium borohydride to afford C-3 alcohol 16 as a single isomer in 71% yield. This two-step sequence is better than the published procedure,21 in which 16 is obtained in five steps from 2 in 42 % overall yield.
Scheme 1.

C3-Esterification of 1 using Various Carboxylic Acids
Scheme 2.

C3-Esterification of 2 and 16
All synthesized compounds, 4-19, along with 1-3, were examined in the EBV-EA activation assay in Raji cells.22 Curcumin, a well-known antitumor promoter, was used as a positive control. As shown in Table 1, all compounds showed low cytotoxicity, as exhibited by high viability (60-70%) of Raji cells, even at a high concentration of TPA (32 nmol, a compound/TPA molar ratio of 1000:1).
Table 1.
| Compound | Percentage EBV-EA positive cells |
||||
|---|---|---|---|---|---|
| Compound concentration (mol ratio/TPAc) | |||||
| 1000 | 500 | 100 | 10 | IC50d | |
| BA (1) | 7.9 ± 0.4 (60) e | 37.2 ± 1.2 | 75.1 ± 2.3 | 100 ± 0.6 | 403 ± 7.6 |
| CA (2) | 8.5 ± 0.5 (60) | 35.7 ± 1.2 | 72.1 ± 2.1 | 100 ± 0.6 | 400 ± 6.9 |
| DSB (3) | 6.1 ± 0.3 (60) | 36.1 ± 1.3 | 74.2 ± 2.2 | 96.5 ± 0.9 | 396 ± 7.9 |
| 4 | 0 ± 0.2 (70) | 26.2 ± 1.1 | 66.7 ± 2.0 | 91.6 ± 0.6 | 299 ± 3.2 |
| 5 | 0 ± 0.2 (70) | 25.7 ± 1.1 | 65.7 ± 2.0 | 90.2 ± 0.6 | 295 ± 3.0 |
| 6 | 0 ± 0.1 (70) | 23.5 ± 1.0 | 62.5 ± 1.9 | 88.7 ± 1.0 | 285 ± 3.0 |
| 7 | 17.0 ± 0.7 (60) | 53.8 ± 1.6 | 83.2 ± 2.5 | 100 ± 0.5 | 546 ± 6.3 |
| 8 | 15.3 ± 0.6(60) | 50.4 ± 1.5 | 80.1 ± 2.5 | 100 ± 0.5 | 523 ± 6.0 |
| 9 | 10.9 ± 0.5 (60) | 43.3 ± 1.4 | 77.8 ± 2.3 | 97.0 ± 0.6 | 445 ± 6.2 |
| 10 | 12.3 ± 0.6 (60) | 46.1 ± 1.4 | 79.0 ± 2.4 | 100 ± 0.5 | 482 ± 6.5 |
| 11 | 7.3 ± 0.5 (60) | 37.9 ± 1.3 | 75.9 ± 2.3 | 94.8 ± 0.6 | 390 ± 7.2 |
| 12 | 4.8 ± 0.5 (60) | 33.7 ± 1.3 | 72.4 ± 3.1 | 92.5 ± 0.8 | 344 ± 7.5 |
| 13 | 5.1 ± 0.6 (60) | 35.0 ± 1.5 | 72.0 ± 2.6 | 93.8 ± 0.7 | 349 ± 7.5 |
| 14 | 5.7 ± 0.6 (60) | 35.6 ± 1.5 | 73.1 ± 2.1 | 95.3 ± 0.7 | 352 ± 7.6 |
| 15 | 10.1 ± 0.6 (60) | 36.4 ± 1.3 | 74.3 ± 2.3 | 100 ± 0.6 | 442 ± 6.1 |
| 16 | 14.2 ± 0.7 (60) | 39.2 ± 1.3 | 79.0 ± 2.5 | 100 ± 0.6 | 475 ± 7.0 |
| 17 | 0 ± 0.1 (70) | 29.3 ± 1.2 | 70.1 ± 2.3 | 93.1 ± 0.7 | 303 ± 7.8 |
| 18 | 0 ± 0.2 (60) | 31.5 ± 1.3 | 71.0 ± 2.2 | 94.2 ± 0.6 | 308 ± 7.1 |
| 19 | 0 ± 0.3 (60) | 31.8 ± 1.3 | 71.2 ± 2.1 | 94.7 ± 0.6 | 309 ± 7.3 |
| Curcuminf | 0 ± 0.2 (60) | 22.8 ± 1.2 | 81.7 ± 2.5 | 100 ± 0.5 | 341 ± 7.8 |
Values represent percentages relative to the positive control value (100%).
For experimental details, see Ref. 23.
TPA concentration is 20 ng/mL (32 pmol/mL).
The molar ratio of compound, relative to TPA, required to inhibit 50% of the positive control activated with 32 pmol TPA
Values in parentheses are viability percentages of Raji cells. In all other experiments, viability was >80%.
Positive control
Compared with 1, most compounds showed stronger inhibitory effects on EBV-EA activity; however, compounds 7-10 conjugated with higher fatty acids, as well as CA analogs, 15 and 16, showed weaker inhibitory effects. Notably, compounds 4-6 conjugated with lower unsaturated fatty acids exhibited significant inhibitory effects with IC50 values of 285-299 mol ratio/32pmol TPA, which are better than those of curcumin (IC50 = 341). Percentages of EBV-EA positive cells were 0, 24-26, 67-63 and 89-92%, meaning 100%, 74-76%, 33-37%, and 8-11% inhibition of activation at concentrations of 1000, 500, 100 and 10 mol ratio/TPA, respectively. Compound 6 showed the most potent inhibitory activity, with 11% inhibition at the lowest tested concentration and an IC50 value of 285 mol ratio/32pmol TPA. Compound 6 contains an isopentenoyl group, which is structurally similar to a prenyl group. The positive effects of prenylated compounds have been discussed previously.23
The data for 4-12 indicate that the chain length and number of double bonds in the C3 ester group affect the inhibitory activity. For example, compounds 4-6 (C4-C6 esters) were more effective inhibitors than 7-12 (C18-C22 esters). Particularly among 7-12, the percentage of EBA-EA positive cells at each concentration as well as IC50 values clearly correlated with the above mentioned structural factors. The inhibitory effects of the compounds decreased as the length of the ester side chain and numbers of double bonds (W), regardless of their position, increased. The rank orders of the three factors were as follows: Activity, 12 > 11 > 9 > 10 > 8 > 7; Length, 12 (C18) = 11 (C18) =9 (C18) > 10 (C20) = 8 (C20) > 7 (C22); Number of double bonds, 12 (W1) >11 (W2) > 9 (W3) >10 (W4) > 8 (W5) >7 (W6).
Compounds 17-19 (IC50 = 303-309), which have (S)- or (R)-monomethyl rather than dimethyl substitution at the C3′ position of the ester side chain, were more potent than 3 (IC50 = 396). These three compounds fully blocked EBV-EA activation at 1000 mol ratio/TPA and showed 6-7% inhibition at the lowest tested concentration. Compounds with hydroxy and carboxylic acid groups at the ester terminus had comparable potency (compare 17 and 18). The stereochemistry of the C3′ methyl group did not affect the activity; 18 and 19 were equipotent.
Regarding SAR of ring-A, unesterified 16 with a five-membered ring-A (IC50 value 475) was less active than the corresponding 1 with a six-membered ring (IC50 value 403). In contrast, when the C3 hydroxyl was esterified, the five-membered ring-A analog (14, IC50 value 352) showed slightly higher activity than the six-membered ring-A analog (3, IC50 value 396). Interestingly, the inhibitory effect of 16 (86% inhibition at highest concentration, IC50 = 475) was comparatively lower than that of 2 (91% inhibition at highest concentration, IC50 = 400), although the corresponding succinylated analogs 14 and 13, respectively, exhibited similar inhibitory effects with 94-95% inhibition at highest tested concentration and IC50 = 349-352. These results indicate that a carboxylic acid at the C2 position of ring-A positively affects the potency when the C-3 OH is not esterified.
In conclusion, sixteen 1-analogs, 4-19, were synthesized and evaluated in an in vitro EBV-EA activation assay, which is well established to correlate with antitumor-promoting activity. Among them, analogs 4-6 and 17-19 showed significant potency as antitumor promoters. Compound 6 modified with a prenyl-like ester group was the most effective. Further structural modifications are in progress, and active compounds are being tested in an in vivo two-stage mouse skin carcinogenesis assay.
Figure 1.

Figure 2.

Structures of 17-19
Acknowledgements
This study was supported in part by grant CA-17625 from the National Cancer Institute, NIH, awarded to K. H. L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Cichewicz RH, Kouzi SA. Med. Res. Rev. 2004;24:90. doi: 10.1002/med.10053. [DOI] [PubMed] [Google Scholar]
- 2.Yogeeswari P, Sriram D. Curr. Med. Chem. 2005;12:657. doi: 10.2174/0929867053202214. [DOI] [PubMed] [Google Scholar]
- 3.Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. J. Nat. Prod. 1994;57:243. doi: 10.1021/np50104a008. [DOI] [PubMed] [Google Scholar]
- 4.Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. Proc. Natl. Acad. Sci. USA. 2003;100:13555. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessler JH, Mullauer FB, de Roo DM, Medema JP. Cancer Lett. 2007;251:132. doi: 10.1016/j.canlet.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee R, Kumar V, Srivastava SK, Agarwal SK, Burman AC. Anti-Cancer Agents in Med. Chem. 2006;6:271. doi: 10.2174/187152006776930846. [DOI] [PubMed] [Google Scholar]
- 7.Fulda S, Friesen M, Los M, Scaffidi C, Mier W, Benedict M, Nunes G, Krammer PH, Peter ME, Debatin KM. Cancer Res. 1997;57:4956. [PubMed] [Google Scholar]
- 8.Schmit ML, Kuzmanoff KL, Ling-Indeck L, Pezzuto JM. Eur. J. Cancer. 1997;33:2007. doi: 10.1016/s0959-8049(97)00294-3. [DOI] [PubMed] [Google Scholar]
- 9.Fulda S, Jeremias I, Steiner HH, Poetsh T, Debatin KM. Int. J. Cancer. 1999;82:435. doi: 10.1002/(sici)1097-0215(19990730)82:3<435::aid-ijc18>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Chandramu C, Manohar RD, Krupadanam DG, Dashavantha RV. Phytother. Res. 2003;27:129. doi: 10.1002/ptr.1088. [DOI] [PubMed] [Google Scholar]
- 11.Schihly W, Heilmann J, Callis I, Sticher O. Planta Med. 1999;65:740. doi: 10.1055/s-1999-14054. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee PK, Saha K, Das J, Pal M, Saha BP. Planta Med. 1997;63:367–369. doi: 10.1055/s-2006-957705. [DOI] [PubMed] [Google Scholar]
- 13.Steele JC, Warhurst DC, Kirby GC, Simmonds MS. Phytother. Res. 1999;13:115. doi: 10.1002/(SICI)1099-1573(199903)13:2<115::AID-PTR404>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 14.Bringmann G, Saeb W, Assi LA, Francois G, Narayanan ASS, Peters K, Petera EM. Planta Med. 1997;63:255. doi: 10.1055/s-2006-957666. [DOI] [PubMed] [Google Scholar]
- 15.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. J. Med. Chem. 1996;39:1016. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 16.Takasaki M, Konoshima T, Kozuka M, et al. Bio. Pharm. Bull. 1995;18:435–438. doi: 10.1248/bpb.18.435. [DOI] [PubMed] [Google Scholar]
- 17.Fukuda Y, Sakai K, Matsunaga S, Tokuda H, Tanaka R. Chem. Biodiversity. 2005;2:421. doi: 10.1002/cbdv.200590021. [DOI] [PubMed] [Google Scholar]
- 18.Akihisa T, Nakamura Y, Tagata M, Tokuda H, Yasukawa K, Uchiyama E, Suzuki T, Kimura Y. Chem. Biodiversity. 2007;4:224. doi: 10.1002/cbdv.200790027. [DOI] [PubMed] [Google Scholar]
- 19.Akihisa T, Nakamura Y, Tokuda H, Uchiyama E, Suzuki T, Kimura Y, Uchikura K, Nishino H. J. Nat. Prod. 2007;70:948. doi: 10.1021/np0780001. [DOI] [PubMed] [Google Scholar]
- 20.Qian K, Nakagawa-Goto K, Yu D, Marris-Natschke SL, Nitz TJ, Kilgore N, Allaway GP, Lee KH. Bioorg. Med. Chem. Lett. 2007;17:6553. doi: 10.1016/j.bmcl.2007.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee SS, Chen WC, Huang CF, Su Y. J. Nat. Prod. 1998;61:1343. doi: 10.1021/np9800856. [DOI] [PubMed] [Google Scholar]
- 22.In vitro EBV-EA activation experiments: EBV-EA positive serum from a patient with nasopharyngeal carcinoma (NPC) was a gift from Professor H. Hattori, Department of Otorhinolaryngology, Kobe University. The EBV genome carrying lymphoblastoid cells (Raji cells derived from Burkitt’s lymphoma) were cultured in 10% fetal bovine serum (FBS) in RPMI-1640 medium (Sigma R8758, USA). Spontaneous activation of EBV-EA in our subline of Raji cells was less than 0.1%. The inhibition of EBV-EA activation was assayed using Raji cells (virus non-producer type) as described below. The cells were incubated at 37 °C for 48 h in 1 mL of medium containing n-butyric acid (4 mM), TPA [32 pM = 20 ng in 2 μL dimethyl sulfoxide (DMSO)] and various amounts of the test compounds dissolved in 2 μL of DMSO. Smears were made from the cell suspension. The EBV-EA inducing cells were stained by the means of an indirect immunofluorescence technique. In each assay, at least 500 cells were counted, and the number of stained cells (positive cells) was recorded. Triplicate assays were performed for each compound. The average EBV-EA induction of the test compound was expressed as a ratio relative to the control experiment (100%), which was carried out with n-butyric acid (4 mM) plus TPA (32 pM). EBV-EA induction was ordinarily around 35%. The viability of treated Raji cells was assayed by the Trypan blue staining method. The cell viability of the TPA positive control was greater than 80%. Therefore, only compounds that induced less than 80% (% of control) of the EBV-active cells (those with a cell viability of more than 60%) were considered able to inhibit the activation caused by promoter substances. Student’s t-test was used for all statistical analysis.
- 23 a).Suzuki M, Nakagawa-Goto K, Nakamura S, Tokuda H, Morris-Natschke SL, Kozuka M, Nishino H, Lee KH. Pharm. Biol. 2006;44:178. [Google Scholar]; b) Itoigawa M, Ito C, Tokuda H, Enjo F, Nishino H, Furukawa H. Cancer Lett. 2004;214:165. doi: 10.1016/j.canlet.2004.05.005. [DOI] [PubMed] [Google Scholar]