

Abstract
Neurons containing the neuropeptide hypocretin (orexin) are localized only in the lateral hypothalamus from where they innervate multiple regions implicated in arousal, including the basal forebrain. HCRT activation of downstream arousal neurons is likely to stimulate release of endogenous factors. One such factor is adenosine (AD), which in the basal forebrain increases with waking and decreases with sleep, and is hypothesized to regulate the waxing and waning of sleep drive. Does loss of HCRT neurons affect AD levels in the basal forebrain? Is the increased sleep that accompanies HCRT loss a consequence of higher AD levels in the basal forebrain? In the present study, we investigate these questions by lesioning the HCRT neurons (hypocretin-2-saporin) and measuring sleep and extracellular levels of AD in the basal forebrain. In separate groups of rats, the neurotoxin HCRT2-SAP or saline were administered locally to the lateral hypothalamus and 80 days later AD and sleep were assessed. Rats given the neurotoxin had a 94% loss of the HCRT neurons. These rats awake less at night, and had more REM sleep, which is consistent with a HCRT hypofunction. These rats also had more sleep after brief periods of sleep deprivation. However, in the lesioned rats, AD levels did not increase with 6h sleep deprivation, whereas such an increase in AD occurred in rats without lesion of the HCRT neurons. These findings indicate that AD levels do not increase with waking in rats with a HCRT lesion, and that the increased sleep in these rats occurs independently of AD levels in the basal forebrain.
Keywords: Adenosine, Orexin, Basal forebrain, Sleep, REM Sleep, Microdialysis
Introduction
The alternation between sleep and wake is hypothesized to result from a coordinated interaction between distributed populations of neurons. Chief among the arousal neurons are ones containing the neuropeptide hypocretin, which is also known as orexin (de Lecea et al, 1998;Elias et al, 1998;Nambu et al, 1999;Peyron et al, 1998;Sakurai et al, 1998). These neurons are located in the lateral hypothalamus (LH) and have both ascending and descending projections to major arousal neurons, including the basal forebrain (Peyron et al, 1998). HCRT receptors are present in the basal forebrain, and administration of HCRT-1 and HCRT-2 into the basal forebrain produces wakefulness in rats (Espana et al, 2001;Thakkar et al, 2001). HCRT depolarizes BF cholinergic neurons (Eggermann et al, 2001) via the HCRT type 2 receptor.
HCRT's influence on the BF may be assessed by measuring endogenous factors in the BF. In the present study, we measure adenosine (AD) a naturally occurring purine nucleoside that is released under a variety of conditions including waking (Braas et al, 1986). In the basal forebrain extracellular concentrations of AD increase with wake and then decrease during sleep (Murillo-Rodriguez et al, 2004;Porkka-Heiskanen et al, 1997). AD levels in the BF increase further in response to prolonged waking presumably reflecting increased energy consumption within the wake-active neurons (Blanco-Centurion et al, 2006b). The hypocretin neurons, which are active during wake and silent during sleep (Lee et al, 2005;Mileykovskiy et al, 2005) may induce waking, in part, by the action of HCRT on BF neurons. We hypothesize that deletion of the HCRT neurons should reduce HCRT's influence onto wake-active BF neurons, resulting in a reduced effect on downstream cascades such as AD. In the present study, we directly test this hypothesis by using the neurotoxin hypocretin-2-saporin (HCRT2-SAP) to lesion hypocretin receptor bearing neurons in the lateral hypothalamus and then measuring sleep and extracellular levels of AD in the BF in response to prolonged waking.
Materials and Methods
Animals
Young male (3 months; 285–370 g) Sprague-Dawley rats (Charles River Labs, MA) were housed at a constant temperature (21 ± 1°C) and under a controlled light-dark cycle (lights on: 07:00–19:00h; 200 lux) throughout the experiment. Food and water were provided ad libitum. All rats were treated in accordance with institutional policy on care and use of laboratory animals. The institutional animal care and use committee (IACUC) reviewed and approved the research protocol.
Overall experimental plan
The neurotoxin HCRT2-SAP (also called Orexin B-SAP; Catalogue # IT-02; Advanced Targeting Systems, San Diego, CA) or pyrogen free saline solution were delivered to the LH and the rats were returned to the vivarium. Two months later the sleep recording electrodes, and the microdialysis guide cannulas were implanted. Two weeks after the surgery a 24 h baseline sleep recording was made. The following morning the sleep drive of the rats was assessed using the rodent sleep pressure test. A week later the microdialysis guide cannula was inserted (when the lights turned off), and the following day extracellular fluid was collected via the microdialysis cannula from the basal forebrain before and during six hours of total sleep deprivation. The rats were then euthanized and the brains extracted, formalin-fixed and processed for immunohistochemistry.
Lateral hypothalamic lesion with HCRT2-SAP
The rats were placed in a stereotaxic apparatus (anesthetic: cocktail of acepromazine (0.75mg/kg), xylazine (2.5 mg/kg), and ketamine (22mg/kg) administered IM) and bilateral microinjections of HCRT2-SAP (200 ng/μL, vol=0.5uL) or pyrogen free saline (vol=0.5 μL) were made to the LH. The HCRT2-SAP conjugate (Advanced Targeting Systems, San Diego, CA), or pyrogen free saline (Sigma-Aldrich, St. Louis MO) were delivered via a glass micropipette with a tip diameter of 20 μm using a Picospritzer (General Valve, Fairfield NJ). After injection, the pipette was left in place for 5 min and then withdrawn slowly. All injections were made in the LH (A=−3.3 mm; L=1.6 to 1.8 mm; V=8.2 mm below the dura) (Paxinos and Watson C, 2006).
Surgery to implant sleep recording electrode
The animals were implanted under deep anesthesia (cocktail of acepromazine (0.75mg/kg), xylazine (2.5 mg/kg), and ketamine (22mg/kg) administered IM) with cortical sleep recording electrodes as previously described (Shiromani et al, 2000). Briefly, four stainless-steel screws were fixed to the skull and were used to record the electroencephalogram (EEG). Two miniature screws were inserted 2 mm on either side of the sagittal sinus and 3 mm anterior to bregma (frontal cortex). The other 2 screws were located 3 mm on either side of the sagittal sinus and 6 mm behind bregma (occipital cortex). The EEG was recorded between a left or right frontal and a contralateral occipital cortex screw (frontal-occipital). Two stainless-steel flexible multiwire electrodes inserted into the nuchal muscles were used to monitor the electromyogram (EMG).
All electrodes were placed in a six-pin plastic plug and secured onto the skull using dental cement. At this time a guide-cannula (IC guide, BioAnalytical Systems, West Lafayette, IN, USA) was placed stereotaxically into the basal forebrain (target coordinates: A= −.35; L= 2.0; H= 7.5). The guide- cannula was then fixed onto the skull with a thin layer of dental cement.
Animals were allowed to recover for at least fourteen days after surgery. During this period, each animal was attached to the electroencephalographic recording leads to habituate to the recording conditions.
Analysis of sleep recordings
Contralateral frontal-occipital EEG screw electrodes were used for EEG acquisition. The EEG analogue data were amplified, filtered at 70 Hz (low-pass filter) and 0.3 Hz (high-pass filter) using a Grass electroencephalograph (Grass Models 9,12,15 West Warwick, RI). The analogue signal was then converted to digital and continuously sampled at 128 Hz. The EEG data recordings were scored manually on a computer (Icelus software, M Opp) in 12-s epochs for wake, non-REM Sleep and REM sleep. Wake state was identified by the presence of desynchronized EEG and high EMG activity. Non-REM sleep consisted of high-amplitude slow waves together with a low EMG tone relative to awake. REM sleep was identified by the presence of regular theta activity coupled with low EMG relative to non-REM Sleep. Because the EEG was recorded on three separate polygraphs, delta power analysis could not be assessed. Attempts to normalize the data were not successful as the variability in delta power was still very high, and it obscured any meaningful interpretation.
Microdialysis sampling procedure
The microdialysis probe (1 mm of length. Polyacrylonitrile, MWCO = 30,000 daltons; 340 micrometer OD; BAS, West Lafayette, IN, USA) was inserted through the guide cannula into the target structure at 1900 h (ZT 12; lights off) and the tissue was allowed to stabilize for 12 h. During this period aCSF (aCSF composition: NaCl (147mM), KCl (3mM), CaCl (1.2mM), MgCl (1.0mM), pH 7.2) was perfused continuously (flow rate of 0.25μl/min). In preliminary experiments, we determined that it took about 6–7h after probe insertion for the AD levels to stabilize.
The next day beginning at ZT 2 (two hours after lights on) extracellular fluid (5 μl) from the microdialysis probe was collected at the end of the hour (last 20 minute sample of the hour) and immediately frozen. Two hours later the rats were kept awake for 6 h, and samples were collected at the end of each hour (last 20 minutes). At the end of the 6h period of prolonged waking the rats were allowed to sleep undisturbed and microdialysis probe samples were collected at the end of the hour for two hours. The rats were kept awake by lightly tapping or shaking the cage and/or introducing novel objects into the cage whenever they displayed behavioral or EEG signs of sleep. A lesioned rat was usually monitored alongside one or two saline (non-lesioned) rats, and if one rat was awakened then the same intervention (light tapping/shaking/novel object) was given to the other rats. Two people were tasked with keeping the rats awake, and they did not know which rats were lesioned or non-lesioned.
Chromatographic analysis of adenosine
The samples were injected into a high-performance liquid chromatograph (HPLC) (BAS, West Lafayette, IN, USA) for purine analysis. The mobile phase consisted of 10 mM NaH2PO4 (pH 4.5) plus 9% of CH3OH. Separation was achieved by a BAS Microbore column (octadecyl silica, 3 μm, 100X1 mm; BAS, West Lafayette, IN, USA) attached directly to the injector (CC-5e; BAS, West Lafayette, IN, USA). Purine concentrations were then measured by a UV detector set to a wavelength of 254 nm (UV-116A; BAS, West Lafayette, IN, USA). Chromatographic data was recorded in a PC computer and peak heights of AD in microdialysis samples were compared to standards using the chromatograph report software (BAS, West Lafayette, IN, USA).
Rodent Sleep Pressure Test
The rats were kept awake (light tapping/novel objects in cage) for 20 min beginning two hours after the start of the light on period (ZT 0) and then allowed to sleep undisturbed for 20 min. The alternating 20 min periods of wake followed by 20 min periods of sleep were continued until the end of the light-on period. Sleep was recorded throughout the test. During the 20 min sleep periods the number of minutes the rats spent in non-REM sleep, and REM sleep were determined. Previously, this test was used to measure REM sleep propensity in a genetic line of rats (Shiromani et al, 1991) and sleep drive in rats with lesions of the cholinergic basal forebrain neurons (Blanco-Centurion et al, 2006b).
Histological verification of probe location and immunohistochemistry
At the end of the experiments the rats were euthanized with a lethal dose of pentobarbital and perfused transcardially with 0.9 % saline solution followed by 10% formaldehyde. The brain was removed and post fixed overnight in 10% formaldehyde followed by 30% sucrose-0.1M PBS for 48h. The brains were cut (frozen sections, 30μm, coronal) and collected in 1:5 serial order. After immunohistochemistry was done, sections were lightly counterstained with Neutral Red to allow for better visualization of the cannula track.
Immunohistochemistry
Five series of coronal sections were cut at 30 μm on a sliding microtome. Each set of coronal brain sections was incubated overnight at room temperature in the primary antibody. After washing, the sections were incubated with the secondary antibody for 1 hr (Chemicon; 1:500 dilution) and then reacted with avidin-biotin complex for 1 hr (Vector Laboratories, Burlingame, CA). The DAB method was used to visualize the reaction product (Vector Laboratories, Burlingame, CA). The specificity of the goat-anti-orexin-A (HCRT-1) antibody was tested in our lab in brain sections from HCRT knockout mice, and no HCRT-1 immunoreactive neurons in the perifornical area were detected.
Antibodies
Goat anti-orexin-A (HCRT1) (SC 8070, 1:10,000, Santa Cruz Biotechnology, Santa Cruz CA), and goat anti-choline acetyltransferase (AB 144P, 1: 2000 dilution; Chemicon, Temecula, CA)
Cell counts
A person who did not know the lesion status of the rats counted clearly stained HCRT-immunoreactive (ir) somata in 24 sections (two non-adjacent wells in 1 in 5 series) that encompassed the perifornical area where the HCRT neurons are located (between −1.6 and −3.8 mm from bregma (Paxinos and Watson C, 2006). The neurons were counted bilaterally within a 0.2 mm2 rectangular grid in 24 sections in the perifornical area where the HCRT neurons are located. Camera Lucida drawings (Nikon microscope) were made to identify the lesioned area.
Statistical analysis
A 2-way repeated measures ANOVA (Sigmastat, version 3.05) followed by the post hoc Holm-Sidak test was used to compare the sleep and adenosine data. Student's t-test (paired and independent) was also used when appropriate. Statistical significance was assessed at the p<0.05 level.
Results
There was a significant reduction (−94%) in the average number of HCRT-ir neurons per section in rats given the neurotoxin, HCRT2-SAP, compared to saline rats (HCRT2-SAP lesioned rats=3.93 ± 0.79; Saline rats=65.0 ± 3.7; independent t-test=12.9; df=16; p<0.001). Photomicrographs of the LH from representative saline and neurotoxin treated rats are shown in Figure 1.
Fig 1.
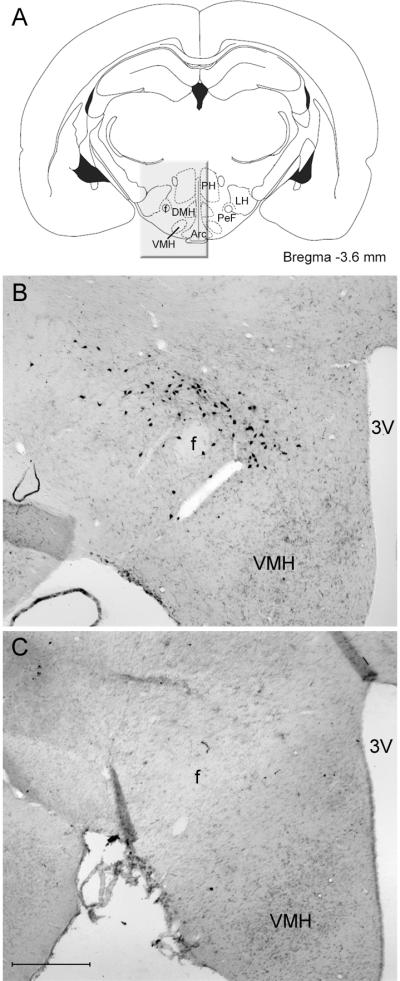
Figure 2 summarizes the changes in wake, non-REM and REM sleep produced by the destruction of the lateral hypothalamic neurons, including the HCRT neurons. In lesioned rats there was a significant increase in REM sleep at night (independent t-test=2.317; df=15; p<0.035), which is consistent with the sleep profile of mice (Chemelli et al, 1999;Hara et al, 2001;Mochizuki et al, 2004) and rats (Beuckmann et al, 2004;Gerashchenko et al, 2001;Gerashchenko et al, 2003;Zhang et al, 2007) with HCRT loss. The lesioned rats also had a significant decrease in waking at night (independent t-test=2.289; df=15; p<0.037) which is consistent with reports in orexin null mice (Chemelli et al, 1999;Mochizuki et al, 2004) and rats with HCRT neuronal loss (Beuckmann et al, 2004).
Fig 2.

To gauge the sleep drive of the LH lesioned rats, the rats were kept awake for brief periods (20 minutes) and then allowed to sleep for 20 minutes (Figure 3). A 2-way repeated measures ANOVA (group × sleep periods) revealed a significant difference between the saline and lesion groups for REM sleep (F (1,15)=11.78; p<0.004), non-REM (F (1,15)=36.71; p<0.001) and total sleep time (F (1,15)=44.62; p<0.001). Post-hoc analysis revealed significant increases in REM sleep, non-REM and total sleep time at the points noted in figure 3 (p<0.05).
Fig 3.

Next, adenosine levels were measured in the basal forebrain (Figure 4). Consistent with published data (Blanco-Centurion et al, 2006b;Murillo-Rodriguez et al, 2004;Strecker et al, 2000), there was a significant increase in AD levels in saline rats in response to 6 h of prolonged waking (group effect; (F (1,12)=49.53; p<0.001). In saline rats, extracellular levels of AD were significantly higher throughout the 6h of prolonged waking compared to pre-deprivation levels (p< 0.01; Holm-Sidak post hoc test) and then there was a rapid decline during the 2 h of undisturbed sleep when the AD levels were not significantly different from pre-TSD conditions (Fig. 4). However, in LH lesioned rats AD levels did not increase during the 6h total sleep deprivation (TSD) period compared to pre-TSD levels (Fig. 4). During the two hour period before the sleep deprivation, AD levels were not significantly different compared to control rats indicating that AD levels in the lesioned rats were within detectable range. Figure 5 identifies the location of the cannula in the basal forebrain region from where the AD was measured. The cannula was located to sample from within the cholinergic zone of the magnocellular preoptic area (MCPO) where previously we (Blanco-Centurion et al, 2006b;Murillo-Rodriguez et al, 2004) and others (Strecker et al, 2000) have shown that AD levels increase in response to W
Fig 4.

Fig 5.

Discussion
The primary finding of this study was that in rats with lesions of the lateral hypothalamus (LH) adenosine (AD) levels in the basal forebrain did not increase with prolonged waking, and yet the rats slept more. These results along with results from our previous study (Blanco-Centurion et al, 2006b) do not support the adenosine-BF hypothesis of sleep regulation which posits that waxing and waning of sleep are regulated by AD that accumulates specifically in the BF with waking and acts to inhibit the neural activity of W-promoting neurons (Strecker et al, 2000). Instead, we suggest that AD represents a downstream cascade in the basal forebrain (BF) that is stimulated by the activity of the arousal neurons in the LH, especially the HCRT neurons. This is analogous to the release of dopamine in the striatum as a marker of activity in the nigrostriatal pathway. However, unlike dopamine whose release in the striatum is required for smooth motor control, the AD release in the BF is not required for regulating sleep.
In the present study, extracellular AD levels increased in response to 6h of sleep deprivation in control rats administered saline and without any lesion of the HCRT neurons. Results that are consistent with reports in cats (Porkka-Heiskanen et al, 2000) and rats (Blanco-Centurion et al, 2006b;Murillo-Rodriguez et al, 2004) and indicates that our methodology is able to detect an increase in extracellular AD levels. We have previously determined that the cholinergic neurons in the BF are the source of the increase in extracellular AD during prolonged waking, because when these neurons are destroyed (192-IgG-saporin) then AD levels do not increase (Blanco-Centurion et al, 2006b). In that study we also determined that sleep drive continued to build without a corresponding increase in AD levels in the BF. In the present study, AD levels in the BF of rats with LH lesion did not increase with 6h prolonged waking, even though the lesioned rats slept more. Thus, we conclude that AD release in the BF is a by-product of the HCRT stimulation of the cholinergic neurons and is not essential in regulating daily levels of wake.
The BF from where the AD was measured is also stimulated by other pathways, most notably the mesopontine neurons (Semba et al, 1988). The mesopontine neurons have been implicated in arousal (Jones, 2003), and the mesopontine-BF circuit should be active during the period of sleep deprivation. Nevertheless, AD levels in the BF did not increase with the sleep deprivation indicating that the LH/HCRT neurons, relative to the mesopontine neurons, provide a stronger signal associated with sleep deprivation onto the BF for release of AD. It remains to be determined whether stimulation other than sleep deprivation can evoke AD in the BF. For instance, electrical stimulation of the mesopontine neurons produces arousal and cortical acetylcholine release (from the BF cholinergic neurons) (Rasmusson et al, 1992;Rasmusson et al, 1994). It remains to be determined whether such stimulation will drive AD release in the BF in the absence of HCRT neurons.
In the present study, the neurotoxin HCRT2-SAP was used to lesion HCRT receptor bearing neurons in the lateral hypothalamus. This neurotoxin lesions both HCRT and non-HCRT LH neurons (Gerashchenko et al, 2001), and the loss of other LH neurons may have also contributed to the decrease in AD in the BF. To understand specifically the contribution of HCRT to the basal forebrain, AD could be measured in the orexin-ataxin-3 rats (Beuckmann et al, 2004) where the HCRT neurons die from the toxic accumulation of polyglutamine in neurons. Similarly, HCRT null and HCRT-ataxin-3 mice could also be utilized.
AD levels were assessed approximately 80 days after lesion of the HCRT neurons, to give ample time for the HCRT neurons to die, and the HCRT hypofunction to stabilize. In the present study, 94% of the HCRT neurons were destroyed by the HCRT2-SAP. Previously, we examined the time course of decline in CSF HCRT-1 levels following HCRT2-SAP lesions of the LH and found a significant decline 64 days after the lesion (Gerashchenko et al, 2003). In the HCRT-ataxin rats, we found that CSF HCRT-1 levels declined once the HCRT neurons died and they remained low throughout the life-span of the rats (Zhang et al, 2007). Taken together, these studies indicate that the HCRT levels do not recover once the HCRT neurons are dead, and that the surviving HCRT neurons are unable to compensate fully for the HCRT neuronal loss. Therefore, in the present study, at the time when sleep and AD levels were measured, the rats had a severe HCRT hypofunction.
We also assessed whether the lesioned rats had increased sleep and REM sleep, as they should after HCRT neuronal loss (see figure 2). Effects of sleep deprivation have been assessed in the HCRT null mice (Mochizuki et al, 2004), but not in the HCRT-ataxin rats or in our previous studies where the HCRT2-SAP neurotoxin was used to lesion the LH. In the HCRT null mice sleep homeostasis was normal even after eight hours of prolonged waking (Mochizuki et al, 2004). Moreover, the HCRT null mice stayed awake as long as wild type mice in an unfamiliar environment. In the present study, we elected to use an ultra-short sleep-wake schedule to ascertain whether the sleep drive was increased in the lesioned rats. Such schedules have been used in humans (Richardson et al, 1978) as it avoids possible confounds (e.g., muscle fatigue) that may be associated with uninterrupted periods of sleep loss (6–12h). Previously, using such an ultra-short sleep-wake paradigm (20 minute awake+20 minute sleep) we found increased REM sleep propensity in a genetic line of rats (Shiromani et al, 1991) and that sleep drive was intact in rats with lesions of the BF cholinergic neurons (Blanco-Centurion et al, 2006b). In the present study, rats with LH lesions slept more (both non-REM and REM sleep) during the 20-minute sleep periods that followed 20 minutes of wake, indicating a heightened tendency to sleep during the normal sleep period. Human narcoleptics when placed in a 90 minute schedule (60 min wake+30 minute sleep repeated for 72h) also have more sleep and REM sleep during the 30 minute sleep periods (Dantz et al, 1994).
It is not clear whether such ultra-short sleep schedules measure sleep homeostasis or excessive sleepiness at certain times of day. Sleep homeostasis is intact in HCRT null mice but these mice do sleep more during their normal active period. In the present study, the LH lesioned rats also were awake less at night, and slept more after brief periods of waking. We suggest that rather than reflecting increasing sleep pressure, the decreased waking in the HCRT null mice and in the LH lesioned rats may reflect reduced activation of downstream arousal neurons, such as the BF. However, the BF contains both cholinergic and non-cholinergic neurons, and both are active during wake and hypothesized to drive arousal. Which of these conveys the HCRT arousal signal? We suggest that the non-cholinergic neurons convey the HCRT arousal signal to the cortex. For instance, several studies have now shown that when the BF cholinergic neurons are lesioned daily levels of wake, sleep homeostasis, or EEG synchrony does not change (Bassant et al, 1995;Blanco-Centurion et al, 2006b;Kapas et al, 1996;Kaur et al, 2008). The BF cholinergic neurons do release AD during waking, but as the present study and previous data (Blanco-Centurion et al, 2006b) show sleep can occur without any corresponding increase in AD levels in the BF.
Instead, the non-cholinergic wake-active neurons are likely to mediate HCRT's influence on the cortex because microinjection of HCRT-1 to a BF devoid of the cholinergic neurons is able to evoke a strong arousal response (Blanco-Centurion et al, 2006a). These neurons might be GABAergic innervating cortical GABA interneurons, and may cause arousal through disinhibition (Duque et al, 2000). When these neurons (identified by parvalbumin) are lesioned (ibotenic acid) cortical EEG becomes more synchronized (higher delta power) (Buzsaki et al, 1988;Kaur et al, 2008).
The BF also contains sleep-active neurons, some of which contain neuropeptide Y (Zaborszky and Duque, 2003) whereas others are GABAergic. These sleep-active neurons would be disinhibited when the wake-active BF neurons become silent (Jones, 2003). The GABAergic neurons increase activity in conjunction with cortical slow waves (Manns et al, 2000). They project to the cortex (Gritti et al, 1997) and to the posterior lateral hypothalamus where the HCRT neurons are located. Their activity would suppress activity of the HCRT wake-active neurons and promote sleep. Interestingly, BF neurons project to the HCRT neurons, whereas the arousal neurons of the locus coeruleus and the histaminergic neurons in the tuberomammillary nucleus do not (Sakurai et al, 2005).
The discovery of hypocretin (orexin) and its linkage with the sleep disorder narcolepsy led to the hypothesis that these neurons may be driving downstream arousal areas. The results from the present study support a link between the LH where the HCRT neurons are located, and the BF, in cortical arousal. We suggest that a weak HCRT drive onto the wake-active neurons in the BF, such as after LH and HCRT lesion, would not adequately arouse the cortex. There are also sleep-active neurons in the BF, which are non-cholinergic, and after HCRT lesions, these neurons might not be adequately inhibited. Other arousal neurons that receive strong HCRT input include the LC and the TMN, and a weak HCRT drive might not adequately stimulate them.
Does the link between the HCRT neurons and the BF regulate sleep homeostasis? We think not since HCRT null mice have intact sleep drive. Similarly, the AD release in the BF is a by-product of the HCRT stimulation and is not necessary for regulating sleep homeostasis. HCRT also stimulates wake-active non-cholinergic neurons and we suggest that these neurons are sufficient to arouse the cortex. We suggest that a weak HCRT drive onto the wake-active neurons in the BF, such as after LH and HCRT lesion, would not adequately arouse the cortex. There are also sleep-active neurons in the BF, which are non-cholinergic, and after HCRT lesions, these neurons might not be adequately inhibited. Other arousal neurons that receive strong HCRT input include the LC and the TMN, and a weak HCRT drive might not adequately stimulate them.
Acknowledgments
We thank Elizabeth Winston for analyzing the sleep data. Supported by NIH grants NS030140, NS052287, MH055772, and Medical Research Service of the Department of Veterans Affairs.
References
- Bassant MH, Apartis E, Jazat-Poindessous FR, Wiley RG, Lamour YA. Selective immunolesion of the basal forebrain cholinergic neurons: effects on hippocampal activity during sleep and wakefulness in the rat. Neurodegeneration. 1995;4:61–70. doi: 10.1006/neur.1995.0007. [DOI] [PubMed] [Google Scholar]
- Beuckmann CT, Sinton CM, Williams SC, Richardson JA, Hammer RE, Sakurai T, Yanagisawa M. Expression of a poly-glutamine-ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J.Neurosci. 2004;24:4469–4477. doi: 10.1523/JNEUROSCI.5560-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion C, Shiromani A, Winston E, Shiromani PJ. Effects of hypocretin-1 in 192-IgG-saporin-lesioned rats. Eur.J.Neurosci. 2006a;24:2084–2088. doi: 10.1111/j.1460-9568.2006.05074.x. [DOI] [PubMed] [Google Scholar]
- Blanco-Centurion C, Xu M, Murillo-Rodriguez E, Gerashchenko D, Shiromani AM, Salin-Pascual RJ, Hof PR, Shiromani PJ. Adenosine and sleep homeostasis in the Basal forebrain. J.Neurosci. 2006b;26:8092–8100. doi: 10.1523/JNEUROSCI.2181-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braas KM, Newby AC, Wilson VS, Snyder SH. Adenosine-containing neurons in the brain localized by immunocytochemistry. J Neurosci. 1986;6:1952–1961. doi: 10.1523/JNEUROSCI.06-07-01952.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J.Neurosci. 1988;8:4007–4026. doi: 10.1523/JNEUROSCI.08-11-04007.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Dantz B, Edgar DM, Dement WC. Circadian rhythms in narcolepsy: studies on a 90 minute day. Electroencephalogr.Clin.Neurophysiol. 1994;90:24–35. doi: 10.1016/0013-4694(94)90110-4. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc.Natl.Acad.Sci.U.S.A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque A, Balatoni B, Detari L, Zaborszky L. EEG correlation of the discharge properties of identified neurons in the basal forebrain. J Neurophysiol. 2000;84:1627–1635. doi: 10.1152/jn.2000.84.3.1627. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, Jones BE, Muhlethaler M. Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience. 2001;108:177–181. doi: 10.1016/s0306-4522(01)00512-7. [DOI] [PubMed] [Google Scholar]
- Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J.Comp Neurol. 1998;402:442–459. [PubMed] [Google Scholar]
- Espana RA, Baldo BA, Kelley AE, Berridge CW. Wake-promoting and sleep-suppressing actions of hypocretin (orexin): basal forebrain sites of action. Neuroscience. 2001;106:699–715. doi: 10.1016/s0306-4522(01)00319-0. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Blanco-Centurion C, Greco MA, Shiromani PJ. Effects of lateral hypothalamic lesion with the neurotoxin hypocretin-2-saporin on sleep in Long-Evans rats. Neuroscience. 2003;116:223–235. doi: 10.1016/s0306-4522(02)00575-4. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Kohls MD, Greco M, Waleh NS, Salin-Pascual R, Kilduff TS, Lappi DA, Shiromani PJ. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J.Neurosci. 2001;21:7273–7283. doi: 10.1523/JNEUROSCI.21-18-07273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti I, Mainville L, Mancia M, Jones BE. GABAergic and other noncholinergic basal forebrain neurons, together with cholinergic neurons, project to the mesocortex and isocortex in the rat. J.Comp Neurol. 1997;383:163–177. [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Jones BE. Arousal systems. Front Biosci. 2003;8:S438–S451. doi: 10.2741/1074. [DOI] [PubMed] [Google Scholar]
- Kapas L, Obal F, Jr., Book AA, Schweitzer JB, Wiley RG, Krueger JM. The effects of immunolesions of nerve growth factor-receptive neurons by 192 IgG-saporin on sleep. Brain Res. 1996;712:53–59. doi: 10.1016/0006-8993(95)01431-4. [DOI] [PubMed] [Google Scholar]
- Kaur S, Junek A, Black MA, Semba K. Effects of ibotenate and 192IgG-saporin lesions of the nucleus basalis magnocellularis/substantia innominata on spontaneous sleep and wake states and on recovery sleep after sleep deprivation in rats. J Neurosci. 2008;28:491–504. doi: 10.1523/JNEUROSCI.1585-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J.Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns ID, Alonso A, Jones BE. Discharge properties of juxtacellularly labeled and immunohistochemically identified cholinergic basal forebrain neurons recorded in association with the electroencephalogram in anesthetized rats. J.Neurosci. 2000;20:1505–1518. doi: 10.1523/JNEUROSCI.20-04-01505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T, Crocker A, McCormack S, Yanagisawa M, Sakurai T, Scammell TE. Behavioral state instability in orexin knock-out mice. J.Neurosci. 2004;24:6291–6300. doi: 10.1523/JNEUROSCI.0586-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo-Rodriguez E, Blanco-Centurion C, Gerashchenko D, Salin-Pascual RJ, Shiromani PJ. The diurnal rhythm of adenosine levels in the basal forebrain of young and old rats. Neuroscience. 2004;123:361–370. doi: 10.1016/j.neuroscience.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Distribution of orexin neurons in the adult rat brain. Brain Res. 1999;827:243–260. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press, INC.; 2006. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J.Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, McCarley RW. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience. 2000;99:507–517. doi: 10.1016/s0306-4522(00)00220-7. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson DD, Clow K, Szerb JC. Frequency-dependent increase in cortical acetylcholine release evoked by stimulation of the nucleus basalis magnocellularis in the rat. Brain Res. 1992;594:150–154. doi: 10.1016/0006-8993(92)91041-c. [DOI] [PubMed] [Google Scholar]
- Rasmusson DD, Clow K, Szerb JC. Modification of neocortical acetylcholine release and electroencephalogram desynchronization due to brainstem stimulation by drugs applied to the basal forebrain. Neuroscience. 1994;60:665–677. doi: 10.1016/0306-4522(94)90495-2. [DOI] [PubMed] [Google Scholar]
- Richardson GS, Carskadon MA, Flagg W, van den HJ, Dement WC, Mitler M. Excessive daytime sleepiness in man: multiple sleep latency measurement in narcoleptic and control subjects. Electroencephalogr.Clin.Neurophysiol. 1978;45:621–627. doi: 10.1016/0013-4694(78)90162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, Kageyama H, Kunita S, Takahashi S, Goto K, Koyama Y, Shioda S, Yanagisawa M. Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron. 2005;46:297–308. doi: 10.1016/j.neuron.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Semba K, Reiner PB, McGeer EG, Fibiger HC. Brainstem afferents to the magnocellular basal forebrain studied by axonal transport, immunohistochemistry, and electrophysiology in the rat. J.Comp Neurol. 1988;267:433–453. doi: 10.1002/cne.902670311. [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Lu J, Wagner D, Thakkar J, Greco MA, Basheer R, Thakkar M. Compensatory sleep response to 12 h wakefulness in young and old rats. Am.J.Physiol Regul.Integr.Comp Physiol. 2000;278:R125–R133. doi: 10.1152/ajpregu.2000.278.1.R125. [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Velazquez-Moctezuma J, Overstreet D, Shalauta M, Lucero S, Floyd C. Effects of sleep deprivation on sleepiness and increased REM sleep in rats selectively bred for cholinergic hyperactivity. Sleep. 1991;14:116–120. [PubMed] [Google Scholar]
- Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW. Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav.Brain Res. 2000;115:183–204. doi: 10.1016/s0166-4328(00)00258-8. [DOI] [PubMed] [Google Scholar]
- Thakkar MM, Ramesh V, Strecker RE, McCarley RW. Microdialysis perfusion of orexin-A in the basal forebrain increases wakefulness in freely behaving rats. Arch.Ital.Biol. 2001;139:313–328. [PubMed] [Google Scholar]
- Zaborszky L, Duque A. Sleep-wake mechanisms and basal forebrain circuitry. Front Biosci. 2003;8:d1146–d1169. doi: 10.2741/1112. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lin L, Kaur S, Thankachan S, Blanco-Centurion C, Yanagisawa M, Mignot E, Shiromani PJ. The development of hypocretin (orexin) deficiency in hypocretin/ataxin-3 transgenic rats. Neuroscience. 2007;148:34–43. doi: 10.1016/j.neuroscience.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]