

Abstract
Activation of muscarinic cholinergic receptors on pyramidal cells of the cerebral cortex induces the appearance of a slow afterdepolarization that can sustain autonomous spiking after a brief excitatory stimulus. Accordingly, this phenomenon has been hypothesized to allow for the transient storage of memory traces in neuronal networks. Here we investigated the molecular basis underlying the muscarinic receptor-induced afterdepolarization using molecular biological and electrophysiological strategies. We find that the ability of muscarinic receptors to induce the inward aftercurrent underlying the slow afterdepolarization is inhibited by expression of a Gαq-11 dominant negative and is also markedly reduced in a phospholipase C β1 (PLCβ1) knock-out mouse. Furthermore, we show, using a genetically encoded biosensor, that activation of muscarinic receptor induces the breakdown of phosphatidylinositol 4,5-bisphosphate in pyramidal cells. These results indicate that the Gαq-11/PLCβ1 cascade plays a key role in the ability of muscarinic receptors to signal the inward aftercurrent. We have shown previously that the muscarinic afterdepolarization is mediated by a calcium-activated nonselective cation current, suggesting the possible involvement of TRPC channels. We find that expression of a TRPC dominant negative inhibits, and overexpression of wild-type TRPC5 or TRPC6 enhances, the amplitude of the muscarinic receptor-induced inward aftercurrent. Furthermore, we find that coexpression of TRPC5 and T-type calcium channels is sufficient to reconstitute a muscarinic receptor-activated inward aftercurrent in human embryonic kidney HEK-293 cells. These results indicate that TRPC channels mediate the muscarinic receptor-induced slow afterdepolarization seen in pyramidal cells of the cerebral cortex and suggest a possible role for TRPC channels in mnemonic processes.
Introduction
The cholinergic input to the cerebral cortex plays an important role supporting processes such as arousal, attention, memory, and learning (Phillis, 2005). This effect is mediated in part by the ability of muscarinic receptors to modulate multiple ion currents and thus sculpture how neurons function within networks (Lucas-Meunier et al., 2003). In this context, one of the most intriguing electrophysiological effects signaled by muscarinic receptors is their ability to induce the appearance of a slow afterdepolarization (sADP) in pyramidal cells (Krnjević et al., 1971; Schwindt et al., 1988; Andrade, 1991; Constanti and Bagetta, 1991; Klink and Alonso, 1997), the principal cell type of the cerebral cortex. This afterpotential, once induced, can sustain persistent spiking that greatly outlasts the original stimulus and therefore may serve as a mechanism for the transient storage of memory traces within neuronal networks (Krnjević et al., 1971; Schwindt et al., 1988; Andrade, 1991; Constanti and Bagetta, 1991; Lisman and Idiart, 1995; Haj-Dahmane and Andrade, 1998; Egorov et al., 2002).
Although the muscarinic receptor-induced sADP was discovered in the context of cholinergic actions in cortex, it is now clear that a variety of neuromodulators acting on heptahelical receptors capable of coupling to Gαq-11 also induce the appearance of an sADP in this region (Araneda and Andrade, 1991; Greene et al., 1994; Sidiropoulou et al., 2009). Furthermore, an sADP with characteristics similar to that observed in cortical pyramidal cells has also been observed in several other regions of the brain (McQuiston and Madison, 1999; Lawrence et al., 2006; Fowler et al., 2007; Pressler et al., 2007; Yamamoto et al., 2007). These results suggest that the ability to induce an sADP may be a generalized action signaled by Gαq-11-coupled receptors in many types of central neurons.
Surprisingly, although the existence and potential functional role of a muscarinic receptor-induced sADP seen in cortical pyramidal cells has been appreciated since the seminal studies of Krnjević and associates in the early 1970s (Krnjević et al., 1971), our understanding of the molecular mechanisms underlying this phenomenon remains limited. There is now a general consensus that the sADP is gated by calcium influx into the cell (Schwindt et al., 1988; Andrade, 1991; Constanti and Bagetta, 1991; Pressler and Strowbridge, 2006) and thus represents a calcium-activated current. However, the identity of the current underlying the sADP (IsADP) has remained uncertain. Thus, IsADP has been reported to represent a calcium-activated nonselective cation current (Haj-Dahmane and Andrade, 1998; Pressler and Strowbridge, 2006), a calcium-induced inhibition of a resting potassium current (Constanti and Bagetta, 1991) or a combination of these (Greene et al., 1994). In addition, little is known at the present time about the signaling cascade linking muscarinic receptor activation to the induction of IsADP. In the current work, we have used molecular biological approaches implemented in organotypic brain slices to address both of these issues.
Materials and Methods
Brain slice preparation and transfection.
Brain slice preparation and transfection was essentially as described previously (Haj-Dahmane and Andrade, 1998; Béïque and Andrade, 2003) except for the use of animals in the third and fourth postnatal week. All experiments were conducted in accordance with the guidelines for care and use of experimental animals established by the Wayne State University Institutional Animal Care and Use Committee. Briefly, rats or mice of either sex [postnatal day 19 (P19) to P24] were killed by decapitation after the induction of deep anesthesia using halothane. The brain was cooled, and cortical slices (300–400 μm) containing the medial prefrontal and anterior sensorimotor cortices immediately anterior to the genu of the corpus callosum were prepared using a vibratome (Series 1000; Lancer). Slices were cut in Ringer's solution of standard composition [in mm: 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose (bubbled to saturation with 95% O2–5% CO2)] containing 10 mm HEPES. Acute slices were allowed to recover in Ringer's for at least 1 h before recording. Alternatively, slices were placed in organotypic culture (Stoppini et al., 1991; Béïque and Andrade, 2003; Villalobos et al., 2004), transfected the same day using particle mediated gene transfer (“gene gun”), and used 2–5 d after transfection. IsADP and other electrophysiological parameters were stable over this time frame in culture. Transfected neurons were identified by the coexpression of green fluorescent protein (GFP). Human embryonic kidney HEK-293 cell culture and transfection were implemented as described previously (Bouhamdan et al., 2006).
Electrophysiological recordings.
For electrophysiological and imaging experiments, acute slices, slices in organotypic culture, or HEK-293 cells plated on coverslips were transferred to a recording chamber on the stage of an upright microscope (Olympus BX50 or Nikon E600) in which they were continuously perfused with Ringer's solution at equilibrium with 95% O2–5% CO2 at 30–31°C. Pyramidal neurons and HEK-293 cells were targeted for recording using differential interference contrast imaging, and transfected cells were identified under fluorescence microscopy by the expression of GFP.
Whole-cell recordings from pyramidal neurons were obtained using potassium-based intracellular solution of standard composition (in mm: 125 KmeSO4, 5 KCl, 5 NaCl, 0.02 EGTA, 10 HEPES, 1 MgCl2, 10 NaH2PO4, 4, ATP Mg salt, and 0.3 GTP Na salt, pH 7.3), whereas recordings from HEK-293 cells used a cesium-based intracellular solution (in mm: 130 Cs gluconate, 5 NaCl, 1 MgCl2, 10 HEPES, 0.02 EGTA, 10 phosphocreatine, 4 ATP Mg salt, and 0.5 GTP Na salt, pH 7.3). In some experiments, 10 mm inositol was added to this intracellular recording solution by reducing KMeSO4 to 120 mm. To confirm the role of calcium in the triggering of IsADP in cultured slices, we conducted paired recordings in which one of the electrodes was filled with an intracellular solution modified to include 10 mm BAPTA and 2.28 mm CaCl2 to buffer intracellular free calcium near 100 nm (Max-chelator; http://www.stanford.edu/∼cpatton/webmaxc/webmaxcS.htm). Patch pipette resistances ranged from 2.5 to 5 MΩ when filled with the appropriate intracellular recording solution. In some experiments, extracellular sodium was reduced by substituting 119 mm NaCl in the Ringer's solution with an equimolar amount of N-methyl-d-glucamine chloride. The solution exchange was monitored using action potentials evoked using constant current pulses. Action potential failure occurred generally within 10 min.
Whole-cell recordings were obtained using EPC7/10 (HEKA) or Axopath/Axoclamp (Molecular Devices) amplifiers, and data were collected using an analog-to-digital converter under the control of pClamp (Molecular Devices) or Patchmaster (HEKA). The liquid junction potential for these recordings was not corrected. Series resistance was monitored using 5–10 mV hyperpolarizing step throughout the experiment, and cells were discarded if series resistance changed more than ∼20% during the experiment. For most experiments, IsADP was elicited using one to three 100- to 200-ms-long depolarizing steps to 0 mV from a holding potential of −60 mV in the presence of 1 μm TTX and 10–30 μm carbachol. The slow afterhyperpolarization current (IsAHP) was measured 300 ms after the end of the first depolarizing step (Villalobos et al., 2004), whereas peak IsADP was measured 0.3–1 s after the third depolarizing step. All agonists and blockers were administered dissolved in the Ringer's solution. Experiments examining the effects of phospholipase C β1 (PLCβ1) gene deletion compared IsADP in cells in slices derived from wild-type and knock-out littermates. Experiments involving transfection of pyramidal cells used two types of controls, either cells transfected with pcDNA3.1:pEGFP–N1 (4:1) or untransfected cells generally adjacent to the transfected neurons (paired recordings).
In preliminary experiments, we noticed that IsADP appears during the third week of postnatal life and grows thereafter. Therefore, in the present experiments, we used animals aged P17–P24. Remarkably, but as found by others (Xiang et al., 2000), the preparation of organotypic slices from these older animals required methods essentially identical to those used when working with much younger animals. The use of animals in the third and fourth postnatal week probably accounts for the smaller amplitude for IsADP in the present study (∼50%) vis à vis the amplitude of IsADP observed previously in adult animals (Haj-Dahmane and Andrade, 1998). The transfection at a time point when IsADP is developing probably contributed to the effectiveness of the TRPC5 dominant negative (DN) as discussed previously for other gene-silencing strategies (Guzowski et al., 2000).
Imaging.
The plectin homology domain of PLCdelta1 fused to GFP (GFP–PHPLCδ1) was expressed in organotypic cortical brain slices and imaged using an Olympus Fluoview laser scanning microscope using the 488 nm line of an argon laser. Live imaging of transfected neurons in organotypic slices was conducted under conditions essentially identical to those used for the electrophysiological experiments, except imaging was conducted at room temperature and carbachol was added as a bolus (10 μl of a 10 mm carbachol solution added directly into the bath) to obtain responses with rapid onset and recovery. For these experiments, we selected cells with large apical dendrites and assayed the redistribution of this reporter construct during administration of carbachol by collecting image stacks (four to six optical slices, 0.5–1 μm apart) every 20–30 s using a 60× objective and focusing on a small section of the proximal apical dendrite. NIH ImageJ was used to analyze the fluorescence intensity over a short segment of dendrite, or over a small area of membrane or cytosol, at a constant optical plane over time.
Statistics.
Data are reported as means ± SEM in both the text and the figures. Statistical analysis used t tests and ANOVAs, followed by Scheffé's post hoc comparisons.
Reagents.
All drugs were obtained from Sigma, except TTX (BMD Bioscience) and SKF96365 [1-[β-[3-(4-methoxyphenyl)propoxy]-4-methoxyphenethyl]-1H-imidazole · HCl] (Biomol). HEK-293 were transfected using Lipofectamine 2000 (Invitrogen) using 1 μg of each of the plasmids and 0.5 μg of pEGFP–N1. “Bullets” for particle-mediated gene transfer (gene gun) were prepared using ∼3–6 mg of 1.6 μm gold particles and 50 μg of DNA. When transfecting TRPC5 or TRPC6, which were untagged, we used 40 μg of plasmid DNA and 10 μg of pEGFP–N1 (Clontech) to identify the transfected cells. The PLCbeta1-carboxy tail (PLCβ-ct) DN (Kammermeier and Ikeda, 1999) was a kind gift from Dr. S. R. Ikeda (National Institute on Alcohol Abuse and Alcoholism, Rockville, MD), GFP–PHPLCδ1 (Stauffer et al., 1998) from Dr. T. Meyer (Stanford University, Stanford, CA), α1G (Perez-Reyes et al., 1998) from Dr. E. Perez-Reyes (University of Virginia, Charlottesville, VA), mTRPC5 (Tang et al., 2001) from Dr. M. X. Zhu (Ohio State University, Columbus, OH), TRPC6 from Dr. S. Kitayama (Hiroshima University School of Dentistry, Hiroshima, Japan) (Mizuno et al., 1999), and the TRPC5 dominant negative (Greka et al., 2003) from Dr. D. E. Clapham (Harvard Medical School, Boston, MA). The PLCβ1 knock-out mouse (Kim et al., 1997) was a kind gift from Drs. R. K. Wong (SUNY Downstate Medica Centre, Brooklyn, NY) and H. S. Shin (Korea Institute of Science and Technology, Seoul, Korea).
Results
As seen in vivo as well as in acute brain slices (Krnjević et al., 1971; Schwindt et al., 1988; Andrade, 1991; Constanti et al., 1993), cortical pyramidal neurons maintained in organotypic culture express a carbachol-induced sADP capable of sustaining repetitive spiking activity (Fig. 1A). In general, layer III–V pyramidal neurons in organotypic slices prepared from animals late in the third or fourth postnatal weeks reliably expresses a robust sADP (or IsADP) during carbachol administration (10–30 μm). In contrast, carbachol induced a detectable ADP/IsADP only infrequently in organotypic slices derived from animals younger than P17. This observation is consistent with the previous observation of a developmental delay in appearance of the muscarinic receptor-dependent sADP in entorhinal cortex (Reboreda et al., 2007). Beyond this age limitation, expression of a carbachol-induced sADP or IsADP was well preserved in standard organotypic culture conditions for at least 5 d, the longest interval tested. Therefore, all experiments reported in the current work were conducted using brain slices derived from P17–P24 pups and maintained in organotypic culture for 2–5 d.
Figure 1.

Muscarinic receptors activate IsADP in organotypic cortical slices. A1, Under basal conditions, a suprathreshold depolarizing pulse delivered to a layer V cortical pyramidal cell of the prefrontal cortex is followed by a slow afterhyperpolarization. A2, Administration of carbachol (30 μm) leads to the replacement of this afterhyperpolarization by an sADP that supports sustained spiking. A3, The current underlying this sADP (IsADP) can be recorded using a hybrid current–voltage-clamp protocol in which it appears as a slow inward aftercurrent following the spike burst. Inset, IsADP can also be triggered by depolarizing steps to 0 mV in voltage clamp. B1, B2, The carbachol-induced IsADP is greatly suppressed when intracellular calcium is buffered near rest using BAPTA. B1 and B2 depict a paired recording from two pyramidal cells in which B1 was recorded using control intracellular solution and B2 using an intracellular solution containing BAPTA to buffer intracellular calcium. C1, C2, Reducing extracellular sodium inhibits IsADP in the control cell depicted in B1 and B2.
As illustrated in Figure 1A, the muscarinic receptor-induced sADP, once triggered by a burst of spikes, could sustain autonomous spiking greatly outlasting the initial stimulus (Fig. 1A2). In organotypic slices, this autonomous spiking generally self-terminated after several seconds (mean spiking duration, 5.6 ± 1.7 s; n = 6 cells), which contrasts with the long-lasting sustained spiking activity seen in acute slices from this same region derived from adult animals (Andrade, 1991; Haj-Dahmane and Andrade, 1998). This difference notwithstanding, these observations indicate that the essential features of the muscarinic receptor-induced sustained spiking are preserved in organotypic culture.
In the present work, we sought to examine the molecular basis for IsADP, the current underlying the sADP and sustained spiking. This current can be recorded directly using a hybrid current-clamp protocol in which the amplifier is switched into voltage clamp immediately after a burst of spikes (Fig. 1A3). An inward aftercurrent can also be induced in voltage clamp using one to three brief depolarizing steps to 0 mV, with three pulses eliciting a near-maximal activation of IsADP at a given carbachol concentration (Fig. 1A3, inset). In the present work, we have used both of these protocols to study IsADP.
Previous studies in acute slices derived from adult animals have reported that IsADP is gated by calcium influx, leading to a rise in intracellular free calcium (Andrade, 1991; Constanti and Bagetta, 1991; Greene et al., 1994; Haj-Dahmane and Andrade, 1998). Consistent with these findings, the muscarinic receptor-dependent inward aftercurrent in organotypic slices is suppressed by buffering intracellular calcium near resting levels (control IsADP = −89 ± 41 pA, n = 5; BAPTA-buffered IsADP = 9 ± 14 pA, n = 7; p < 0.05) (Fig. 1B). Similarly, previous studies have also shown that IsADP in adult prefrontal cortex is mediated by a nonselective cation current (Andrade, 1991; Haj-Dahmane and Andrade, 1998). Again, consistent with these findings, the carbachol-induced inward aftercurrent recorded in pyramidal cells in organotypic slices is also inhibited by lowering extracellular sodium (Fig. 1C) (residual current in low sodium = 40 ± 11% of control; p < 0.05; n = 6 cells). These results indicate that the basic mechanism underlying the carbachol-induced sADP in adult cortex is also preserved in organotypic slices.
Signaling of IsADP
The ability of carbachol (10–30 μm) to induce IsADP is significantly inhibited by administration of 100 nm of the muscarinic antagonist pirenzepine (n = 5 cells; p < 0.05) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and completely blocked by administration of 1 μm of this antagonist (n = 4 cells) (Fig. 2A). In contrast, administration of the muscarinic antagonist PD102807 [3,6a,11,14-tetrahydro-9-methoxy-2-methyl-(12H)-isoquino[1,2-b]pyrrolo[3,2-f][1,3]benzoxazine-1-carboxylic acid, ethyl ester] (1 μm) has no detectable effect on the ability of carbachol to induce IsADP in pyramidal cells (n = 5 cells) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Pirenzepine at ≤100 nm can be expected to preferentially inhibit muscarinic M1 receptors, whereas PD102807 at a concentration of 1 μm can be expected to be essentially without effect on M1 receptors but to inhibit M2–M4 receptors (Caulfield and Birdsall, 1998). These observations suggest the involvement of muscarinic M1 receptors, which are robustly expressed in the cerebral cortex (Buckley et al., 1988), in the signaling of IsADP.
Figure 2.

Carbachol elicits IsADP by activating Gαq-11. A1–A3, The ability of carbachol (30 μm) to induce the appearance of IsADP in a pyramidal cell of prefrontal cortex in organotypic culture is blocked by administration of pirenzepine (1 μm), an antagonist of Gαq-11-coupled M1 muscarinic receptors. B1, A burst of three depolarizing steps delivered in the presence of carbachol trigger IsADP in a different pyramidal cell recorded in a cultured organotypic cortical brain slice (gray trace, control; black trace, 30 μm carbachol). B2, The same stimuli fail to elicit an IsADP in a pyramidal cell transfected with the Gαq-11 dominant-negative PLCβ-ct.
Because muscarinic M1 receptors signal many of their effects through heterotrimeric G-proteins of the Gαq-11 subtype, we next tested for the involvement of these G-proteins in the signaling of IsADP. To that effect, we used particle-mediated gene transfer (gene gun) to express in pyramidal cells a PLCβ1 fragment fused to GFP (PLCβ-ct) that functions as a dominant negative against this class of G-proteins (Kammermeier and Ikeda, 1999). Expression of this dominant negative significantly inhibited the ability of carbachol (30 μm) to induce the appearance of IsADP (Fig. 2B) (control IsADP = −21.3 ± 5.2 pA, n = 14; PLCβ-ct transfected IsADP = −6.7 ± 3.9 pA, n = 12; p < 0.05). Importantly, this suppression did not reflect a nonspecific effect of the transfection because expression of GFP alone had no detectable impact on the ability of carbachol to induce IsADP (p = 0.97) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Furthermore, the effect of the PLCβ-ct dominant negative was selective because it failed to suppress the ability of the GABAB agonist baclofen (30 μm) to elicit a potassium current in these cells, a response known to be mediated by heterotrimeric G-proteins of the Gαi/o family (p = 0.52) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These experiments indicated that IsADP is activated downstream from Gαq-11.
Activation of muscarinic receptors coupling to Gαq-11 leads to the activation of PLC and the breakdown of inositol phospholipids in the cerebral cortex (Brown et al., 1984). To determine whether this effect occurs in pyramidal cells, we took advantage of the availability of a genetically encoded GFP-based biosensor (GFP–PHPLCδ1) that detects phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] breakdown/IP3 production by redistributing from the plasma membrane to the cytosol (Stauffer et al., 1998). Under basal conditions, GFP–PHPLCδ1 localizes to the plasma membrane of pyramidal cells, including their apical dendrites (Fig. 3A2), a site of muscarinic receptor expression (Vogt and Burns, 1988). However, during administration of carbachol, this biosensor redistributes away from the membrane into the cytoplasm, indicating the breakdown of PtdIns(4,5)P2 and the liberation of IP3 in pyramidal cells (Fig. 3A2). This results in a significant reduction in the GFP fluorescence associated with the membrane and a concomitant increase in the GFP fluorescence in the cytosol (n = 6 cells) (Fig. 3A3). During removal of the carbachol from the perfusate, GFP–PHPLCδ1 recovers its membrane localization (Fig. 3A2,A3). These results indicate that carbachol induces the breakdown of PtdIns(4,5)P2 in pyramidal cells.
Figure 3.

Muscarinic receptors elicit the breakdown of PtdIns(4,5)P2 in pyramidal cells and signal IsADP through PLCβ1. A1, Flattened confocal image stack illustrating a group of cortical pyramidal cells in an organotypic brain slice transfected with GFP–PHPLCδ1. Scale bar, 200 μm. A2, Top, Close up of the proximal apical dendrite of a pyramidal cell transfected with GFP–PHPLCδ1 at rest (left), in the presence of carbachol (middle), or after recovery from the agonist (right). Notice the redistribution of GFP–PHPLCδ1 away from the membrane during the application of carbachol. Scale bar, 5 μm. Bottom, Quantification of the redistribution of the fluorescent reporter in the top during administration of carbachol. The region sampled for these plots is denoted by a box in the left top. A3, Graph summarizing the changes in GFP–PHPLCδ1 fluorescence at the membrane (Fmemb) and in the cytosol (Fcyt) during carbachol administration. n = 6 cells; *p < 0.05. B1, Administration of carbachol (30 μm) to a prefrontal cortex pyramidal neuron in a slice derived from a wild-type mouse induces IsADP. B2, In contrast, administration of carbachol to a pyramidal cell derived from PLCβ1 knock-out mice fails to induce IsADP under the same experimental conditions. The difference in IAHP between the cells illustrated here is within the range of cell-to-cell variability expected for slow and medium afterhyperpolarization currents in pyramidal cells of prefrontal cortex (Villalobos et al., 2004).
Previous studies have shown that PLCβ1 is the predominant PLCβ isozyme expressed in the cerebral cortex (Watanabe et al., 1998) and that its deletion results in a marked inhibition in the ability of carbachol to induce the breakdown of inositol phospholipids (Kim et al., 1997). Therefore, to test whether this enzyme played an essential role in the signaling of IsADP, we examined the muscarinic-induced IsADP in PLCβ1 knock-out mice (Kim et al., 1997). As illustrated in Figure 3B, the ability of carbachol to induce an IsADP was markedly reduced in PLCβ1−/− mice with respect to their wild-type (PLCβ1+/+) littermates (wild type, −43 ± 5.5 pA, n = 33; PLCβ1−/−, −9 ± 3.2 pA, n = 7; p < 0.01). Overall, we observed an ∼80% reduction in IsADP mean amplitude, consistent with the previously observed incomplete suppression in the turnover of inositol phospholipids in PLCβ1−/− mice (Kim et al., 1997). The residual response likely reflects the expression of PLCβ4, which is also expressed, albeit at lower levels, in cortex (Watanabe et al., 1998). The amplitude of IsAHP, another calcium-activated current also expressed in these cells, was not significantly affected in the PLCβ1−/− mice (p = 0.22; n = 18 PLCβ1+/+ and 20 PLCβ1−/−). These results support the idea that that muscarinic receptors induce IsADP through a mechanism involving Gαq-11 and the activation of PLCβ1 leading to the breakdown of PtdIns(4,5)P2.
Coexpression of TRPC and calcium channels reconstitute a muscarinic receptor-regulated inward aftercurrent in HEK-293 cells
We have shown previously that, in pyramidal neurons of the prefrontal cortex, the muscarinic receptor-dependent sADP is mediated by a nonselective cation current. Among nonselective cation channels, the TRPC family of TRP channels has been shown to be activated by heptahelical receptors through a mechanism involving Gαq-11 and PLC (Clapham, 2003; Montell, 2005). TRPC channels formed by TRPC5 and TRPC6 subunits can also be regulated by intracellular free calcium (Strübing et al., 2001, 2003; Ramsey et al., 2006). Therefore, these observations raised the possibility that TRPC channels could mediate IsADP. One problem with this idea is that, although some TRPC can be regulated by intracellular calcium, such channels are generally not thought as mediators of calcium-activated currents. Therefore, to determine whether TRPC channels can generate calcium-activated currents, we cotransfected TRPC5 and the voltage-dependent α1G T-type calcium channel into HEK-293 cells and tested for the appearance of muscarinic-dependent outward aftercurrent. The TRPC5 subunit was selected for these experiments because previous studies have documented its positive regulation by intracellular calcium (for review, see Plant and Schaefer, 2003). The α1G calcium channel was chosen for this experiment because small depolarizations capable of activating T-type calcium currents in cortex are sufficient to trigger IsADP in pyramidal cells (Andrade, 1991).
As illustrated in Figure 4, brief depolarizing steps to −30 mV capable of activating T-type calcium channels elicits the appearance of a modest inward aftercurrent in cells cotransfected with TRPC5 and α1G calcium channels but fail to elicit any significant aftercurrent in mock-transfected cells (empty plasmid) or in cells transfected with either TRPC5 or α1G calcium channels alone (Fig. 4A4,B) (p < 0.01). Furthermore, this small inward aftercurrent is greatly facilitated by application of carbachol (30 μm) (Fig. 4A4,B) (p < 0.01), which in these cells activates endogenous muscarinic receptors (Schaefer et al., 2000). In contrast, no carbachol-induced aftercurrent develops in cells transfected with either TRPC5 or α1G alone. Importantly, administration of carbachol, however, does induce the expected inward current in cells transfected with TRPC5 alone as well as TRPC5 and α1G. This confirms the expression of TRPC5 channels in both of these experimental groups and emphasizes the essential role played by calcium channels in the generation of the aftercurrent (Fig. 5A1, inset). These results support the idea that TRPC channels can generate a muscarinic receptor-dependent inward aftercurrent. The small inward aftercurrent seen in the absence of carbachol is consistent with the previous observation that expression of TRPC5 in HEK-293 cells can result in the expression of spontaneously active channels (Watanabe et al., 1998).
Figure 4.
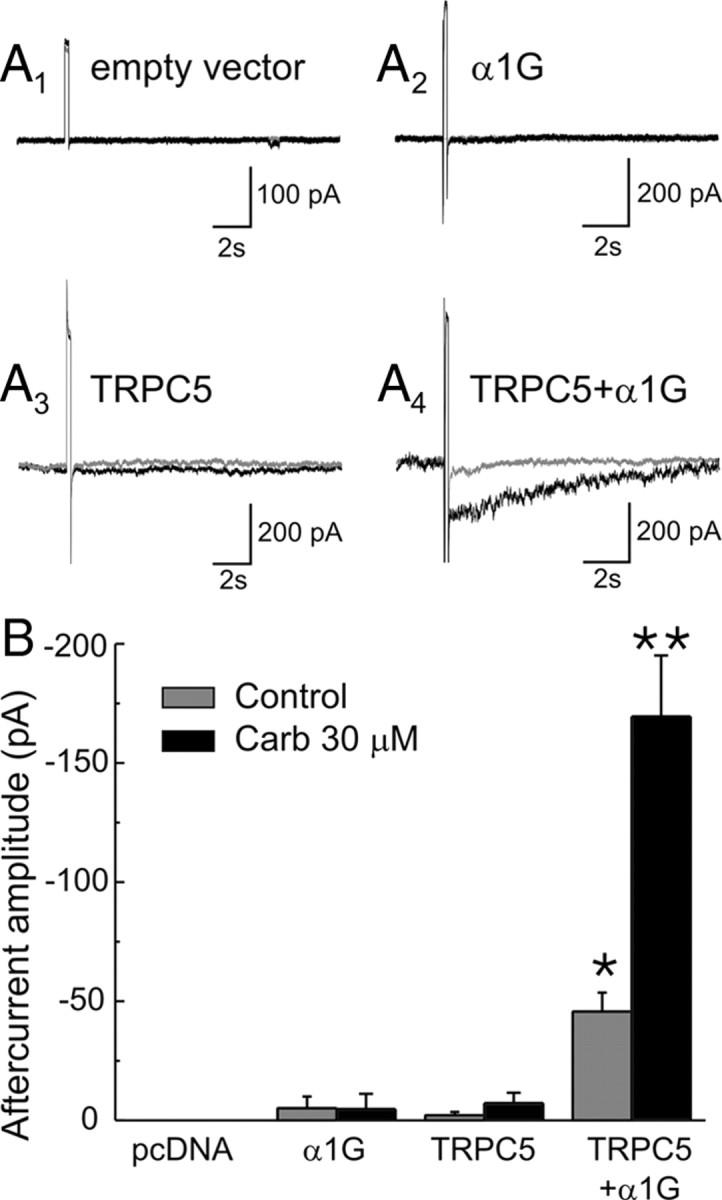
Coexpression of TRPC5 and T-type calcium channels in HEK-293 cells reconstitutes a muscarinic-receptor-regulated inward aftercurrent. A1–A4, Traces depicting the aftercurrents evoked by depolarizing steps from −80 to −30 mV in cells transfected with pcDNA3.1, the T-type calcium channel α1G, TRPC5, and TRPC5 plus α1G under resting conditions (gray traces) and in the presence of carbachol (30 μm; black traces). B, Graph summarizing the results of this experiment in 6 cells transfected with empty vector, 8 cells transfected α1G, 7 cells transfected with TRPC5, and 19 cells transfected with TRPC5 plus α1G. *p < 0.01 with respect to pcDNA3, α1G, and TRPC5. **p < 0.01 with respect to α1G plus TRPC5 alone.
Figure 5.

Properties of the carbachol-induced inward current and aftercurrent in HEK-293 cells transfected with TRPC5 and α1G. A, Administration of carbachol (30 μm) induces an inward current in HEK-293 cells transfected with TRPC5 and α1G. This inward current is inhibited by reducing extracellular sodium and replacing it with N-methyl-d-glucamine. In these same cells, reducing extracellular sodium also inhibits the inward aftercurrent. *p < 0.01; n = 7 cells. B, The carbachol-induced inward current observed in HEK-293 cells transfected with TRPC5 and α1G is inhibited by cotransfection with a pore-dead TRPC5 dominant negative (TRPC5-DN). This dominant negative also inhibits the inward aftercurrent. *p < 0.01; n = 19 cells cotransfected with empty vector (pcDNA3.1) and 10 cells cotransfected with TRPC5-DN.
If the carbachol-induced inward aftercurrent were mediated directly by TRPC5 channels, it should be reduced by lowering extracellular sodium. Consistent with this prediction, both the inward current and the slow inward aftercurrent present in cells cotransfected with TRPC5 and α1G calcium channels were suppressed when most extracellular sodium was replaced by N-methylglucamine (Fig. 5A). Similarly, the inward current and inward aftercurrent should both be inhibited by a pore-dead TRPC5 subunit capable of acting as a dominant negative against TRPC channels. Again, consistent with this prediction, cotransfection with a pore-dead TRPC5 fused to GFP shown previously to act as an effective dominant negative (TRPC5-DN) (Greka et al., 2003; Strübing et al., 2003) blocked both the inward current and the inward aftercurrent in cells expressing wild-type TRPC5 and α1G calcium channels (Fig. 5B). These results indicate that the muscarinic receptor-modulated inward aftercurrent seen in HEK-293 cotransfected with TRPC5 and α1G channels is carried through TRPC channels and thus that TRPC channels can mediate calcium-activated inward aftercurrents.
IsADP is facilitated by overexpression of TRPC5 and inhibited by a pore-dead TRPC5 dominant negative
Previous studies have reported the expression of most TRPC subunits in the cerebral cortex (Mizuno et al., 1999; Fowler et al., 2007), and we confirmed the expression of TRPC1 and TRPC3–TRPC6 mRNA in the rat prefrontal cortex (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). To test for the possible involvement of these TRPC subunits in mediating IsADP, we first used a pharmacological approach. Because SKF96365, 2-APB, and flufenamic acid have been used widely as TRPC blockers, we first examined their effectiveness in HEK-293 cells transfected with TRPC5 and α1G. All three of these compounds blocked the TRPC5 currents induced by carbachol but also simultaneously inhibited the T-type calcium channel currents (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material). This limited their usefulness for identifying TRPC currents in situ. The best selectivity was seen with flufenamic acid, which at a concentration of 10 μm only marginally inhibited T-type calcium currents while still producing a robust suppression of the carbachol/TRPC5-dependent inward aftercurrent (Fig. 6A2). Therefore, we tested the effect of 10 μm flufenamic acid on IsADP induced by the administration of carbachol in acute cortical brain slices. As illustrated in Figure 6, A1 and A2, administration of 10 μm flufenamic acid significantly inhibited IsADP but had no significant effect on IsAHP (control, 19.2 ± 3.2 pA; flufenamic, 15.5 ± 4.2 pA; n = 9 cells; p = 0.5). These results are consistent with previous observations indicating that flufenamic acid, albeit at considerably higher concentrations, inhibits IsADP (Haj-Dahmane and Andrade, 1999; Zhang and Arsenault, 2005; Fowler et al., 2007). The results of these pharmacological experiments support the involvement of TRPC channels in the generation of IsADP in pyramidal neurons but are inconclusive as a result of the weak selectivity of the available TRPC blockers.
Figure 6.

The carbachol-induced IsADP is inhibited by the TRP channel blocker flufenamic acid and by a TRPC dominant negative. A1, Administration of 10 μm flufenamic acid inhibits the IsADP induced by 30 μm carbachol in pyramidal cells recorded in acute brain slices. The gray traces depict the aftercurrents recorded under resting conditions, and the black traces depict the aftercurrent recorded in the presence of 30 μm carbachol. A2, Graph summarizing the results of experiments assessing the ability of 10 μm flufenamic acid to inhibit the muscarinic receptor-induced inward aftercurrent seen in HEK-293 cells transfected with TRPC5 plus α1G and to inhibit IsADP in acute brain slices. n = 8 HEK-293 cells and 9 pyramidal neurons; *p < 0.01. B1, IsADP recorded in a control (GFP transfected) pyramidal neuron and a pyramidal neuron transfected with TRPC5-DN. B2, Summary graph depicting the effect of expressing the TRPC5-DN on IsADP and IsAHP in pyramidal cells recorded in organotypic slices. n = 14 control cells and 15 cells transfected with TRPC5-DN. *p < 0.01.
To further test for the involvement of TRPCs in the generation of IsADP, we took advantage of the effectiveness of pore-dead TRPC subunits to function as dominant negatives against homomultimeric and heteromultimeric TRPC channels (Hofmann et al., 2002; Strübing et al., 2003). The six TRPC subunits expressed in the brain are thought to fall into two broad classes comprising TRPC1/4/5 and TRPC3/6/7, and previous studies have shown that these two groups can form heteromultimeric TRP channels in the presence of TRPC3 (Strübing et al., 2001, 2003). Because TRPC3 is abundantly expressed in the prefrontal cortex (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material), the TRPC5-DN has the potential to serve as a general TRPC dominant negative in this region. Therefore, we expressed the TRPC5-DN, which is GFP tagged, in organotypic slices and confirmed its expression in pyramidal cells by visualizing the GFP (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material). As illustrated in Figure 6B, expression of the TRPC5-DN essentially blocked the carbachol-induced IsADP in pyramidal cells (n = 14 cells; p < 0.01). Importantly, expression of the TRPC5-DN had no detectable effect on IsAHP (p = 0.9), a second calcium-activated current also expressed in pyramidal cells. This indicates that the inhibition of IsADP is not simply secondary to a nonspecific inhibition of calcium influx. These results support the involvement of TRPC channels in the generation of IsADP.
If TRPC subunits played an essential part in the formation of the channel carrying IsADP, it could be expected that overexpression of TRPC subunits should facilitate this current. To test this conjecture, we expressed TRPC5 and TRPC6 in pyramidal cells and examined their effect on the carbachol-induced IsADP. Both of these subunits have been shown previously to form channels facilitated by intracellular calcium (for review, see Ramsey et al., 2006) and are representative of the two main groups of TRPC subunits expressed in the brain. Remarkably, under unstimulated conditions, pyramidal neurons expressing TRPC5 or TRPC6 appeared remarkably normal in terms of their basic electrophysiology. They exhibited membrane potentials/holding currents comparable with those of control untransfected or GFP-transfected neurons, exhibited no detectable change in the amplitude of IsAHP (p > 0.5), and fired in a manner indistinguishable for control pyramidal cells (Fig. 7B). Administration of carbachol (30 μm) to these cells, however, resulted in the appearance of an extraordinarily large IsADP (Fig. 7) (TRPC5, n = 10 cells; TRPC6, n = 8 cells). This facilitated IsADP was frequently many times larger than the largest IsAHP ever observed in control cells and reached in some cases a nanoampere or more. These results further support the idea that TRPC subunits mediate the carbachol-induced IsADP in pyramidal neurons.
Figure 7.

Expression of TRPC5 and TRPC6 facilitates IsADP. A1–A3, IsADP recorded in a control (untransfected) pyramidal neuron, a pyramidal neuron transfected with TRPC6, and a third pyramidal neuron transfected with TRPC5. B1, Flattened confocal image stack depicting a pyramidal neuron transfected with TRPC6. Scale bar, 100 μm. B2, B3, Neuronal firing elicited by depolarizing constant current pulses (0, 100, 350 pA) in a control (untransfected) pyramidal neuron and an adjacent cell transfected with TRPC6 (paired recording). During administration of carbachol the TRP6-transfected pyramidal neuron developed an IsADP in excess of 1 nA. C, Graph summarizing the effect of expressing TRPC5 or TRPC6 on the amplitude of IsADP in pyramidal cells of the prefrontal cortex. *p < 0.05.
Discussion
Previous studies have shown that activation of muscarinic receptors signals the appearance of a calcium-activated sADP in cortical pyramidal cells and that this afterpotential can sustain regenerative spiking after a brief suprathreshold triggering stimulus (Krnjević et al., 1971; Schwindt et al., 1988; Andrade, 1991; Constanti and Bagetta, 1991; Lisman and Idiart, 1995; Haj-Dahmane and Andrade, 1998; Egorov et al., 2002). Here we have used a combination of dominant-negative, overexpression, gene deletion, and reconstitution strategies to address the molecular mechanisms by which muscarinic receptors induce this afterpotential. We find that the current underlying the sADP is carried through TRPC channels and is signaled by muscarinic receptors through a mechanism that involves Gαq-11 and PLCβ1.
Activation of muscarinic receptors coupling to Gαq-11, such as M1 receptors, activate PLCβ in cerebral cortex to increase the breakdown of inositol phospholipids (Kim et al., 1997). However, recent studies suggest that PLCβ activation is just one of two signaling branches downstream of Gαq-11, with the second branch using p63RhoGEF and its homologs to activate RhoA (Lutz et al., 2007; Williams et al., 2007). Furthermore, it has also been reported that M1 receptors can also signal in a manner that is independent of heterotrimeric G-proteins (Heuss and Gerber, 2000). Thus, it was important to examine the specific signaling mechanism used by muscarinic receptors to activate IsADP. In the current work, we find that the ability of muscarinic receptors to induce IsADP is blocked by a Gαq-11 dominant negative, thus identifying this G-protein as a key component of the cascade signaling this response. Furthermore, we use a GFP-based biosensor to demonstrate that activation of muscarinic receptors results in the breakdown of PtdIns(4,5)P2 and the generation of IP3 in pyramidal neurons. Finally, we show that the ability of muscarinic receptors to elicit IsADP is greatly inhibited by the deletion of PLCβ1, the predominant PLCβ isoform expressed in cortex. These results identify Gαq-11 and PLCβ1 as key molecules involved in the signaling of IsADP. However, additional studies will be required to precisely delineate the molecular steps involved in the activation of this current.
TRPC channels have emerged as ubiquitous cellular effectors downstream from surface receptors capable of activating PLC (Clapham, 2003; Montell, 2005; Putney, 2007). In the current work, we show that TRPC channels mediate the slow calcium-activated afterdepolarization elicited by muscarinic and presumably also other Gαq-11-coupled receptors in pyramidal neurons. The evidence supporting this conclusion is twofold. First, we show that coexpression of TRPC5 and the α1G calcium channel is sufficient to reconstitute a muscarinic receptor-activated inward aftercurrent in HEK-293 cells. Although these experiments used T-type calcium channels, it seems likely that the key factor is calcium influx, independent of the specific calcium channel involved. Second, we show that IsADP in pyramidal neurons is facilitated by expression of TRPC5 or TRPC6 and is inhibited by low concentration of flufenamic acid, a widely used TRPC blocker, as well as by expression of a pore-dead TRPC5 subunit capable of acting as a dominant negative against homomeric and heteromeric TRPC channels. Admittedly, individually each of these experiments has limitations. However, in combination, these results point to a key role for TRPC channels in the generation of IsADP in pyramidal cells.
Previous studies have shown that TRPC channels mediate some of the effects of neurotrophins and of guidance molecules such as Netrin-1, at least in part through their ability to flux calcium (Talavera et al., 2008). However, it is worth noting that the facilitation of IsADP by TRPC overexpression and suppression by TRPC inhibition observed here are unlikely to have resulted simply from changes in calcium influx. IsADP was not triggered by calcium influx secondary to TRPC activation but rather by calcium influx secondary to membrane depolarizations capable of activating voltage-dependent calcium channels. Furthermore, these depolarizing steps are not only temporally uncoupled from activation of TRPC channels but also would be expected to reduce rather than enhance calcium influx through TRPC channels. Thus, the current results identify TRPC channels as essential components of the channel carrying IsADP.
TRPCs expressed in the brain fall into two main groups: one group composed of TRPC3, TRPC6, and TRPC7 and the other composed of TRPC1, TRPC4, and TRPC5 (Clapham, 2003; Montell, 2005; Putney, 2007). IsADP exhibits a voltage dependence reminiscent of that exhibited by TRPC channels containing TRPC1 and TRPC4/5 subunits (Haj-Dahmane and Andrade, 1998; Strübing et al., 2001), thus hinting at a possible role for these subunits in mediating IsADP. However, it is worth noting that expression of TRPC4 or TRPC5 alone can also give rise to currents exhibiting a voltage dependence similar to that of IsADP, especially at low levels of channel activation (Plant and Schaefer, 2003). Furthermore, previous studies have shown that activation of PLC modulates TRPV1 currents (Chuang et al., 2001) to generate a current that exhibits a voltage dependence very similar to that of IsADP. These considerations argue for caution in using electrophysiological characteristics to identify the molecular composition of the TRPC channel mediating IsADP. Equally important, although in the current experiments we find that IsADP is inhibited by a TRPC5 dominant negative, TRPC channels show a complex pattern of heteromultimerization, especially in the presence of TRPC3. Accordingly, this experiment does not provide clear-cut support for the involvement of TRPC1/4/5. Finally, in the current experiments, we find that expression of either TRPC5 or TRPC6, which are representative of the two main TRPC subfamilies, greatly facilitates the amplitude of IsADP. Thus, the physiological and molecular approaches used so far do not yet allow for the unambiguous identification of the subunit composition of the IsADP channel.
The identification of TRPC channels as the molecular basis of the muscarinic receptor-induced sADP suggests that these channels function in central neurons as coincidence detectors encoding concurrent Gαq-11/PLCβ activation and calcium influx, a proxy for neuronal activity, into long-lasting changes in excitability. In the brain, this property could allow for the transient storage of memory traces either through its ability to maintain sustained spiking after a suprathreshold stimulus (Krnjević et al., 1971; Schwindt et al., 1988; Andrade, 1991; Constanti and Bagetta, 1991; Lisman and Idiart, 1995; Haj-Dahmane and Andrade, 1998; Egorov et al., 2002) or through its ability to integrate the firing history of the cell and “code” this history onto sustained membrane depolarizations (Andrade, 1991). If this conjecture is correct, then TRPC channels could play an important role supporting mnemonic processes in the CNS.
Footnotes
This work was supported by National Institutes of Health Grants MH49355 and MH43985. We thank Dr. R. Bianchi for help securing the PLCβ1 KO mice, Dr. K. Azatian for help with some of the initial experiments, and Dr. S. Haj-Dahmane for many helpful discussions.
References
- Andrade R. Cell excitation enhances muscarinic cholinergic responses in rat association cortex. Brain Res. 1991;548:81–93. doi: 10.1016/0006-8993(91)91109-e. [DOI] [PubMed] [Google Scholar]
- Araneda R, Andrade R. 5-Hydroxytryptamine 2 and 5-hydroxytryptamine 1A receptors mediate opposing responses on membrane excitability in rat association cortex. Neuroscience. 1991;40:399–412. doi: 10.1016/0306-4522(91)90128-b. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhamdan M, Yan HD, Yan XH, Bannon MJ, Andrade R. Brain-specific regulator of G-protein signaling 9-2 selectively interacts with α-actinin-2 to regulate calcium-dependent inactivation of NMDA receptors. J Neurosci. 2006;26:2522–2530. doi: 10.1523/JNEUROSCI.4083-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E, Kendall DA, Nahorski SR. Inositol phospholipid hydrolysis in rat cerebral cortical slices. I. Receptor characterisation. J Neurochem. 1984;42:1379–1387. doi: 10.1111/j.1471-4159.1984.tb02798.x. [DOI] [PubMed] [Google Scholar]
- Buckley NJ, Bonner TI, Brann MR. Localization of a family of muscarinic receptor mRNAs in rat brain. J Neurosci. 1988;8:4646–4652. doi: 10.1523/JNEUROSCI.08-12-04646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Constanti A, Bagetta G. Muscarinic receptor activation induces a prolonged post-stimulus afterdepolarization with a conductance decrease in guinea-pig olfactory cortex neurones in vitro. Neurosci Lett. 1991;131:27–32. doi: 10.1016/0304-3940(91)90329-r. [DOI] [PubMed] [Google Scholar]
- Constanti A, Bagetta G, Libri V. Persistent muscarinic excitation in guinea-pig olfactory cortex neurons: involvement of a slow post-stimulus afterdepolarizing current. Neuroscience. 1993;56:887–904. doi: 10.1016/0306-4522(93)90135-3. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransén E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Fowler MA, Sidiropoulou K, Ozkan ED, Phillips CW, Cooper DC. Corticolimbic expression of TRPC4 and TRPC5 channels in the rodent brain. PLoS ONE. 2007;2:e573. doi: 10.1371/journal.pone.0000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CC, Schwindt PC, Crill WE. Properties and ionic mechanisms of a metabotropic glutamate receptor-mediated slow afterdepolarization in neocortical neurons. J Neurophysiol. 1994;72:693–704. doi: 10.1152/jn.1994.72.2.693. [DOI] [PubMed] [Google Scholar]
- Greka A, Navarro B, Oancea E, Duggan A, Clapham DE. TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat Neurosci. 2003;6:837–845. doi: 10.1038/nn1092. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Ionic mechanism of the slow afterdepolarization induced by muscarinic receptor activation in rat prefrontal cortex. J Neurophysiol. 1998;80:1197–1210. doi: 10.1152/jn.1998.80.3.1197. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Andrade R. Muscarinic receptors regulate two different calcium-dependent non-selective cation currents in rat prefrontal cortex. Eur J Neurosci. 1999;11:1973–1980. doi: 10.1046/j.1460-9568.1999.00612.x. [DOI] [PubMed] [Google Scholar]
- Heuss C, Gerber U. G-protein-independent signaling by G-protein-coupled receptors. Trends Neurosci. 2000;23:469–475. doi: 10.1016/s0166-2236(00)01643-x. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier PJ, Ikeda SR. Expression of RGS2 alters the coupling of metabotropic glutamate receptor 1a to M-type K+ and N-type Ca2+ channels. Neuron. 1999;22:819–829. doi: 10.1016/s0896-6273(00)80740-0. [DOI] [PubMed] [Google Scholar]
- Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH, Ryu SH, Suh PG, Shin HS. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature. 1997;389:290–293. doi: 10.1038/38508. [DOI] [PubMed] [Google Scholar]
- Klink R, Alonso A. Ionic mechanisms of muscarinic depolarization in entorhinal cortex layer II neurons. J Neurophysiol. 1997;77:1829–1843. doi: 10.1152/jn.1997.77.4.1829. [DOI] [PubMed] [Google Scholar]
- Krnjević K, Pumain R, Renaud L. The mechanism of excitation by acetylcholine in the cerebral cortex. J Physiol. 1971;215:247–268. doi: 10.1113/jphysiol.1971.sp009467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, Statland JM, Grinspan ZM, McBain CJ. Cell type-specific dependence of muscarinic signalling in mouse hippocampal stratum oriens interneurones. J Physiol. 2006;570:595–610. doi: 10.1113/jphysiol.2005.100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Idiart MA. Storage of 7 +/− 2 short-term memories in oscillatory subcycles. Science. 1995;267:1512–1515. doi: 10.1126/science.7878473. [DOI] [PubMed] [Google Scholar]
- Lucas-Meunier E, Fossier P, Baux G, Amar M. Cholinergic modulation of the cortical neuronal network. Pflugers Arch. 2003;446:17–29. doi: 10.1007/s00424-002-0999-2. [DOI] [PubMed] [Google Scholar]
- Lutz S, Shankaranarayanan A, Coco C, Ridilla M, Nance MR, Vettel C, Baltus D, Evelyn CR, Neubig RR, Wieland T, Tesmer JJ. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science. 2007;318:1923–1927. doi: 10.1126/science.1147554. [DOI] [PubMed] [Google Scholar]
- McQuiston AR, Madison DV. Muscarinic receptor activity induces an afterdepolarization in a subpopulation of hippocampal CA1 interneurons. J Neurosci. 1999;19:5703–5710. doi: 10.1523/JNEUROSCI.19-14-05703.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno N, Kitayama S, Saishin Y, Shimada S, Morita K, Mitsuhata C, Kurihara H, Dohi T. Molecular cloning and characterization of rat trp homologues from brain. Brain Res Mol Brain Res. 1999;64:41–51. doi: 10.1016/s0169-328x(98)00296-4. [DOI] [PubMed] [Google Scholar]
- Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- Phillis JW. Acetylcholine release from the central nervous system: a 50-year retrospective. Crit Rev Neurobiol. 2005;17:161–217. doi: 10.1615/critrevneurobiol.v17.i3-4.30. [DOI] [PubMed] [Google Scholar]
- Plant TD, Schaefer M. TRPC4 and TRPC5: receptor-operated Ca2+-permeable nonselective cation channels. Cell Calcium. 2003;33:441–450. doi: 10.1016/s0143-4160(03)00055-1. [DOI] [PubMed] [Google Scholar]
- Pressler RT, Strowbridge BW. Blanes cells mediate persistent feedforward inhibition onto granule cells in the olfactory bulb. Neuron. 2006;49:889–904. doi: 10.1016/j.neuron.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Pressler RT, Inoue T, Strowbridge BW. Muscarinic receptor activation modulates granule cell excitability and potentiates inhibition onto mitral cells in the rat olfactory bulb. J Neurosci. 2007;27:10969–10981. doi: 10.1523/JNEUROSCI.2961-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here) Cell Calcium. 2007;42:103–110. doi: 10.1016/j.ceca.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- Reboreda A, Raouf R, Alonso A, Séguéla P. Development of cholinergic modulation and graded persistent activity in layer v of medial entorhinal cortex. J Neurophysiol. 2007;97:3937–3947. doi: 10.1152/jn.01233.2006. [DOI] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Chubb MC, Crill WE. Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol. 1988;59:450–467. doi: 10.1152/jn.1988.59.2.450. [DOI] [PubMed] [Google Scholar]
- Sidiropoulou K, Lu FM, Fowler MA, Xiao R, Phillips C, Ozkan ED, Zhu MX, White FJ, Cooper DC. Dopamine modulates an mGluR5-mediated depolarization underlying prefrontal persistent activity. Nat Neurosci. 2009;12:190–199. doi: 10.1038/nn.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- Talavera K, Nilius B, Voets T. Neuronal TRP channels: thermometers, pathfinders and life-savers. Trends Neurosci. 2008;31:287–295. doi: 10.1016/j.tins.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem. 2001;276:21303–21310. doi: 10.1074/jbc.M102316200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos C, Shakkottai VG, Chandy KG, Michelhaugh SK, Andrade R. SKCa channels mediate the medium but not the slow calcium-activated afterhyperpolarization in cortical neurons. J Neurosci. 2004;24:3537–3542. doi: 10.1523/JNEUROSCI.0380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Burns DL. Experimental localization of muscarinic receptor subtypes to cingulate cortical afferents and neurons. J Neurosci. 1988;8:643–652. doi: 10.1523/JNEUROSCI.08-02-00643.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Nakamura M, Sato K, Kano M, Simon MI, Inoue Y. Patterns of expression for the mRNA corresponding to the four isoforms of phospholipase Cbeta in mouse brain. Eur J Neurosci. 1998;10:2016–2025. doi: 10.1046/j.1460-9568.1998.00213.x. [DOI] [PubMed] [Google Scholar]
- Williams SL, Lutz S, Charlie NK, Vettel C, Ailion M, Coco C, Tesmer JJ, Jorgensen EM, Wieland T, Miller KG. Trio's Rho-specific GEF domain is the missing Galpha q effector in C. elegans. Genes Dev. 2007;21:2731–2746. doi: 10.1101/gad.1592007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Hrabetova S, Moskowitz SI, Casaccia-Bonnefil P, Young SR, Nimmrich VC, Tiedge H, Einheber S, Karnup S, Bianchi R, Bergold PJ. Long-term maintenance of mature hippocampal slices in vitro. J Neurosci Methods. 2000;98:145–154. doi: 10.1016/s0165-0270(00)00197-7. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Ueta Y, Kato N. Dopamine induces a slow afterdepolarization in lateral amygdala neurons. J Neurophysiol. 2007;98:984–992. doi: 10.1152/jn.00204.2007. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Arsenault D. Gain modulation by serotonin in pyramidal neurones of the rat prefrontal cortex. J Physiol. 2005;566:379–394. doi: 10.1113/jphysiol.2005.086066. [DOI] [PMC free article] [PubMed] [Google Scholar]