

Abstract
Background
Reactivation of Cytomegalovirus (CMV) is frequently observed in recipients of solid organs and bone marrow transplants and is associated with increased risk of acute and chronic allograft rejection, opportunistic infection, graft failure, and patient mortality. The molecular mechanisms by which reactivation occurs are not well understood. Previous studies have suggested that Tumor Necrosis Factor alpha (TNF-α), which is induced by allogeneic transplantation, may have a role in reactivation of CMV through activation of NF-κB and subsequent transcriptional reactivation of Immediate Early (ie) gene expression.
Methods and Results
We have tested the role TNF-α in reactivation of CMV directly by testing whether TNF-α is required to initiate transcription of ie gene expression in a murine model of allogeneic transplantation of kidneys latently infected with mouse CMV (MCMV).
Conclusions
Our studies show that although TNF-α seems to be sufficient, it is not required for initiating transcription of ie gene expression in this model suggesting that both TNF-α-dependent and TNF-α-independent pathways play an important role in reactivation of latent CMV infection.
Keywords: Cytomegalovirus, TNF, Viral Reactivation, Transplantation
Introduction
Human CMV (HCMV) is a ubiquitous Herpes virus that has the ability to establish a lifelong latent infection. Reactivation of latent virus is frequently observed in recipients of solid organs and bone marrow transplants and is associated with increased risk of acute and chronic allograft rejection, opportunistic infection, graft failure, and patient mortality (1). Due to the species specificity of HCMV, we and others have used MCMV as a model to study CMV latency and reactivation in vivo. MCMV is similar to HCMV in its ability to establish latency and reactivate, and in regulation of its gene expression. The molecular mechanisms by which CMV establishes a latent infection and avoids clearance by the host immune system, and the mechanisms leading to reactivation of virus are not well understood. The complexities of the issues surrounding latency and reactivation have been recently reviewed (2). Latency has been defined operationally as the presence of viral DNA in the absence of detectable virus. However, ie-1 transcripts are sometimes detectable in latently infected mice (3-6), and thus, it has not been clear whether there is a true state of latency, in which the viral genome is transcriptionally silent with respect to expression of genes involved in lytic replication, or whether latency is maintained solely as a result of elimination of productively infected cells by immunosurveillance. The very low ratio of RNA to DNA (5, 7, 8), and the observation that both the HCMV and MCMV enhancers are associated with deacetylated histones in latently infected cells (9, 10) suggest that there is a true latency in which the ie genes are transcriptionally inactive and that occasional detection of ie-1 transcripts is due to sporadic low level reactivation. Because the ie proteins are essential for viral replication, these observations further suggest that reactivation of ie gene expression is a critical first step in reactivation of the virus.
HCMV and MCMV ie gene expression is controlled by the enhancer region of the major immediate early promoter (MIEP) (11, 12). Transcriptional activation of the MIEP results in ie-1 and ie-2 (HCMV) and ie-1 and ie-3 (MCMV) gene expression through alternative splicing. The HCMV MIEP enhancer has been analyzed extensively and contains binding sites for many cellular transcription factors, including NF-κB and ATF, and AP-1. The MCMV ie enhancer also has multiple putative sites for NF-κB and AP-1. NF-κB and AP-1 are not active in resting cells. They are activated by growth factors, inflammatory cytokines, including TNF–α and Interleukin-1 (IL-1), Toll-like receptor ligands, viral infection and by oxidative stress (13). Thus, factors that activate these transcription factors are likely to be important in driving transcriptional reactivation of CMV ie gene expression.
In prior studies, we showed that TNF-α is sufficient to induce transcriptional reactivation of ie genes in MCMV latent mice and to induce activation of the HCMV enhancer in MIEP-lacZ transgenic mice carrying a β-galactosidase reporter gene under the control of the HCMV MIEP enhancer. We also developed a mouse kidney transplant model for reactivation of viral gene expression. Using the contralateral donor kidney as a pre-transplant control, we showed that allogeneic, but not syngeneic transplantation induces transcriptional reactivation of ie gene expression (14, 15). The peak of ie gene expression was observed at 2 days post-transplant and was accompanied by activation of NF-κB and AP-1 and increased expression of TNF-α. With the transgenic model, we investigated the requirement for TNF-α by breeding MIEP-lacZ mice to mice deficient in TNFR1 and TNFR2 and using these mice as donors in allogeneic transplants. In addition, we examined the role of TNF-α in a surgical model of ischemia/reperfusion (I/R) injury in the transgenic model. Our studies showed that the HCMV enhancer in MIEP-lacZ mice is activated in both the transplant and I/R models (16, 17) independently of TNFR signaling. Furthermore, activation of the enhancer in this model was the same in both allogeneic and syngeneic transplants. Although there are similarities between mice transgenic for HCMV MIEP and mice latently infected with MCMV, the observed differences between the two models caused us to test directly for the requirement for TNF-α in reactivation of latent MCMV.
Materials and Methods
Virus infection
Smith strain MCMV was originally obtained from the ATCC. The MCMV mutant Δm157 virus (18) was obtained from Ulrich Koszinowski (Max von Pettenkofer-Institute, Ludwig Maximilians-University, Munich, Germany). Virus stocks were derived from salivary glands of BALB/c mice two weeks post-infection as previously described (19). Stocks were titered by plaque assay on murine embryo fibroblasts using standard techniques. Mice were injected i.p. with 1×105 plaque forming units (pfu) of virus, and sacrificed four days post-infection for analysis of acute infection, or after more than 120 days for analysis of latent infection or as transplant donors.
Mice
All mice were obtained from Jackson Labs (Bar Harbor, ME). BALB/c, mice were latently infected with MCMV (Smith strain) for use as donors for analysis of reactivation of latently infected wild type mice. C57BL/6 or B6;129S6-TNFtm1Gkl/J mice were used as allogeneic wild type or TNF-α-deficient recipients, respectively for BALB/c donor kidneys. B6;129S-Tnfrsflatm1ImxTnfrsf1btm1Imx mice were infected with the Δm157 virus for use as donors to test the requirement for TNFR signaling in reactivation. BALB/c mice were used as allogeneic recipients for TNFR1/TNFR2-deficient donor kidneys.
Transplants
Kidney transplants were performed as previously described (20). Donor kidneys were removed after 48 hr. Our previous studies have shown that this is the peak time for reactivation of ie gene expression (15). Contralateral donor kidneys were removed at the time of the transplant for use as a matching, pre-transplant control. Control and transplanted kidneys were immediately frozen in liquid nitrogen after harvest. Mice were maintained in isolation cages and were fed and watered ad libitum. This study protocol was reviewed and approved by the Northwestern University Institutional Animal Care and Use Committee.
Antibody treatment
Recipient mice were injected i.p. with 200 ug 1 hr prior to surgery and again after 24 hr. Low endotoxin anti-TNF-α (clone XT22; Pierce Endogen, Rockford, IL) was used to block TNF-α. Low endotoxin KLH/G1-2-2 rat IgG1kappa was used as an isotype control antibody (Southern Biotech, Birmingham, AL).
Spleen explants
Spleens from latently infected mice were minced into small pieces and cultured in six-well dishes for 17 days in Dulbecco's Modification of Eagle's Medium, with change of medium twice a week. Supernatants were harvested and frozen for titering by plaque assay on murine embryo fibroblasts.
RNA analysis
RNAs were isolated from frozen kidneys by homogenization in TriZol as directed by the manufacturer (Invitrogen). ie-1 RNA was detected by RT-PCR using primers CH16 and CH17 and Southern blot hybridized with CH15 probe and ie-3 RNA was amplified from the cDNA using primers CH17 and IE3R and detected with probe CH15 as previously described (15).
For real time PCR analysis, 7.5 ug of RNA was reverse transcribed in a 100 μl reaction with M-MLV reverse transcriptase (Applied Biosystems) and random primers. TNF-α RNA was quantified from 2 μl of the reverse transcription reaction by relative real time PCR analysis using a TNF-α gene expression assay (Mm00443258_m1, Applied Biosystems). Assays were run on a 7500 Fast Real Time PCR System (Applied Biosystems) using default cycling conditions in the standard mode. TNF-α expression was normalized to HPRT (assay Mm01545399_m1, Applied Biosystems), which was analyzed in parallel. The fold change for each mouse was calculated as 2-ΔΔCt, where ΔΔCt=ΔCt transplant-ΔCt control, ΔCt transplant=Ct (TNF-α in transplanted kidney)-Ct (HPRT transplanted kidney), and ΔCt control=Ct (TNF-α in contralateral kidney)-Ct (HPRT contralateral kidney).
Statistical analysis
A student's t-test was used to determine statistical significance. P values < 0.05 were considered to be significant.
Results
TNF-α expression is both donor and recipient-derived
We previously observed increased expression of TNF-α in our transplants (15, 21). This TNF-α RNA could have been derived from the donor cells as a result of injury, and/or from infiltrating cells of the recipient. To determine the source of TNF-α RNA, we analyzed TNF-α expression in kidneys transplanted into TNF-α-deficient or TNF-α-sufficient (wild type; WT) recipients. In the case of TNF-α-deficient recipients, expression of TNF-α RNA must come from cells of the donor. The results (Fig. 1) show that TNF-α expression was induced in both sets of mice, but greater induction of TNF-α was observed in kidneys transplanted into TNF-α-sufficient recipients than in kidneys transplanted into TNF-α-deficient recipient mice (p < 0.001). These studies suggest that the majority of TNF-α RNA is expressed by cells of the recipient, but that some TNF-α RNA is also derived from cells of the donor. These data suggest that although damage to the donor resulting from ischemia/reperfusion injury may contribute some TNF-α, the host immune response of the recipient and resulting inflammation may play a more important role in activating gene expression in Day 2 allografts. These findings led us to develop specific strategies to block TNF-α in both the donor and recipient in an effort to determine whether TNF-α from either source is required in MCMV ie transcriptional reactivation.
Fig. 1. TNF-α RNA is derived from both donor and recipient cells in kidney allografts.

RNAs from control and transplanted kidneys of latently infected TNF-α-sufficient BALB/c mice transplanted into TNF-sufficient C57BL/6 mice treated with control antibody (n=7) or TNF-deficient (n=5) recipients treated with anti-TNF-α were analyzed for TNF-α expression by real time PCR. Expression was normalized to HPRT. The fold change for each mouse was calculated as the ratio of the transplanted kidney to the contralateral control as described in the methods. Results shown are the average fold change plus standard error. *, p < 0.001.
TNF-α deficiency in the recipient and treatment of the recipient with anti-TNF-α antibody to block donor-derived TNF-α shows that TNF-α is not required for transcriptional reactivation of MCMV ie-1
We used TNF-α KO mice as recipients to prevent expression of any TNF-α that might be produced by the recipient and treated the recipient with anti-TNF-α antibody to block TNF-α produced by the donor. We used this combined strategy in an effort to block the induction of MCMV-ie gene expression by TNF-α from any source. In the control group, WT mice were treated with an isotype control antibody to control for the potential effects of the antibody itself on MCMV reactivation through potentially unrelated pathways. We compared MCMV-ie gene expression in the transplanted (G) kidneys to that in the control (C) kidneys. While MCMV-ie gene expression can be occasionally detected in control kidneys, our analyses focus on TNF-induced induction of expression. MCMV ie -1 gene expression was induced in transplanted kidneys relative to the contralateral control kidneys in 3/6 WT mice treated with control antibody (Figure 2A) and in 2/6 TNF-α KO mice treated with anti-TNF-α (Figure 2B). In a separate experiment, MCMV ie-1 and ie-3 expression was induced in 3/3 and 1/3 WT mice treated with control antibody respectively and in 2/4 and 1/4 in TNF-α KO mice treated with anti-TNF-α (Figure 2C and 2D) These results suggest that TNF–α is not required for transcriptional reactivation of ie gene expression in response to allogeneic transplantation. However, these results leave open the possibility that the anti-TNF–α antibody was insufficient to block TNF–α produced by the donor.
Fig. 2. Role of TNF–α in transcriptional reactivation of ie-1 expression.
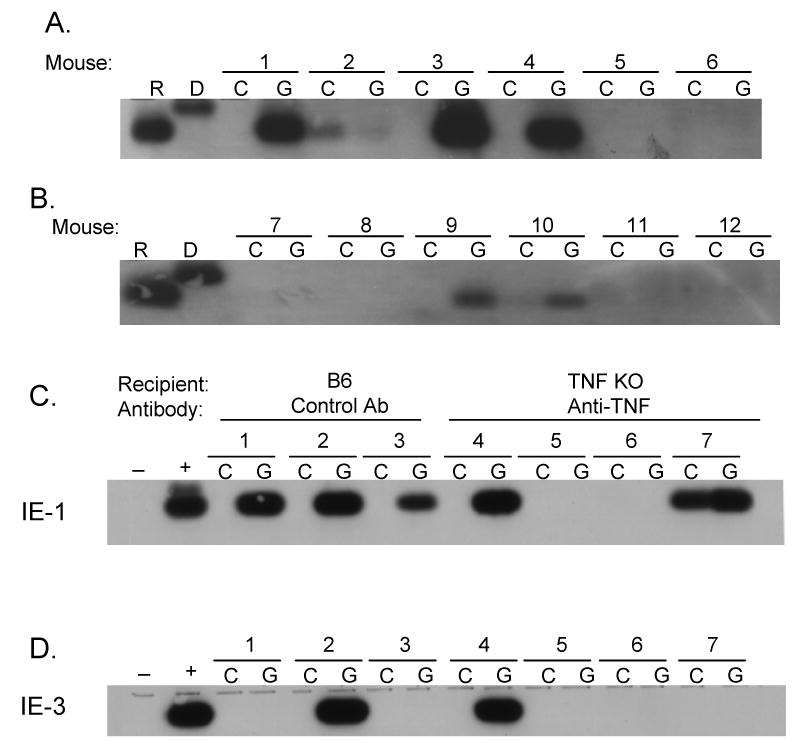
RT-PCR and Southern blot hybridization analysis of RNA extracted from kidneys of six MCMV (Smith strain) latently infected BALB/c mice transplanted into allogeneic C57BL/6 recipient mice treated with control antibody (A) or into TNF–α-deficient mice treated with anti-TNF–α (B). Pairs of contralateral control (C) and transplanted (G) kidneys from each mouse are shown grouped together. ie-1 RNA was amplified from the cDNA with ie-1 specific primers CH16 and CH17 and detected by hybridization with CH15 (6) as previously described (15). ie-1 DNA and RNA are distinguishable by size. R, RNA size marker; D, DNA size marker.
Fig. 2C and 2D. In a separate experiment, ie-1 and ie-3 are induced in 3/3 and 1/3 WT mice (control antibody) respectively, compared to 2/4 and 1/4 respectively in TNF-α KO mice treated with anti-TNF-α antibody.
Blocking TNFR signaling in donor kidneys latently infected with MCMV also shows that TNFR signaling is not required for transcriptional activation of MCMV ie-1
In order to test the requirement for TNF–α signaling in reactivation of ie gene expression more definitively, we adopted the strategy of using latently infected TNF–α receptor-deficient mice as donors for allogeneic transplants. In this case, the cells of the donor kidney would be unable to respond to TNF–α produced either by the donor or the recipient. There are two receptors for TNF–α, TNFR1 and TNFR2. TNFR1 is the major receptor for soluble TNF–α and is also a receptor for lymphotoxin alpha. Although the biological role of TNFR2 has not been well defined, TNFR2 is thought to mediate the effects of membrane-bound TNF–α (22). We therefore used mice deficient in both TNFR1 and TNFR2 to block TNF–α signaling. Commercially available TNFR1/TNFR2-deficient mice are derived from embryos of B6;129S mice. B6 mice are resistant to MCMV infection due to recognition of the ligand encoded by the MCMV m157 gene by the NK cell receptor Ly49H (23). Deletion of this gene results in gain of virulence in Ly49H+ mouse strains (18). We therefore used the Δm157 MCMV virus to test the ability of MCMV to reactivate in allogeneic kidney transplants of TNFR1/TNFR2-deficient mice.
We first tested the ability of this virus to replicate during acute infection. Our studies show that the Δm157 virus was able to replicate during acute infection in multiple organs of TNFR1/TNFR2-deficient mice at levels similar to that of Smith strain virus in wild type BALB/c mice (Fig. 3). We then examined the ability of this virus to establish latent infection. Using nested PCR, we were able to detect MCMV DNA in the lungs and kidneys of Δm157-infected TNFR1/TNFR2-deficient mice six months after infection (Fig. 4A). DNA from Smith-infected BALB/c mice was analyzed in parallel as a positive control. In addition, we were able to induce reactivation of infectious virus from explants of spleen tissue from Δm157-infected TNFR1/TNFR2-deficient mice six months after infection. The titer of virus recovered from these mice was not significantly different from that of explants of Smith-infected BALB/c mice, which were analyzed as a positive control (Fig. 4B).
Fig. 3. Acute infection of Δm157 virus in TNFR1/TNFR2-deficient mice is similar to that of Smith virus in wild type BALB/c mice.

Organs from infected mice (n=3) were homogenized and analyzed for the presence of infectious virus by plaque assay 4 days after infection. WT, BALB/c mice infected with Smith strain virus; TNFR-/-, B6;129S-Tnfrsflatm1ImxTnfrsf1btm1Imx mice infected with Δm157 virus. Results shown are the average plus standard deviation.
Fig. 4. Δm157 virus establishes latency in TNFR1/TNFR2-deficient mice.
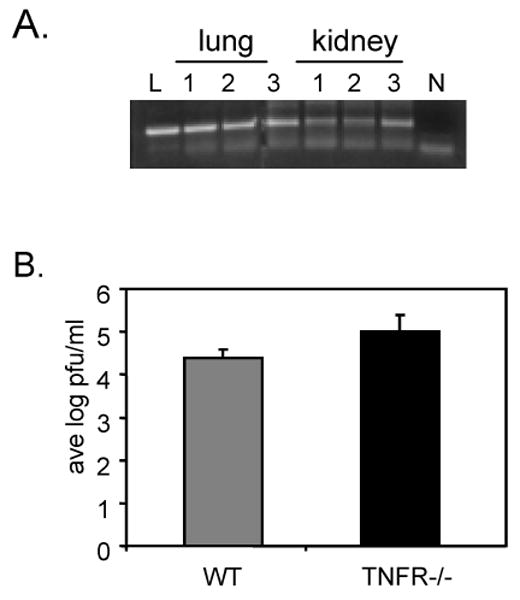
(A) Nested PCR analysis of MCMV ie-1 DNA in lungs and kidneys of Δm157-infected B6;129S-Tnfrsflatm1ImxTnfrsf1btm1Imx mice. Lanes: L, nested PCR analysis of lung DNA from a Smith-infected BALB/c mouse; 1,2,3, nested PCR analysis of DNA isolated from lungs and kidneys of three Δm157 latent B6;129S-Tnfrsflatm1ImxTnfrsf1btm1Imx mice; N, no template control. (B) Titers of reactivated virus in supernatants derived from spleen explants of Smith infected BALB/c mice (WT) or Δm157-infected B6;129S-Tnfrsflatm1ImxTnfrsf1btm1Imx (TNFR-/-) mice. Results shown are the average titers of explants from three mice analyzed in duplicate plus standard deviation.
Having thus demonstrated that the Δm157 virus is able to establish latent infection in TNFR1/TNFR2-deficient mice, we used TNFR1/TNFR2-deficient mice latently infected with the Δm157 virus as donors for allogeneic transplants to assess the requirement for TNFR-mediated signaling in transcriptional reactivation of MCMV. Transcriptional reactivation of ie gene expression was observed to varying degrees in 5/8 kidney transplants from latently infected TNFR1/TNFR2-deficient mice (Fig. 5). MCMV ie-1 induction appeared weak (Lanes 1, 5, 6 and 7) in 4/5 mice suggesting that alternative signaling pathways driving expression of MCMV ie-1 gene expression in allogeneic transplants. Nonetheless, these results also suggest that TNF-α is not required for transcriptional reactivation of ie gene expression in response to allogeneic transplantation since TNF-α signaling cannot occur in the absence of both TNFR1 and TNFR2 in the donor kidney.
Fig. 5. TNFR signaling in donor kidneys is not required for transcriptional reactivation of ie gene expression.
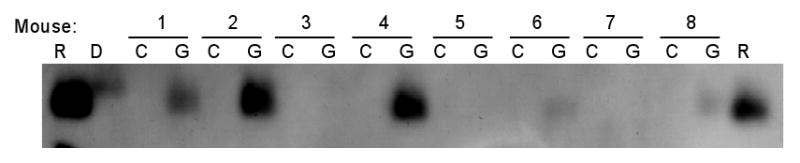
RNA extracted from control (C) and transplanted (G) kidneys of eight Δm157 latently infected B6;129S-Tnfrsflatm1ImxTnfrsf1btm1Imx mice transplanted into allogeneic BALB/c recipients was analyzed by RT-PCR and Southern blot hybridization for expression of ie-1 RNA. R, RNA size marker; D, DNA size marker.
Discussion
Reactivation of CMV in immunosuppressed patients correlates with use of anti T-cell therapy, which induces TNF expression (24-29), and with the onset of TNF-α associated complications such as in sepsis, allograft rejection, or GVHD (30-32). Reactivation of CMV is also temporally associated with sepsis in non-immunosuppressed patients (33-35). Furthermore, in vitro studies have shown that the HCMV enhancer is responsive to TNF-α in an NF-κB-dependent manner in transient transfection assays (36, 37). Studies with MCMV latent mice have shown that TNF-α is sufficient to induce reactivation of ie gene expression or viral replication (4, 15, 38).
We previously hypothesized that expression of TNF-α induced by allogeneic transplantation could play a critical role in transcriptional reactivation of ie gene expression (14, 15). Although TNF-α is sufficient to induce reactivation of ie gene expression in some cells, our studies here show that TNF-α is not required in the context of allogeneic transplantation. This observation is consistent with our previous studies using MIEP-lacZ transgenic mice carrying a β-galactosidase reporter gene under the control of the HCMV major immediate early enhancer. These studies showed that the HCMV enhancer is activated by I/R, syngeneic transplantation, or allogeneic transplantation independently of TNFR signaling (16, 17). However, there are many differences between the HCMV MIEP-lacZ transgenic model and mice latently infected with MCMV that led us to question whether our findings in the transgenic model were reflective of latent MCMV. First, although we observed activation of the HCMV enhancer in transgenic mice in response to I/R injury (16), we were not able to induce reactivation of MCMV ie gene expression in this model (data not shown). Second, although there are many similarities between the HCMV and MCMV enhancer regions, they also differ in sequence. The HCMV enhancer appears to have more complexity in terms of the number of potential transcription factors that may control its regulation (12, 15). Third, activation of the HCMV enhancer in MIEP-lacZ mice does not result in production of viral gene products, which may have both positive and negative regulatory effects on other viral and cellular genes. Fourth, the MCMV genome is likely to be maintained as an episome as are the genomes of other herpesviruses (39), whereas the MIEP transgene is integrated into the cellular genome (40). We have recently shown that the MIEP enhancer region is bound to cellular repressors of transcription (10) in latently infected mice. The context of the DNA may be important in recruitment of these repressors, and thus, they may not be associated with the MIEP when the region is integrated into cellular DNA. Therefore, we felt it was important to define the requirement for TNF-α in reactivation of MCMV ie gene expression induced by transplantation, especially in view of our published observations and those of others regarding the association between TNF-α and CMV reactivation.
Our results showed that TNF-α is expressed by cells of both the donor kidney and the recipient in Day 2 murine allografts. To study the requirement for TNF-α-signaling in reactivation of ie gene expression in the donor kidney, it was therefore necessary to block signaling from both donor and recipient-derived TNF-α. We chose two complimentary approaches to achieve this goal. The first involved blocking interaction of TNF-α with its receptor through the use of TNF-α-deficient recipients treated with anti-TNF-α antibody. The second approach involved use of TNF-α receptor-deficient donor mice latently infected with MCMV, so that the cells harboring latent virus would not be able to respond to TNF-α produced by either the donor or the recipient. In both cases, we were able to demonstrate that ie gene transcription still occurred as a result of allogeneic kidney transplantation. These studies provide the important insight that TNF-α is not required in the transcriptional activation of latent MCMV in a model of kidney allotransplantation.
Expression of both the HCMV and MCMV ie genes is controlled by the enhancer region of the MIEP, which contains potential binding sites for many transcription factors, including NF-κB and ATF, and AP-1 (12). Mutations of these binding sites will help better understand the role of these transcription factors in response to activation by a variety of inflammatory stimuli in addition to TNF-α (13). Currently available mutants have not been helpful in this regard given that they are attenuated for viral replication (data not shown). Previous studies have shown that LPS, a ligand for TLR4, is sufficient to induce reactivation of MCMV and HCMV (9, 38). We have found up-regulation of several endogenous ligands for Toll-like receptors, including fibronectin, fibrinogen, biglycan, and heat shock proteins in murine Day 2 allografts as well as increased expression of several Toll-like receptors and other pathogen recognition receptors (21). Thus, the innate immune response to damaged cellular ligands may play an important role in transcriptional reactivation of MCMV in this model. Determining which of the many possible ligands and receptors in addition to TNF-α mediate activation of the MCMV enhancer in response to transplantation remains a significant challenge for the future. A better understanding of the factors that control reactivation of ie gene expression in the context of transplantation would be valuable in guiding the development of novel therapies targeting initiation of reactivation.
Acknowledgments
The authors thank Ulrich Koszinowski for providing the Δm157 virus.
This study was supported by Public Health Service grant R01 AI42898 (MA) from the NIH.
Abbreviations
- ATF
activating transcription factor
- AP-1
activating protein-1
- HCMV
human cytomegalovirus
- ie-1
immediate early gene 1
- ie-2
immediate early gene 2
- ie-3
immediate early gene 3
- I/R
ischemia reperfusion
- LPS
lipopolysaccharide
- MCMV
murine cytomegalovirus
- MIEP
major immediate early promoter
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- TNF-α
tumor necrosis factor, alpha
- TNFR1
TNF receptor 1
- TNFR2
TNF receptor 2
- WT
wild type
Footnotes
Zheng Zhang: Research design, Performance of research, and Data analysis
Zhigao Li: Performance of research and Data analysis
Shixian Yan: Performance of research
Xueqiong Wang: Performance of research
Michael Abecassis: Research design, Writing of the paper, and Data analysis
The authors have no conflicts of interest to disclose.
References
- 1.Razonable RR, Paya CV. Beta-Herpesviruses in transplantation. Reviews in Medical Microbiology. 2002;13:163. [Google Scholar]
- 2.Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. Murine model of cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol. 2008;325:315. doi: 10.1007/978-3-540-77349-8_18. [DOI] [PubMed] [Google Scholar]
- 3.Kurz SK, Rapp M, Steffens HP, Grzimek NK, Schmalz S, Reddehase MJ. Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J Virol. 1999;73(1):482. doi: 10.1128/jvi.73.1.482-494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simon CO, Seckert CK, Dreis D, Reddehase MJ, Grzimek NK. Role for tumor necrosis factor alpha in murine cytomegalovirus transcriptional reactivation in latently infected lungs. J Virol. 2005;79(1):326. doi: 10.1128/JVI.79.1.326-340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grzimek NK, Dreis D, Schmalz S, Reddehase MJ. Random, asynchronous, and asymmetric transcriptional activity of enhancer-flanking major immediate-early genes ie1/3 and ie2 during murine cytomegalovirus latency in the lungs. J Virol. 2001;75(6):2692. doi: 10.1128/JVI.75.6.2692-2705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henry SC, Hamilton JD. Detection of murine cytomegalovirus immediate early 1 transcripts in the spleens of latently infected mice. J Infect Dis. 1993;167(4):950. doi: 10.1093/infdis/167.4.950. [DOI] [PubMed] [Google Scholar]
- 7.Yuhasz SA, Dissette VB, Cook ML, Stevens JG. Murine cytomegalovirus is present in both chronic active and latent states in persistently infected mice. Virology. 1994;202(1):272. doi: 10.1006/viro.1994.1343. [DOI] [PubMed] [Google Scholar]
- 8.Simon CO, Seckert CJ, Grzimek N, Reddehase M. Murine Model of Cytomegalovirus Latency and Reactivation: the Silencing/Desilencing and Immune Sensing Hypothesis. In: Reddehase M, editor. Cytomegaloviruses: Molecular Biology and Immunology. Norfolk, UK: Caister Academic Press; 2006. p. 483. [Google Scholar]
- 9.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A. 2005;102(11):4140. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu XF, Yan S, Abecassis M, Hummel M. Establishment of Murine Cytomegalovirus Latency In Vivo Is Associated with Changes in Histone Modifications and Recruitment of Transcriptional Repressors to the Major Immediate-Early Promoter. J Virol. 2008;82(21):10922. doi: 10.1128/JVI.00865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorsch-Hasler K, Keil GM, Weber F, Jasin M, Schaffner W, Koszinowski UH. A long and complex enhancer activates transcription of the gene coding for the highly abundant immediate early mRNA in murine cytomegalovirus. Proc Natl Acad Sci U S A. 1985;82(24):8325. doi: 10.1073/pnas.82.24.8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meier JL, Stinski MF. Major Immediate-early Enhancer and its Gene Products. In: Reddehase MJ, editor. Cytomegaloviruses Molecular Biology and Immunology. Norfolk, UK: Caister Academic Press; 2006. p. 151. [Google Scholar]
- 13.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336(15):1066. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 14.Hummel M, Abecassis MI. A model for reactivation of CMV from latency. J Clin Virol. 2002;25:S123. doi: 10.1016/s1386-6532(02)00088-4. [DOI] [PubMed] [Google Scholar]
- 15.Hummel M, Zhang Z, Yan S, et al. Allogeneic transplantation induces expression of cytomegalovirus immediate-early genes in vivo: a model for reactivation from latency. J Virol. 2001;75(10):4814. doi: 10.1128/JVI.75.10.4814-4822.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SJ, Varghese TK, Zhang Z, et al. Renal ischemia/reperfusion injury activates the enhancer domain of the human cytomegalovirus major immediate early promoter. American Journal of Transplantation. 2005;5(7):1606. doi: 10.1111/j.1600-6143.2005.00912.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Kim SJ, Varghese T, Thomas G, Hummel M, Abecassis M. TNF receptor independent activation of the cytomegalovirus major immediate early enhancer in response to transplantation. Transplantation. 2008;85:1039. doi: 10.1097/TP.0b013e318168449c. [DOI] [PubMed] [Google Scholar]
- 18.Bubic I, Wagner M, Krmpotic A, et al. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78(14):7536. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brune W, Hengel H, Koszinowski UH. A mouse model for cytomegalovirus infection. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. New York, NY: John Wiley and Sons, Inc.; 1999. p. 19.7.1. [Google Scholar]
- 20.Zhang Z, Schlachta C, Duff J, Stiller C, Grant D, Zhong R. Improved techniques for kidney transplantation in mice. Microsurgery. 1995;16(2):103. doi: 10.1002/micr.1920160212. [DOI] [PubMed] [Google Scholar]
- 21.Hummel M, Kurian SM, Lin S, et al. Intragraft TNF receptor signaling contributes to activation of innate and adaptive immunity in a renal allograft model. Transplantation. 2009;87(2):178. doi: 10.1097/TP.0b013e3181938971. [DOI] [PubMed] [Google Scholar]
- 22.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 23.Lodoen MB, Lanier LL. Viral modulation of NK cell immunity. Nature Reviews Microbiology. 2005;3(1):59. doi: 10.1038/nrmicro1066. [DOI] [PubMed] [Google Scholar]
- 24.Abramowicz D, Schandene L, Goldman M, et al. Release of tumor necrosis factor, interleukin-2, and gamma-interferon in serum after injection of OKT3 monoclonal antibody in kidney transplant recipients. Transplantation. 1989;47(4):606. doi: 10.1097/00007890-198904000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Chatenoud L, Ferran C, Legendre C, et al. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation. 1990;49(4):697. doi: 10.1097/00007890-199004000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Debets JM, Leunissen KM, van Hooff HJ, van der Linden CJ, Buurman WA. Evidence of involvement of tumor necrosis factor in adverse reactions during treatment of kidney allograft rejection with antithymocyte globulin. Transplantation. 1989;47(3):487. doi: 10.1097/00007890-198903000-00018. [DOI] [PubMed] [Google Scholar]
- 27.Grant SC, Lamb WR, Brooks NH, Brenchley PE, Hutchinson IV. Serum cytokines in human heart transplant recipients. Is there a relationship to rejection? Transplantation. 1996;62(4):480. doi: 10.1097/00007890-199608270-00010. [DOI] [PubMed] [Google Scholar]
- 28.Hibberd PL, Tolkoff-Rubin NE, Cosimi AB, et al. Symptomatic cytomegalovirus disease in the cytomegalovirus antibody seropositive renal transplant recipient treated with OKT3. Transplantation. 1992;53(1):68. doi: 10.1097/00007890-199201000-00013. [DOI] [PubMed] [Google Scholar]
- 29.Portela D, Patel R, Larson-Keller JJ, et al. OKT3 treatment for allograft rejection is a risk factor for cytomegalovirus disease in liver transplantation. J Infect Dis. 1995;171(4):1014. doi: 10.1093/infdis/171.4.1014. [DOI] [PubMed] [Google Scholar]
- 30.Mutimer D, Mirza D, Shaw J, O'Donnell K, Elias E. Enhanced (cytomegalovirus) viral replication associated with septic bacterial complications in liver transplant recipients. Transplantation. 1997;63(10):1411. doi: 10.1097/00007890-199705270-00007. [DOI] [PubMed] [Google Scholar]
- 31.Fietze E, Prosch S, Reinke P, et al. Cytomegalovirus infection in transplant recipients. The role of tumor necrosis factor. Transplantation. 1994;58(6):675. [PubMed] [Google Scholar]
- 32.Razonable RR, Rivero A, Rodriguez A, et al. Allograft rejection predicts the occurrence of late-onset cytomegalovirus (CMV) disease among CMV-mismatched solid organ transplant patients receiving prophylaxis with oral ganciclovir. J Infect Dis. 2001;184(11):1461. doi: 10.1086/324516. [DOI] [PubMed] [Google Scholar]
- 33.Docke WD, Prosch S, Fietze E, et al. Cytomegalovirus reactivation and tumour necrosis factor. Lancet. 1994;343(8892):268. doi: 10.1016/s0140-6736(94)91116-9. [DOI] [PubMed] [Google Scholar]
- 34.Heininger A, Jahn G, Engel C, Notheisen T, Unertl K, Hamprecht K. Human cytomegalovirus infections in nonimmunosuppressed critically ill patients. Crit Care Med. 2001;29(3):541. doi: 10.1097/00003246-200103000-00012. see comments. [DOI] [PubMed] [Google Scholar]
- 35.Kutza AS, Muhl E, Hackstein H, Kirchner H, Bein G. High incidence of active cytomegalovirus infection among septic patients. Clin Infect Dis. 1998;26(5):1076. doi: 10.1086/520307. see comments. [DOI] [PubMed] [Google Scholar]
- 36.Prosch S, Staak K, Stein J, et al. Stimulation of the human cytomegalovirus IE enhancer/promoter in HL-60 cells by TNF alpha is mediated via induction of NF-kB. Virology. 1995;208:197. doi: 10.1006/viro.1995.1143. [DOI] [PubMed] [Google Scholar]
- 37.Stein J, Volk HD, Liebenthal C, Kruger DH, Prosch S. Tumour necrosis factor alpha stimulates the activity of the human cytomegalovirus major immediate early enhancer/promoter in immature monocytic cells. J Gen Virol. 1993;74(Pt 11):2333. doi: 10.1099/0022-1317-74-11-2333. [DOI] [PubMed] [Google Scholar]
- 38.Cook CH, Trgovcich J, Zimmerman PD, Zhang Y, Sedmak DD. Lipopolysaccharide, tumor necrosis factor alpha, or interleukin-1beta triggers reactivation of latent cytomegalovirus in immunocompetent mice. J Virol. 2006;80(18):9151. doi: 10.1128/JVI.00216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolovan-Fritts CA, Mocarski ES, Wiedeman JA. Peripheral blood CD14+ cells from healthy subjects carry a circular conformation of latent cytomegalovirus genome. Blood. 1999;93:394. [PubMed] [Google Scholar]
- 40.Baskar JF, Smith PP, Nilaver G, et al. The enhancer domain of the human cytomegalovirus major immediate-early promoter determines cell type-specific expression in transgenic mice. J Virol. 1996;70(5):3207. doi: 10.1128/jvi.70.5.3207-3214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]