

Abstract
Objective
A novel indole-ethyl isothiocyanate derivative (7Me-IEITC) was defined as a potent growth-suppressing agent to cell lines derived from ovarian cancers. Key mechanisms of the cellular response in vitro were studied and suggest a potential of 7Me-IEITC as a therapeutic drug.
Methods
The viability of ovarian cancer cell lines (SKOV-3, OVCAR-3) in comparison to pancreatic and prostate cancer cell lines, primary fibroblast and immortalized trophoblasts after treatment with 7Me-IEITC was analyzed. Morphological and apoptotic responses of SKOV-3 were studied by fluorescence microscopy (DAPI staining, TUNEL assay). SKOV-3 proliferation was estimated by a standardized BrdU incorporation assay. The phosphorylation of MAP-Kinases, pro-survival factors and the activation of caspases and PARP-1 were analyzed by western blotting. Changes of the mitochondrial transmembrane-potential and in cell cycle progression were studied by FACS analysis. MAP-Kinase and caspase inhibitors were employed in cytotoxicity studies.
Results
7Me-IEITC selectively reduced the viability of SKOV-3, OVCAR-3, BXPC-3 and PC-3 cells (IC50 values ≤5μM), while the viability of fibroblasts or trophoblasts remained unaffected at concentrations below 20μM. 7Me-IEITC treatment down-regulated pro-survival kinases and transcription factors (STAT-3, IKKα and NF-κB), caused rapid loss of the mitochondrial transmembrane-potential and inactivation of PARP-1 along with activation of caspases. The use of p38 MAP-Kinase- and caspase- inhibitors suppressed the cytotoxicity of the drug. 7Me-IEITC acted as an anti-proliferative agent and arrested the cell-cycle progression of SKOV-3 in G2/M phase.
Conclusion
7Me-IEITC is a potent and growth-suppressing agent to cell lines derived from ovarian cancers by causing de-activation of survival signals, apoptosis, and cell-cycle arrest.
Keywords: Isothiocyanate, ovarian cancer, cytotoxicity, MAP Kinase, Caspase, cell cycle
Introduction
In the United States, epithelial ovarian cancer (EOC) is the leading cause of death from gynecologic malignancies and the fourth most common cause of death due to cancer among women. In 2007, there will be an estimated twenty-two thousand new cases and an estimated fifteen-thousand deaths secondary to ovarian cancer [1]. Although most patients initially respond to cytoreductive surgery and adjuvant paclitaxel and platinum-based chemotherapy, the majority will experience disease recurrence. There is only a 53% 5-year overall survival rate for all stages of ovarian cancer [2]. Therefore, research has to focus on the discovery of novel chemotherapeutics with increased activity and response rates to assist in the management of ovarian cancer.
The main cause of therapeutic failures in various cancers is drug resistance. Isothiocyanates (ITC) represent a new class of suppressors of multi-drug resistance (MDR) in cancer. ITC such as phenethyl ITC (PEITC), benzyl ITC (BITC) and naphthyl ITC (NITC) have been reported to inhibit function of ABC efflux proteins, P-glycoproteins and other factors [3,4] implicated in emergence of MDR [5,6,7]. Further mechanisms of ITC activity in cancer prevention have been proposed, such as induction of G2/M cell-cycle arrest and apoptosis (8) or suppression of angiogenesis with a disruption of microtubulin polymerization and mitotic progression of endothelial cells [9,10].
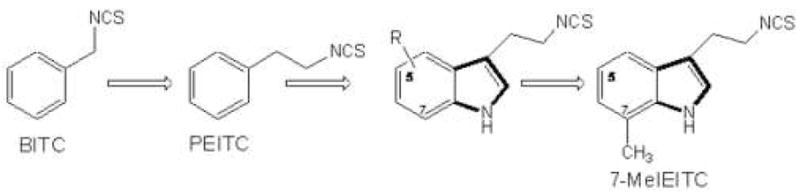
The primary objective of the present study was to identify an improved ITC derivative to treat drug resistant ovarian cancer cells and to define key mechanisms of the cellular response. Novel ITC were designed [11] on the basis of their close structural analogy with PEITC and other isothiocyanate with anti-tumor activity in vitro. We converted commercially available tryptamine derivatives to the corresponding ITCs which were screened for cytotoxicity against neuroblastoma cell lines [11]. Replacing a phenyl ring (PEITC) with an indole ring and inserting non-polar groups on the indole moiety (e.g. at the 7-position) resulted in increased cytotoxicity leading to the use of 7Me-IEITC (Figure 1) in the present study. The therapeutic potential of 7Me-IEITC to treat ovarian cancer cells was investigated by i) analyzing its cytotoxic effects on platinum-resistant ovarian cancer cell lines SKOV-3 and OVCAR-3 in comparison to other cancer and non-cancer cell lines and by ii) describing the drugs anti-proliferative and apoptotic effects in conjunction with key signaling changes in SKOV-3 cells.
Figure 1. Design concept of 7Me-IEITC.
Materials and Methods
Synthesis of 7Me-IEITC
7Me-IEITC was synthesized in one step [11]. Yield: 234 mg (72%), IR(DCM): 3145, 2940, 1629, 1479, 1045 cm-1, 1H NMR(CDCl3) δ7.39–7.41 (d, 1H, J=6.6 Hz, Ar), 7.25–7.26( d, 1H, J= 2.7 Hz, Ar), 7.041–7.13 (m, 2H, Ar), 3.76–3.77 (t, 2H, J=5.1Hz, CH2-Ar), 3.15–3.17 (t, 2H, J=5.1, CH2NCS), 2.52 (t, 3H, CH3); MS: m/z = 217[M+1]), checked on silica gel 60F254 TLC plates, visualized by exposure to iodine-vapor and UV light and by spraying with ninhydrine. IR spectra (Perkin Elmer Paragon-1000PC) was recorded as DCM solution and NMR (300MHz) spectra were recorded using Bruker 300-DRX and chemical shifts expressed in δ units relative to TMS. Mass spectra were recorded (API-3000 LCMS/MS; direct inlet system under positive ion electrospray ionization).
Cell Lines (human)
SKOV-3, OVCAR-3 (ovarian epithelial adenocarcinomas), PC-3 (prostate adenocarcinoma), BxPC-3 (pancreatic adenocarcinoma), and A-431 (epidermoid skin carcinoma were purchased from ATCC (Manassas, VA). TCL-1 (immortalized trophoblasts) and HTR-8/SVneo (first-trimester cytotrophoblasts with extended lifespan) were provided by Dr. Sharma (Providence, RI, USA). Cells were grown T75 cell culture flasks (Corning, New York, NY) in complete medium (Gibco, Rockville, MD) according to the suppliers recommendation. For all assays cells after seeding were allowed to attach overnight and treated in complete medium.
Cell viability assay
Viability of cells was determined by the 96® Aqueous-One-Solution Assay (Promega, Madison, WI) following the manufacturer’s recommendations. This colorimetric assay is based on the ability of mitochondria to reduce a substrate (MTS) into a soluble formazan product with an absorbance at 490nm (ELISA plate reader; (Thermo Labsystems, Waltham, MA) directly proportional to the number of living cells [12]. Cells (5×103) were plated into 96 well flat bottom plates (Corning, Inc., Corning, NY) and treated with 7Me-IEITC as indicated (result section). For assays with inhibitors (p38/SB203580, JNK/SP600125, caspase-3/Z-DEVD-FMK, caspase-8/Z-IETD-FMK, caspase-9/Z-LEHD-FMK, pan-caspase/Z-VAD-FMK; all from Calbiochem, La Jolla, CA) the cells were pre-incubated with 40μM inhibitor for 2h prior to drug addition. Following incubation for 46hrs MTS reagent was added for an additional 2hrs and absorbance measured at 490nm. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % of absorbance of samples with untreated cells [100%].
Cell proliferation assay
Cell proliferation was determined by a BrdU assay (Roche Applied Science, Indianapolis, IN) measuring incorporation of a pyrimidine analogue (BrdU) during DNA synthesis according to the manufacturer recommendations. Cells (5×103) were plated into 96 well flat bottom plates (Corning, Inc., Corning, NY), treated for 42hrs with 7Me-IEITC as indicated (result section) and the assay carried out as described elsewhere [13]. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % of absorbance of samples with untreated cells [100%].
Mitochondrial Transmembrane Potential
Cells (1×106) were seeded in a 100mm petri-dish and treated with 3μM of 7Me-IEITC for 3 or 24hrs. After treatment cells were washed with PBS, resuspended in fresh medium (5×105 cells/ml) and incubated with 15nM DiOC6(3) for 30min at 37°C. The cells were washed twice with DPBS, resuspended in an equal volume of DPBS, and DiOC6(3) and measured by flow cytometry (excitation - 488nm, emission - 520nm). Data was acquired on a BD FACSort flow cytometer using CellQuest software (BD Immunocytometry-Systems, San Jose, CA) and analyzed (ModFit LT software, Verity Software House, Inc., Topsham, ME). Ten thousand cells were analyzed for each sample.
Morphological studies
Cells were seeded (1×104/chamber) into a Lab-Tek Chamber-Slide System. (Nalge Nunc., Naperville, IL) and treated for 24hrs with 4μM of 7Me-IEITC. Following two wash-steps in PBS the cells were fixed in PBS, 2% PFA, 0.2% Triton X for 20min at RT and stained for 10min with 200ng/ml 4′-6-Diamidino-2-Phenylindole (DAPI) in PBS before mounting. Representative images were taken with an inverted microscope (Nikon Eclipse TE2000-E, CCD camera) and 20X objective.
Cell-cycle analysis (by FACS)
Cell-cycle analysis was carried out by flow cytometry. Cells were seeded into 100 mm2 tissue culture dishes (7.5×105 cells/dish) (Corning Inc., Corning, NY), treated with 3μM of 7Me-IEITC for 12 or 48hrs and the assay carried out as described elsewhere [13]. Appropriate gating was used to select the single cell population. The same gate was used on all samples, ensuring that the measurements were made on a standardized cell population. Experiments were performed in duplicate.
TUNEL assay
DNA fragmentation was detected using the DeadEnd™ Fluorometric TUNEL System assay (Promega, Madison, WI) according to the manufacturer’s recommendations. Cells (5×103/well) were plated into 96well flat bottom plates (Corning, Inc., Corning, NY), treated with 4μM of 7Me-IEITC or 25μM Actinomycin D for 48hrs, washed with PBS, fixed with PBS, 2% PFA, 0.2% Triton X for 30min at RT, and labeled with fluorescein-12-dUTP using terminal deoxynucleotidyl-transferase. After rinsing with PBS, nuclei were counterstained with propidium iodide (5μg/mL) for 20min. The localized green fluorescence of apoptotic cells (fluorescein-12-dUTP) was detected by fluorescence microscopy with an inverted microscope (Nikon Eclipse TE2000-E) and a 10x objective. Four randomly chosen microscopic fields were captured. Experiments were performed in triplicate.
Western blot analysis
Cells were seeded into 100mm2 tissue culture dishes (5×105 cells/dish) and treated with 4μM of 7Me-IEITC for various time intervals as indicated (result section). Preparation of cell lysates, PAGE and immunoblotting was carried out as described elsewhere [13]. Primary antibodies were all purchased from (Cell Signaling Technology, Beverly, MA). The bands were visualized using horseradish peroxidase-conjugated secondary antibodies (Amersham-Pharmacia Biotech, Piscataway, NJ), followed by enhanced chemiluminescence (Upstate, Waltham, MA) and documented autoradiography (F-Bx810 Film, Phoenix, Hayward, CA). As a size standard pre-stained Precision Plus Protein Kaleidoscope (Biorad, Hercules, CA) marker was used. Experiments were performed in triplicate.
Data analysis
Statistically significant differences between groups were determined by one-way ANOVA, using the Newman-Keuls test to account for multiple comparisons in post hoc analyses. Software used for these analyses was GraphPad Prism 3.0 (San Diego, CA).
Results
7Me-IEITC displays differential effects on the viability of various human cell lines, including platinum-resistant ovarian cancer cells
In an initial approach to analyze the cytotoxic effects of 7Me-IEITC (Figure 1) on ovarian cancer cells we performed a viability assay using SKOV-3 and OVCAR-3 (ovarian epithelial adenocarcinomas) along with adenocarcinoma cell lines from different tissues (PC-3, prostate; BxPC-3, pancreatic). Furthermore, we tested the effect of 7Me-IEITC on A-431 cancer cells (epidermoid skin) and immortalized cell lines (TCL-1, trophoblasts; HTR-8, first trimester cytotrophoblasts). 7Me-IEITC proved to be highly and selectively cytotoxic to all 4 adenocarcinoma cell lines studied including ovarian cancer cell lines SKOV-3 and OVCAR-3 with IC50 values ≤5μM after 44hrs of treatment (Figure 2). In contrast, the viability of A-431 (skin carcinoma) or TCL-1 and HTR-8 (trophoblasts) were only affected at higher drug concentrations ≥20μM and was not changed within the same time course (48hrs, Figure 2) or over longer time periods (unpublished data) by 7Me-IEITC at 10μM.
Figure 2. Comparative analysis of the cytotoxic effect of 7Me-IEITC in various human cancer and control cell lines.

SKOV-3, OVCAR-3 (ovarian epithelial adenocarcinomas), PC-3 (prostate adenocarcinoma), BxPC-3 (pancreatic adenocarcinoma), A-431 (epidermoid skin carcinoma), TCL-1 (trophoblasts) and HTR-8 (first trimester cytotrophoblasts) human cell lines were treated with various concentrations (2.5 to 20μM) of 7Me-IEITC for 48 hrs. The MTS viability assay was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % cell viability of samples with untreated cells [100%].
7Me-IEITC affects the mitochondrial membrane depolarization potential of SKOV-3 cells and causes morphological hallmarks of apoptosis
In a first approach to understand the effect of 7Me-IEITC on cellular functions we tested the effect of 7Me-IEITC treatment on the integrity/loss of the mitochondrial transmembrane depolarization potential (ΔΨm). 7Me-IEITC treatment of SKOV-3 cells resulted in a time-dependent increase in the number of cells with ΔΨm. After 3hrs, 20% of the cells and by 24hrs, 39% of the cells had lost their Ψm (Figure 3B, bar diagram). Even within the population of remaining cells (61%), fluorescent staining with DiOC6(3) showed a broader distribution of intensity as compared to the non-treated cells indicating an altered mitochondrial transmembrane depolarization potential (Figure 3B, insert at top of bar diagram).
Figure 3. 7Me-IEITC causes apoptosis in platinum-resistant SKOV-3 ovarian cancer cells.

Panel A: Membrane depolarization analysis after 7Me-IEITC treatment. SKOV-3 cells were treated for 3 or 24hrs with 3μM 7Me-IEITC, fixed and stained with DiOC6(3) as described (Materials and Methods). Fluorescence of the single cell population was measured by flow cytometry (excitation at 488nm, emission at 520nm; insert at top of panel B) and the transmembrane depolarization potential of the single cell populations plotted (bar chart). Ten thousand cells were analyzed in each sample.
Panel B: Morphological changes after 7Me-IEITC treatment. SKOV-3 cells were treated for 24hrs with 4μM 7Me-IEITC, fixed and stained with 4′-6-Diamidino-2-Phenylindole (DAPI) as described (Materials and Methods) before mounting. Microscopy was carried out (Nikon Eclipse TE2000-E inverted microscope, 10x objective) and representative images were taken. Bar, 5 μM.
Panel C: Caspase activation after 7Me-IEITC treatment. SKOV-3 cells were treated with 4μM 7Me-IEITC for 1hr, 6hrs, or 18hrs. Analysis of the expression of proteins in the lysates of treated and untreated cells was carried out by PAGE and Western Blot analysis as described (Material and Methods). Primary antibodies against activated caspase-3, -8, -9, and inactivated/cleaved PARP-1 were used. As an internal standard for equal loading the blots were probed with an anti-β-Actin antibody. As a size standard pre-stained Kaleidoscope (Biorad, Hercules, CA) marker was used.
Panel D: Effect of caspase inhibitors on SKOV-3 viability after 7Me-IEITC treatment. SKOV-3 cells were pre-incubated with specific inhibitors (40μM) against pan-caspase, capase-3, caspase-8, or caspase-9 for 2hrs and treated with 7Me-IEITC (0, 2.5 or 5μM) in the continued presence of the inhibitors (40μM) for additional 48hrs. The MTS viability assay was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % cell viability of samples with untreated cells.
Panel E: TUNEL assay. SKOV-3 cells were treated with either 4μM 7Me-IEITC or 25μM Actinomycin D for 48hrs. Labeling of DNA nicks with fluorescein-12-dUTP and chromatin counterstaining with propidium iodide was carried out as described (Materials and Methods) using a TUNEL assay (Promega, Madison, WI) according to the manufacturer’s recommendations. During fluorescent microscopy (Nikon Eclipse TE2000-E inverted microscope, 10x objective), representative images were taken, apoptotic stain (FL-dUTP, green) and nuclear stain (Pi, red) overlaid; TUNEL positive nuclei due to DNA fragmentation appear as yellow areas. Bar 10 μM.
Treatment of SKOV-3 cells with 7Me-IEITC resulted in a drastic change in morphologic features. Untreated cells or cells treated for 24 hrs with 4μM of 7Me-IEITC after fixation and chromatin staining with DAPI were analyzed by light- and fluorescence microscopy. The population of untreated cells displayed a homogenous morphology with nuclei lightly and evenly stained by DAPI (Figure 3B, no drug) and occasional appearances of dividing cells in mitosis (bright blue staining of chromosomes lined up along the metaphase plate). In contrast, after treatment with 7Me-IEITC only ~40% of cells showed characteristics similar to the untreated controls, some revealed swelling/increased diameter with homogenous staining of the chromatin, indicating necrotic events, while the majority of cells (~60%) displayed fragmented nuclei, densely stained nuclear granular bodies of highly condensed chromatin (“apoptotic bodies”) (Figure 3B).
Induction of apoptosis in SKOV-3 ovarian cancer cells by 7Me-IEITC
To define the cellular response of SKOV-3 cells upon 7Me-IEITC treatment we next analyzed the activation of caspases, characteristic for induction of apoptosis, as well as the inactivation of PARP-1 by immunoblotting. 7Me-IEITC treatment of SKOV-3 cells resulted in strong activation/cleavage of caspase-9 within 1hr, of caspase-3 within 1–6hrs, and of caspase-8 within 6hrs, while PARP-1 was inactivated/cleaved within 1hr after drug treatment (Figure 3C). The proof that reduction of SKOV-3 viability through 7Me-IEITC was a direct consequence of the induction of apoptosis is presented in Figure 3D. We employed various caspase inhibitors which were added to the viability assay 2hrs before and during treatment with 7Me-IEITC. Cytotoxicity of the drug (at 5μM) was reduced by ~50% using a pan-caspase inhibitor, and by ~30% using a caspase-3, -8, or -9 inhibitors (Figure 3D).
We carried out a TUNEL assay, which is a common method for detecting DNA fragmentation that results from apoptotic signaling cascades. The assay relies on the presence of nicks in the DNA of apoptotic (and some necrotic) cells which can be identified by terminal transferase that will catalyze the addition of labeled dUTP (fluorescein). SKOV-3 cells were treated with either 4μM of 7Me-IEITC or 25μM Actinomycin D (positive control for drug-induced DNA fragmentation) for 48hrs. To identify nuclei of the entire cell population, nuclei were counterstained with propidium iodide (Pi) intercalating in the DNA. TUNEL-positive nuclei were identified by yellow spots, resulting from an overlay of the image with apoptotic stain (FL-dUTP) and nuclear stain (Pi). As shown in (Figure 3E), half of the population in 7Me-IEITC treated cells were TUNEL-positive cells indicating fragmented DNA, which directly correlates with the data obtained in our viability study (Figure 2) and by DAPI staining (Figure 3B) at similar concentrations of 7Me-IEITC.
7Me-IEITC effect on cell proliferation and cell-cycle progression
As described in the previous sections 7Me-IEITC is a cytotoxic drug which activates apoptotic processes in SKOV-3 ovarian cancer cells. To investigate if 7Me-IEITC affects the proliferation of SKOV-3 cells (particularly at drug concentrations at or below 2.5μM when viability is not affected or partially reduced; Figure 2) we performed a BrdU incorporation assay. 7Me-IEITC dose-dependently reduced SKOV-3 proliferation (Figure 4A). At a drug concentration of 1.2μM (for 48hrs) proliferation of treated SKOV-3 was inhibited by 70% compared to untreated cells. Even at drug concentrations in the nanomolar range, BrdU incorporation into the DNA was reduced. Thus, 7Me-IEITC is a potent pro-apoptotic agent as well as an anti-proliferative agent.
Figure 4. Effect of 7Me-IEITC on proliferation and cell-cycle progression of SKOV-3 cells .

Panel A: Cell Proliferation/BrdU Incorporation SKOV-3 cells were treated with 3μM 7Me-IEITC for 48hrs. The proliferation assay was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) in % cell proliferation of untreated cells.
Panel B and C: Cell-cycle Analysis by FACS. SKOV-3 cells were treated with various concentrations of 7Me-IEITC (300nM to 2.4μM) for 12hrs or 48hrs. Cell-cycle analysis of treated and untreated cells was carried out as described (Materials and Methods). Data are presented as the relative fluorescence intensity of cell subpopulations in the 2 dimensional FACS profile (panel B) or table (panel C).
Cell-cycle analysis of SKOV-3 cells after 7Me-IEITC treatment revealed an increase in the count of sub-diploidal/2n cells after 48hrs of treatment (Figure 4B). This cell population represents cells with significant DNA damage, indicating a late apoptotic stage. With respect to cycling cells, 7Me-IEITC treatment for 12hrs caused a slight increase and treatment for 48hrs caused a prominent increase of cells in S-phase (Figure 4C). Treatment for 12hrs also leads to an increase of cells in the G2/M phase, which is not apparent after 48hrs of treatment, since cells in G2/M likely became apoptotic (sub-diploidal, 20%). Accordingly, the increase in S-phase and G2/M phase resulted in a sharp decrease of cells in the G0/G1 phase (Figure 4B,C). Apparently, in this asynchronous cell culture, 7Me-IEITC treatment affected cell-cycle checkpoints in G2/M and S-phases causing a reduction of cell-cycle progression along with transition into the apoptotic stage. Even though not the objective of the present report, further studies could analyze the specific effect of sub-cytotoxic concentrations of 7Me-IEITC on specific cell-cycle checkpoints, cell-cycle regulators (cyclin-dependent kinases, cyclins) and replication-start and -progression signals of the S-phase [14,15] in synchronized SKOV-3 cultures.
7Me-IEITC treatment causes MAP-Kinase activation and suppression of pro-survival markers in SKOV-3 ovarian cancer cells
To define hallmark responses of SKOV-3 cells to treatment with 7Me-IEITC we analyzed the expression and activation/phosphorylation of cellular markers involved in pro-survival or pro-apoptotic signaling. Immunoblotting of PAGE-separated cellular lysates revealed that 7Me-IEITC (at 4μM) caused a rapid (within 1hr), strong and sustained activation of p38 and JNK mitogen-activated protein kinases (MAPK) (Figure 5A). We directly addressed the role of p38 and JNK activation in the lethal response of SKOV-3 to 7Me-IEITC by performing the viability assay in the presence of inhibitors against these specific MAPKs (JNK/420119, p38/SB203580; Calbiochem) (Figure 5B): A dramatic suppression of the cytotoxicity of 7Me-IEITC (100% viability at 0 μM, 60% at 2.5μM, 39% at 5μM) was achieved by interfering with the activity of p38 MAPK. The p38 inhibitor itself was slightly cytotoxic to the cells but could almost completely counteract 7-MeIEITC cytotoxicity at lower concentrations and significantly at higher concentrations (89% viability at 0 μM, 81% at 2.5μM, 65% at 5μM 7Me-IEITC) (Figure 5B). As a positive control for the maximal potential effect of inhibition of p38 activity on drug-induced cytotoxicity, SKOV-3 cells were treated with the calcidiol derivative B3CD (13). Here, at the same inhibitor concentration (40μM) the drug effect (8% viability) could be completely suppressed by the p38 inhibitor (93%). Hence, the p38 signaling pathway plays a pivotal role in the response of SKOV-3 ovarian cancer cells to 7Me-IEITC. Since the cytotoxic effect of the drug was not entirely suppressed by the p38 inhibitor, other signaling pathways contribute (evident at high concentrations of 7Me-IEITC; ≥5μM). Figure 5B also depicts a similar study employing a JNK inhibitor. Despite the fact that the JNK inhibitor itself appeared to have cytotoxic effects on SKOV-3 cells (71% viability), viability following treatment with 7Me-IEITC alone (60% at 2.5μM, 33% at 5μM 7Me-IEITC) was not reduced in the presence of the inhibitor (61% at 2.5μM, 31% at 5μM 7Me-IEITC). Thus, this inhibitor exerted unspecific negative effects which were counteracted by specific pro-survival effects during drug treatment.
Figure 5. Expression of pro-survival markers and MAP-Kinase in SKOV-3 after 7Me-IEITC treatment; effect of MAP-Kinase inactivation on cell viability.

Panel A: Activation of MAP Kinases. SKOV-3 were treated with 4μM 7Me-IEITC for 1hr, 6hrs, or 18hrs. Analysis of the expression of proteins in the lysates of treated and untreated cells by PAGE and Western Blot analysis was carried out as described (Material and Methods) using primary antibodies against pro- and activated/phosphorylated (P-) SAP/JNK, p38 and ERK1/2. As an internal standard for equal loading the blots were probed with an anti-beta-Actin antibody. As a size standard pre-stained Kaleidoscope (Biorad, Hercules, CA) marker was used.
Panel B: Effect of JNK and p38 inactivation on cell viability. SKOV-3 cells were pre-incubated with specific inhibitors (40μM) against JNK or p38 MAP-Kinase for 2hrs and treated with 7Me-IEITC (0, 2.5 or 5μM) in the continued presence of the inhibitors for additional 48hrs. As a positive control for the maximal potential effect of inhibition of p38 activity on of drug induced cytotoxicity, SKOV-3 cells were treated with a calcidiol derivative (+Ctr., 1μM B3CD) (Lange et al., in preparation). The MTS viability assay was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % cell viability of samples with untreated cells.
Panel C: Inactivation of survival signaling proteins and transcription factors. SKOV-3 were treated with 4μM 7Me-IEITC for 6hrs, 18hrs, or 36hrs. Analysis of the expression of proteins in the lysates of treated and untreated cells by PAGE and Western Blot analysis was carried out using primary antibodies against pro- and activated/phosphorylated PI-3K, STAT-3, IKKα, or NF-kB. As an internal standard for equal loading blots were probed with an anti-βActin antibody. As a size standard pre-stained Kaleidoscope (Biorad, Hercules, CA) marker was used.
Immunoblotting not only revealed an up-regulation of activated p38 and JNK MAPK, the basal level of inactive JNK also appeared to increase slightly after 6hrs of treatment. In contrast, the expression of inactive p38 was strong even in untreated cells and did not change after 7Me-IEITC treatment (Figure 5A). The expression profile of Erk1/2 MAPK was similar to that of p38 both for the inactive as well as the active phosphorylated forms. Activation of Erk1/2 occurred within 1hr following incubation of SKOV-3 cells with 7Me-IEITC. Both Erk1 and 2 (p44 and p42 MAPK) generally participate in a protein kinase cascade that plays a critical role in the regulation of cell growth and differentiation but can be found activated during survival and apoptotic events [16].
In addition, we studied the survival response of SKOV-3 cells after treatment with 7Me-IEITC by analyzing the expression levels of survival kinases (PI-3K, IKKα), transcription factor NF-κB and signal transducer STAT-3. All these factors are key-players in various cellular events such as cell growth, cell-cycle entry, cell migration, or anti-apoptotic responses and cell survival. 7Me-IEITC treatment (at 4μM) of SKOV-3 cells resulted in rapid (within 6hrs) and sustained (36hrs) deactivation of PI-3K and STAT-3 while the phosphorylation level remained elevated in untreated cells (Figure 5C). IKKα and NF-κB were activated during the first 18hrs of treatment and inactivated by 36hrs, indicating that critical signaling cascades were preceded by various upstream kinases (e.g. PI-3K). A strong basal expression of all of these factors in their inactive (non-phosphorylated) state in SKOV-3 ovarian cancer cells was observed.
Discussion
The low overall survival rate for woman suffering ovarian cancer is related to the development of resistance to standard chemotherapeutic agents, most notably platinum analogs. While re-treatment with a platinum-based drug is possible for some women the response rate to current second line chemotherapy is 15–30% [1,2] and therefore new agents need to be developed. For the present report we choose to study the therapeutic potential of the newly designed isothiocyanate (ITC) derivative 7Me-IEITC [11] to treat ovarian cancer. ITCs have been shown to inhibit chemically induced tumorigenesis in animal models of lung, gastric, colon, liver, esophagus, bladder, and mammary glands cancers [3,17]. The choice for 7Me-IEITC was based on a structure/activity relationship screening in a viability assay in comparison to other new compounds designed in our lab using neuroblastoma cell lines [11] or OVCAR-3 ovarian cancer cells (unpublished data). 7Me-IEITC proved to be cytotoxic at lower concentrations than other derivatives or parent compounds such as PEITC or BITC [18,19]. The two ovarian cancer cell lines used in the current study are multi-drug resistant (see ATCC, Manassas, VA; www.atcc.org); OVCAR-3 cells (ovarian epithelial adenocarcinoma) are resistant to clinically relevant concentrations of adriamycin, melphalan and Cisplatin. SKOV-3 cells (ovarian adenocarcinoma) are resistant to several cytotoxic drugs including Cisplatin and Adriamycin.
The present study demonstrates that 7Me-IEITC is highly cytotoxic to these two ovarian cancer cell lines and to two adenocarcinoma cell lines from different tissues (prostate, pancreas) at concentrations less than 5μM, while A-431 cancer cells (epidermoid skin) and TCL-1 or HTR-8 trophoblastic cell lines are affected only at significantly higher concentrations. Adenocarcinoma is a type of cancer that develops in cells lining glands of internal organs, such as the lung, breast, prostate, stomach, pancreas, and cervix. Apparently, the cytotoxitity of 7Me-IEITC in-vitro is highly dependent on the type of cancer cell or cell line treated. This observation is confirmed by our previous results where 7Me-IEITC selectively reduced the viability of three neuroblastoma cell lines, while the viability of lung fibroblasts (passage 10) was not significantly affected at drug concentrations as high as 20μM [11]. Similar to ovarian cancer cell lines SKOV-3 and OVCAR-3, primary fibroblast at early passages and immortalized trophoblastic cell lines with primary features used as controls in the current study, possess a high metabolism and growth rate. The relative resistance of these three control cell lines to 7Me-IEITC treatment now sets the stage for the testing of this compound in an ovarian cancer animal model.
Morphological changes of cells after drug treatment in vitro are a first indicator for potential drugs effects on tumor metastasis and cell physiology including cell death in vivo. 7Me-IEITC caused apoptosis in SKOV-3 cells indicated by nuclear fragmentation and chromatin condensation (Figure 3B), a classic hallmark of apoptosis [20] and DNA fragmentation (TUNEL assay, Figure 3E; sub-diploidal cell population, Figure 4B). Induction of apoptosis (caspase activation) occurred as early as 1hr after treatment. Within 3hrs of 7-MeIEITC treatment we observed a loss of mitochondrial transmembrane depolarization potential (ΔΨm) in SKOV-3 cells as reported for other ITC derivatives [21]. The ADP:ATP ratio and ΔΨm can be used as an indicator of apoptosis [22,23]. Moreover, the loss of ΔΨm due to chemical agents for other drug-treated cell types has been reported to be indicator of early apoptosis and as the first irreversible step in the induction of apoptosis [24]. Accordingly, loss of the ΔΨm within 3hrs in SKOV-3 following 7-MeIEITC treatment may be the first irreversible step in the induction of apoptosis by this agent. Apparently, the early onset of caspase activation and PARP-1 inactivation (Figure 3C) in SKOV-3 ovarian cancer cells by 7Me-IEITC resulted in the morphological changes observed (Figure 3B).
Apoptosis is executed by initiator caspases which upon activation cleave and activate downstream effector caspases that are responsible for the cleavage of many intracellular proteins, leading to the morphological and biochemical changes associated with apoptosis [25,26]. 7Me-IEITC treatment of SKOV-3 cells resulted in strong activation/cleavage of initiator caspase-8 and -9 and of effector caspase-3, while PARP-1 (involved in DNA repair) [27] was inactivated following drug treatment. 7-Me-IEITC induced both major signaling pathways (intrinsic, extrinsic) described for activation of initiator caspases in mammalian cells. The intrinsic pathway mediates apoptotic responses to stress signals such as drugs, DNA damage or growth factor deprivation. Mitochondrial damage can initiate the intrinsic pathway, leading to the activation of pro-apoptotic members of the Bcl-2 family and results in the mitochondrial release of cytochrome C which activates initiator caspase-9 [28,29] as seen in SKOV-3 cells following 7Me-IEITC treatment. The extrinsic pathway is initiated by interaction of specific ligands with their corresponding “death” receptors, or by receptor oligomerization and caspase-8 activation [28,30] as observed in SKOV-3 cells following 7Me-IEITC treatment. The involvement of both pathways in the execution of apoptosis in SKOV-3 cells following 7Me-IEITC treatment is proven by the partial suppression of its cytotoxicity by either a caspase-8 or caspase-9 inhibitors in viability assays.
The present report suggests the participation of activated/phosphorylated mitogen-activated protein kinases (MAPK) p38, JNK, and Erk1/2 in the induction of apoptosis in SKOV-3 cells upon treatment with 7Me-IEITC. Erk1 and 2 (p44 and p42) generally participate in a protein kinase cascade that regulates cell growth and differentiation as survival factors but have also been reported to be activated in apoptotic events [15,28]. Similarly to 7Me-IEITC, Cisplatin induced apoptosis of renal cells requires Erk1/2 activation [31]. Similarly to the treatment of SKOV-3 cells by 7-MeIEITC, other ITCs caused significant elevations in the phosphorylation of Erk1/2 and JNK in human prostate cancer PC-3 cells [32].
We report that JNK and p38 MAPK pathways were activated in SKOV-3 ovarian cancer cells upon 7Me-IEITC treatment. Both, JNK and p38 are crucial anti-survival factors in signaling cascades responding to inflammatory cytokines, UV light, cytotoxic drugs and diverse other pro-apoptotic stimuli [28,30,33]. Similarly to our experiments, these pathways were activated in the apoptotic response of various carcinoma cells following treatment with drugs such as Doxorubicin, Paclitaxel, Etoposide [34,35], Aplidin™ [36], Tamoxifen [37] or Cisplatin (in human ovarian cancer cells) [38]. In the present study we show, by specific inhibition of the activity of p38 MAPK, that the apoptotic response of SKOV-3 ovarian cancer cells to 7Me-IEITC is strongly mediated by the p38 signaling pathway. Addition of a JNK inhibitor upon SKOV-3 treatment with 7Me-IEITC exerted minor specific pro-survival effects on cells during drug treatment and, thus, the JNK pathway may contribute to the response of SKOV-3 ovarian cancer cells to 7Me-IEITC.
In addition, we studied the survival response of SKOV-3 cells following treatment with 7Me-IEITC by analyzing the expression level of PI-3K, as well as survival kinase IKKα and transcription factor NF-κB which both act downstream of PI-3K. 7Me-IEITC treatment led to the inactivation of PI-3K within hours following treatment. Inactivation of IKKα and NF-κB was observed after 36hrs, following the initial activation of these two factors to promote survival and counteract drug effect. PI-3K, IKKα and NF-κB are known to contribute to tumor growth by promoting cell-cycle entry, cell proliferation, cell migration, or anti-apoptotic responses and are implicated in resistance to radio- and chemotherapy [39]. Activation of the PI3K/Akt pathway has been linked to chemotherapeutic resistance in human tumors [40], contributes to Cisplatin resistance in ovarian cancer cell lines [ 41 ] and to microtubule-directed agents including Vincristine in ovarian cancer [42]. Consequently, our observations that 7Me-IEITC inactivates the PI-3K pathway and downstream factors IKKα and NF-κB suggests that 7Me-IEITC could be used to enhance the effectiveness of chemotherapy in drug resistant cancers.
Interestingly, we also observed inactivation of STAT-3 following 7Me-IEITC treatment of SKOV-3. STAT-3, generally known as an anti-oncogene by promoting apoptosis can however contribute to inflammation-mediated tumor development by enhancing tissue injury and remodeling [43]. STAT-3 is constitutively activated in many types of human cancer [44], including ovarian cancer (SKOV-3). Furthermore, the STAT-3 pathway has been reported to be involved in cross-talk with the PI-3 pathway and NF-kB signaling [45,46]. Finally, several natural compounds and synthetic drugs that are able to inhibit the IKK/NF-kB activation pathway have been shown to either prevent cancer or to inhibit cell growth in animal models [47]. The selectivity of 7Me-IEITC on cell viability of cancer cells together with its effect on the IKK/NF-kB pathway and STAT-3 suggests that the further exploration of this compound in an ovarian cancer animal model is warranted.
The present report also shows that 7Me-IEITC is a potent inhibitor of SKOV-3 ovarian cancer cell proliferation at sub-cytotoxic concentrations. 7Me-IEITC treatment affects cell-cycle checkpoints in G2/M and S-phases causing reduction of cell-cycle progression along with transition into the apoptotic stage. Similarly, another plant-derived molecule, Guggulsterone, caused cell-cycle arrest in S-phase by the suppression of cyclin D1 and cdc2 and increased cyclin-dependent kinase inhibitor p21 and p27 expression in a wide variety of human tumor cell types [48]. Apparently transformed cells can be more sensitive to the CDK inhibition during S-phase due to the fact that components of the cell-cycle machinery are frequently altered in human cancer and [49,50], and thus, can be specifically targeted. Even though not the objective of the present report, future studies will examine the effects of 7Me-IEITC on specific cell-cycle checkpoints in ovarian cancer cells. Targeting cell cycle checkpoints has been suggested as an alternative approach to anti-cancer therapies [51,52].
In summary, the present report suggests that 7Me-IEITC is a potent and growth-suppressing agent to cell lines derived from ovarian cancers and a potential therapeutic drug to treat such tumors in vivo.
Acknowledgments
This work was supported by a Brown University Seed Grant and a NICHD, K12 HD043447 BIRCWH Scholar Grant to Dr. Brard. The authors thank NIH COBRE Grant 1-P20RR018728 for providing instrumentation support and Dr. Paonessa (Roswell Park, USA) for technical advice.
Footnotes
Presented at the 38th Annual Meeting on Women’s Cancer, Society of Gynecologic Oncologists, San Diego, CA, March 3-7, 2007.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heintz APM, Odicino F, Maisonneuve P, Beller U, Benedet JL, Creasman WT, Nga HYS, Pecorelli S. FIGO Annual Report/Carcinoma of the Ovary; 1996–1998. [Google Scholar]
- 2.American Cancer Society. Cancer statistics. 2007 www.cancer.org.
- 3.Barecki RM, Wang EJ, Johnson WW. Quantitative evaluation of isothiocyanates as substrates and inhibitors of P-glycoprotein. J Pharm Pharmacol. 2003;55:1251–7. doi: 10.1211/0022357021666. [DOI] [PubMed] [Google Scholar]
- 4.Tseng E, Kamath A, Morris ME. Effect of Organic Isothiocyanates on the P-Glycoprotein-and MRP1-Mediated Transport of Daunomycin and Vinblastine. Pharm Res. 2002;19:1509–15. doi: 10.1023/a:1020460700877. [DOI] [PubMed] [Google Scholar]
- 5.Gottesman MM. Multi-drug transporter P-glycoprotein. Ann Rev Med. 2002;53:615–7. [Google Scholar]
- 6.Stein U, Lage H, Jordan A, Walther W, Bates SE, Litman T, Hohenberger P, Dietel M. Impact of BCRP/MXR, MRP1 and MDR1/P-Glycoprotein on thermoresistant variants of atypical and classical multidrug resistant cancer cells. Int J Cancer. 2002;97:751–60. doi: 10.1002/ijc.10131. [DOI] [PubMed] [Google Scholar]
- 7.Vidal DO, Lopes LF, Valera ET. Drug resistance and methylation in myelodysplastic syndrome. Curr Pharm Biotechnol. 2007;8:77–81. doi: 10.2174/138920107780487483. [DOI] [PubMed] [Google Scholar]
- 8.Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L, Baskaran R. Sulforaphane-induced G2/M phase cell-cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle. J Biol Chem. 2004;279:25813–22. doi: 10.1074/jbc.M313538200. [DOI] [PubMed] [Google Scholar]
- 9.Jackson SJ, Singletary KW, Venema RC. Sulforaphane suppresses angiogenesis and disrupts endothelial mitotic progression and microtubule polymerization. Vascul Pharmacol. 2007;46:77–84. doi: 10.1016/j.vph.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Xiao D, Singh SV. Phenethyl isothiocyanate inhibits angiogenesis in vitro and ex vivo. Cancer Res. 2007;67:2239–46. doi: 10.1158/0008-5472.CAN-06-3645. [DOI] [PubMed] [Google Scholar]
- 11.Singh RK, Lange TS, Kim K, Zou Y, Lieb C, Sholler GL, Brard L. Effect of indole ethyl isothiocyanates on proliferation, apoptosis and MAPK signaling in neuroblastoma cell lines. Bioorg Med Chem Lett. 2007;17:5846–5852. doi: 10.1016/j.bmcl.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malich G, Markovic B, Winder C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology. 1997;124:179–92. doi: 10.1016/s0300-483x(97)00151-0. [DOI] [PubMed] [Google Scholar]
- 13.Lange TS, Singh RK, Kim KK, Zou YP, Kalkunte SS, Scholler GL, Swamy N, Brard L. Antiproliferative and pro-apoptotic properties of 3-bromoacetoxy calcidiol in high risk neuroblastoma. Chem Biol Drug Des. 2007;70:302–310. doi: 10.1111/j.1747-0285.2007.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pines J. Four-dimensional control of the cell-cycle. Nature Cell Biol. 1999;1:73–9. doi: 10.1038/11041. [DOI] [PubMed] [Google Scholar]
- 15.Stillman B. Cell-cycle control of DNA replication. Science. 1996;274:1659–64. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed-Choudhury J, Williams KT, Young LS, Adams DH, Afford SC. SCCD40 mediated human cholangiocyte apoptosis requires JAK2 dependent activation of STAT3 in addition to activation of JNK1/2 and ERK1/2. Cell Signal. 2006;18:456–68. doi: 10.1016/j.cellsig.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 17.Hecht SS. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J Nutr. 1999;129:768S–74S. doi: 10.1093/jn/129.3.768S. [DOI] [PubMed] [Google Scholar]
- 18.Satyan KS, Swamy N, Dizon DS, Singh RK, Granai CO, Brard L. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: Role of caspase and MAPK activation. Gynecol Oncol. 2006;103:261–70. doi: 10.1016/j.ygyno.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Kalkunte S, Swamy N, Dizon DS, Brard L. Benzyl isothiocyanate (BITC) induces apoptosis in ovarian cancer cells in vitro. J Exp Ther Oncl. 2006;5:287–300. [PubMed] [Google Scholar]
- 20.Earnshaw WC. Nuclear changes in apoptosis. Curr Opin Cell Biol. 1995;7:337–43. doi: 10.1016/0955-0674(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol Cancer Ther. 2003;2:1045–52. [PubMed] [Google Scholar]
- 22.Bradbury A, Simmons TD, Slater KJ, Crouch SPM. Measurement of the ADP:ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J Immunol Meth. 2000;240:79–92. doi: 10.1016/s0022-1759(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 23.Kim R, Emi M, Tanabe K, Murakami S, Uchida Y, Arihiro K. Regulation and interplay of apoptotic and non-apoptotic cell death. J Pathol. 2006;208:319–26. doi: 10.1002/path.1885. [DOI] [PubMed] [Google Scholar]
- 24.Petit PX, Lecoeur H, Zorn E, Dauguet C, Mignotte B, Gougeon M. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol. 1995;130:157–67. doi: 10.1083/jcb.130.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salvesen GS, Abrams JM. Caspase activation - stepping on the gas or releasing the brakes? Lessons from humans and flies. Oncogene. 2004;23:2774–84. doi: 10.1038/sj.onc.1207522. [DOI] [PubMed] [Google Scholar]
- 26.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 27.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem. 1998;73:33533–9. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 28.Pearson G, Robinson F, Beers GT, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 29.Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–53. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Birkenkamp KU, Dokter WH, Esselink MT, Jonk LJ, Kruijer W, Vellenga E. A dual function for p38 MAP kinase in hematopoietic cells: involvement in apoptosis and cell activation. Leukemia. 1999;13:1037–45. doi: 10.1038/sj.leu.2401447. [DOI] [PubMed] [Google Scholar]
- 31.Kim K, Kim JU, Kwon CH, Kim JH, Woo S, Jin SJ, Kim JM. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J Appl Toxicol. 2005;25:374–82. doi: 10.1002/jat.1081. [DOI] [PubMed] [Google Scholar]
- 32.Chang JX, Shen G, Yuan X, Kim J, Gopalkrishnan A, Keum YS, Nair S, Kong ANT. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–45. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 33.Lin A, Dibling B. The true face of JNK activation in apoptosis. Aging Cell. 200;21:112–6. doi: 10.1046/j.1474-9728.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- 34.Brantley FC, Lyle CS, Du L, Goodwin ME, Hall T, Szwedo D, Kaushal GP, Chambers TC. The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem Pharmacol. 2003;66:459–69. doi: 10.1016/s0006-2952(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 35.Wang TH, Chan YH, Chen CW, Kung WH, Lee YS, Wang ST, Chang TC, Wang HS. Paclitaxel (Taxol) upregulates expression of functional interleukin-6 in human ovarian cancer cells through multiple signaling pathways. Oncogene. 2006;25:4857–66. doi: 10.1038/sj.onc.1209498. [DOI] [PubMed] [Google Scholar]
- 36.Cuadrado A, Fernandez LFG, Gonzalez L, Suarez Y, Losada A, Alcaide V, Mart nez T, Fernandez-Sousa JM, Puelles JMS, Munoz A. Aplidin™ Induces Apoptosis in Human Cancer Cells via Glutathione Depletion and Sustained Activation of the Epidermal Growth Factor Receptor, Src, JNK, and p38 MAP-KINASE. J Biol Chem. 2003;278:241–50. doi: 10.1074/jbc.M201010200. [DOI] [PubMed] [Google Scholar]
- 37.Zhang C-C, Shapiro DJ. Activation of the p38 Mitogen activated protein kinase pathway by estrogen or by 4-hydroxytamoxifen is coupled to estrogen receptor-induces apoptosis. J Biol Chem. 2000;275:475–89. doi: 10.1074/jbc.275.1.479. [DOI] [PubMed] [Google Scholar]
- 38.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAP-Kinase pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–56. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 39.Greten FR, Karin M. The IKK/NF-kB activation pathway-a target for prevention and treatment of cancer. Cancer Letters. 2004;206:193–9. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 40.Kip A, West S, Sianna C, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resistance Updates. 2002;5:234–48. doi: 10.1016/s1368-7646(02)00120-6. [DOI] [PubMed] [Google Scholar]
- 41.Lee S, Choi EJ, Jin C, Kim DH. Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol. 2005;97:26–34. doi: 10.1016/j.ygyno.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 42.Vander Weele DJ, Rudin CM. Mammalian target of rapamycin promotes vincristine resistance through multiple mechanisms independent of maintained glycolytic rate. Mol Cancer Res. 2005;3:635–44. doi: 10.1158/1541-7786.MCR-05-0063. [DOI] [PubMed] [Google Scholar]
- 43.Yoshimura A. Signal transduction of inflammatory cytokines and tumor development. Cancer Sci. 2006;97:439–47. doi: 10.1111/j.1349-7006.2006.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darnell JE., Jr Validating Stat3 in cancer therapy. Nature Medicine. 2005;11:595–96. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 45.Squarize CH, Castilho RM, Sriuranpong V, Pinto DS, Gutkind JS. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 2006;8:733–46. doi: 10.1593/neo.06274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abell K, Bilancio A, Clarkson RW, Tiffen PG, Altaparmakov AI, Burdon TG, Asano T, Vanhaesebroeck B, Watson CJ. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat Cell Biol. 2005;7:392–8. doi: 10.1038/ncb1242. [DOI] [PubMed] [Google Scholar]
- 47.Bharti AC, Aggarwal BB. Chemopreventive agents induce suppression of nuclear factor-kappaB leading to chemosensitization. Ann N Y Acad Sci. 2002;973:392–5. doi: 10.1111/j.1749-6632.2002.tb04671.x. [DOI] [PubMed] [Google Scholar]
- 48.Shishodia S, Sethi G, Ahn KS, Aggarwal BB. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem Pharm. 2007;74:118–30. doi: 10.1016/j.bcp.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hartwell LH, Kastan MB. Cell-cycle control and cancer. Science. 1994;266:1821–8. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 50.Gladden AB, Diehl JA. Cell-cycle progression without cyclin E/CDK2: breaking down the walls of dogma. Cancer Cell. 2003;4:160–2. doi: 10.1016/s1535-6108(03)00217-4. [DOI] [PubMed] [Google Scholar]
- 51.Mazumder S, DuPree EL, Almasan A. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr Cancer Drug Targets. 2004;4:65–75. doi: 10.2174/1568009043481669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shapiro GI, Harper JW. Anticancer drug targets: cell-cycle and checkpoint control. J Clin Invest. 1999;104:1645–53. doi: 10.1172/JCI9054. [DOI] [PMC free article] [PubMed] [Google Scholar]