

Abstract
Peroxiredoxins (Prxs) are abundant cellular antioxidant proteins which help control intracellular peroxide levels. These proteins may also function, in part through an evolved sensitivity of some Prxs toward peroxide-mediated inactivation, in hydrogen peroxide signaling in eukaryotes. This review summarizes recent progress in our understanding of the catalytic and regulatory mechanisms of “typical 2-Cys” Prxs and of the biological roles that these important enzymes play in oxidative stress and non-stress related cellular signaling.
Keywords: peroxiredoxins, redox signaling, hydrogen peroxide, hydroperoxides, oxidative stress, signal transduction, antioxidants, sulfenic acid, mechanisms, NADPH oxidase
Introduction and scope
Peroxiredoxins (Prxs1, EC 1.11.1.15) are ubiquitous antioxidant enzymes found in all organisms with the single exception, to our knowledge, of Borrelia burgdorferi (and other Borrelia species). The broad distribution of Prxs and the high levels of expression [1] suggest they are both an ancient and important enzyme family. Prxs are assumed to have evolved from a thioredoxin-like precursor protein and a model for the evolutionary path has been presented [2]. The initial publication defining this family of enzymes introduced the term “peroxidoxin” [3], but shortly thereafter “peroxiredoxin” was suggested [4]. While some publications used peroxidoxin, it is the latter name that has been widely adopted.
Prx research has expanded rapidly in recent years, with a PubMed search for the term “peroxiredoxin” yielding 2, 43, 227, and 542 papers in 1992-95, 1996-99, 2000-03 and 2004-07, respectively. Despite this recent increase in study, Prxs are still not well characterized relative to glutathione peroxidase and catalase. For comparison, 6523 papers were published on catalase and 4205 on glutaredoxin from 2004-2007.
A number of excellent reviews have summarized much of the work on Prx structure, function, and biology [1, 5-8], and on the developing view of hydrogen peroxide signaling [5, 9-11]. The purpose of this mini-review is to highlight recent advances in our understanding of the chemistry of “typical 2-Cys” Prxs and evidence for the role(s) of the sensitivity to inactivation by hydrogen peroxide seen in some members of this group.
Typical 2-Cys Prxs
All Prxs have in common an overall fold and catalytic mechanism involving a conserved, fully folded active site and an unfolding event [6] (Figure 1a and Figure 2). The enzymatic mechanism relies on a conserved cysteine residue, the peroxidatic cysteine (CP), to reduce various peroxide substrates with catalytic efficiencies on the order of 106 - 107 M−1s−1 [7]. Following oxidation of CP by the peroxide substrate, regions of the protein around the active site change conformation, allowing for subsequent reactivation steps (Figure 2a). A second free thiol (CR for the resolving cysteine or sulfur atom, respectively) then forms a disulfide with the CP and is required to complete the catalytic cycle. 1-Cys and 2-Cys Prxs are differentiated by whether the CR (or corresponding thiol group) comes from another molecule (1-Cys) or from a Prx (2-Cys).
Figure 1.
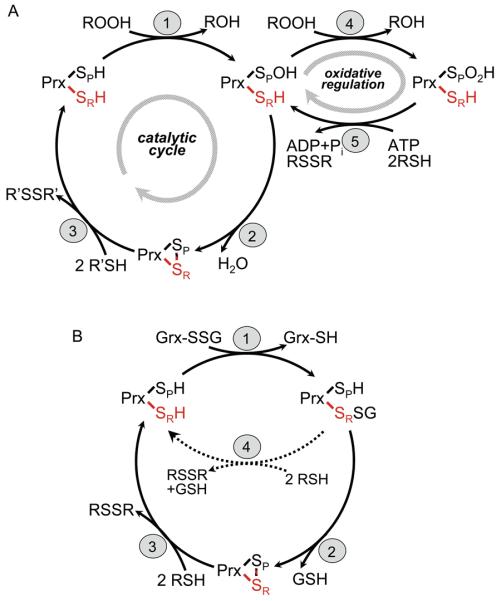
Mechanisms of catalysis by typical 2-Cys Prxs. (A) The peroxidatic catalytic cycle of typical 2-Cys Prxs involves three main steps: (1) peroxidation, (2) resolution and (3) recycling. Not shown is the local unfolding event that occurs in both the CP-loop and C-terminus during step 2 so that the disulfide bond can form (see Figure 2a). The protein is represented as one of two active sites within a functional dimer, with SP and SR (red) designating the sulfur atoms of the peroxidatic and resolving cysteines, respectively, from different subunits. “2 R′SH” in step 3 represents a thioredoxin-like protein or domain. Overoxidation of the CP (step 4) and reduction of the Cys-SPO2H by Srx (step 5) depict redox regulation and repair occurring in some eukaryotic typical 2-Cys Prxs. (B) Mutants of AhpC which suppress the growth defect (dithiothreitol dependence) of the trxB gor mutant from E. coli were shown to catalyze the deglutathionylation of Grx1 (using the C14S mutant) in vitro [24]. Although the catalytic intermediate is shown with glutathione attached in a mixed disulfide to the Prx, the alternative mechanism with Grx attached to Prx is also possible. The truncated cycle shown with dotted lines (step 4 in b) illustrates the finding that only the “resolving cysteine” (with the sulfur depicted as SR) is required for this activity.
Figure 2.

Structural aspects of Prx sensitivity and resurrection. (A) The active site of a sensitive 2-Cys Prx in the fully folded (FF) and locally unfolded (LU) conformations. Both chains of the dimer are colored grey. The C-terminal helix containing the YF motif (cyan) and the loop associated with the GGLG motif (yellow) that are characteristic of sensitive Prxs can be seen to pack against each other and cover the active site CP loop (pink) in the FF conformation (PDB code 1QMV). Comparing the LU structure (PDB code 1QQ2) with the FF structure shows how the CP loop and the protein C-terminus unfold for disulfide formation and how the C-terminal helix hinders this required local unfolding. In robust Prxs, the C-terminal helix containing the YF motif is absent, allowing for more facile unfolding (see Figure 2 in Wood et al, 2003). In the LU form, a star indicates the presence of the additional disordered C-terminal residues. In both the FF and LU images, the four residues conserved in all Prxs (CP, Arg, Thr and Pro) and the CR are colored green with sulfur, oxygen and nitrogen atoms colored dark yellow, red and blue, respectively. (B) The typical 2-Cys dimer (magenta and dark blue) associates with other dimers (light blue) as part of the normal catalytic cycle to form higher order oligomers. The overoxidized state is stabilized in this form [20]. Modeling of Srxs (green) on a Prx decamer shows that Srx can associate with such a structure without significant changes to the decamer, consistent with the role of Srx in the reduction of the overoxidized Prx. Figure modified from [32].
All of the early characterized Prxs were “typical 2-Cys” Prxs. In this subclass of 2-Cys Prxs, the basic active unit is a dimer, with the catalytically relevant disulfide bond being formed between the CP on one chain with the CR from near the C-terminus of the other chain [6] (Figure 2). The subclass of “atypical 2-Cys” Prxs are differentiated from the typical 2-Cys Prxs in that the catalytic disulfide bond is intramolecular in most cases, and the resolving cysteine is not at the “typical” conserved position in the C-terminus. Despite the different positions observed for CR, the catalytic cycle for peroxidase activity in all Prxs can be broken into three steps: (1) peroxidation, (2) resolution and (3) recycling (Figure 1a). Peroxidation occurs in the fully folded active site which contains four conserved residues: the CP, an Arg and a Thr residue presumed to stabilize the thiolate anion, and a Pro that shields the active site from water (Figure 2a). This active site environment lowers the pKa of the CP, which has recently been shown to be between 5 and 6 for the few Prxs thus studied [12-14]. The thiolate anion attacks the peroxide substrate to generate water (or alcohol) and a Cys-sulfenic acid (Cys-SPOH) at the active site [7]. Resolution occurs when the CR attacks the Cys-SPOH to release water and form an intersubunit disulfide bond. The catalytic cycle is completed when the disulfide bond is recycled, typically by a thioredoxin-like molecule, regenerating the free thiol forms of the CP and the CR.
For these enzymes, catalysis not only involves chemical transformations, but also requires the protein to undergo some conformational gymnastics. The form of the enzyme carrying out peroxidation is fully folded; in typical 2-Cys Prxs the CR side chain is buried and roughly 14 Å away from the CP. For the CP and CR to form a disulfide, both the active site region (known as the CP-loop) and the C-terminal region must locally unfold (Figure 2a). Details of the structural changes occurring with catalysis for various Prxs have been recently reviewed by Karplus and Hall [6].
An additional mechanistic complexity of these proteins is that during catalysis they shift quaternary structure between a homodimer and a doughnut-shaped decamer (which consists of a pentamer of dimers)2 (Figure 2b). Studies with the AhpC Prx from Salmonella typhimurium suggested that the decameric form is stabilized in all catalytic states of the enzyme except for the disulfide form (between steps 2 and 3 in Figure 1a); the decamer falls apart upon disulfide formation because the unfolding of the CP-loop destabilizes the decamer-building (dimer-to-dimer) interface [15]. Analogous redox-dependent oligomerization effects are now known to extend to mammalian and plant-derived typical 2-Cys Prxs as well [16-18]. The physiological role of the dimer/decamer transition remains unclear, however studies indicating that decamers are better peroxidases than dimers [19] but are less amenable to reduction by thioredoxins [18] suggest that the quaternary structure transition aids efficient catalysis. There is also evidence for some Prxs that the decamers associate with membranes [20, 21], so it is possible that cellular localization is influenced by oligomeric state.
Recent mechanism-relevant discoveries for Prxs
Although Prxs have been described as broad specificity peroxidases which reduce substrates such as hydrogen peroxide, lipid hydroperoxides and peroxynitrite, recent data suggests that at least some of the typical 2-Cys Prxs are much more active with hydrogen peroxide than with bulkier hydroperoxide substrates [22], and that the reactivity with other oxidants and alkylating agents is remarkably limited [13]. The thermodynamic driving force for peroxide reduction by thiols (step 1 in Figure 1a) is highly favorable, so the relative reactivity for thiol-based peroxidases is, instead, dominated by kinetic factors [11]. On the other hand, turnover of Prxs with reductants (step 3 in Figure 1a) may be subject to greater influence by their midpoint reduction (redox) potentials. In this regard, it is perhaps surprising that the redox potentials for typical 2-Cys Prxs from plant chloroplasts are in the range of −300 mV, similar to or even lower than their physiological reductants [23]. As pointed out by Dietz and colleagues, however, these low potentials likely reflect the unique regulatory environment of this photosynthetic organelle. Recent data for the bacterial antioxidant AhpC indicates a redox potential of −178 mV, sufficiently high for AhpC to remain predominantly reduced even under conditions in which the cell is oxidatively stressed [22].
Also, an alternative catalytic activity that may be exhibited by a subset of Prxs has recently come to light using Escherichia coli mutants with thioredoxin reductase and glutathione reductase gene deletions (the trxB gor mutant) that are compromised in cytoplasmic disulfide reduction, resulting in growth defects. This condition leads to frequent selection of a mutation causing the insertion of one amino acid between residues 37 and 38 of AhpC, converting it from a peroxidase to a disulfide reductase that acts as a glutaredoxin deglutathionylating enzyme [24]. Other single point mutations were also able to confer this activity without eliminating peroxidase activity. Surprisingly, CR (Cys165) rather than CP (Cys46) of the E. coli AhpC is critical for the suppression of the growth defect of the trxB gor mutant, and for the disulfide reductase activity measured in vitro (Figure 1b). Although only speculative at present, it is conceivable that this activity may be more constitutively present in wild-type forms of other Prxs where the CR is particularly reactive, or switched on in some Prxs by protein modifications.
Robust and sensitive typical 2-Cys Prxs
The catalytic cycle explained above is all that is needed to describe the peroxidase activity of the Prxs. However, beginning with a yeast Prx (now known as Tpx1), a number of Prxs have been shown to be quickly inactivated by sub-millimolar concentrations of both hydrogen peroxide and alkyl hydroperoxides [4, 25]. Yang et al. [26] reported that for human Prx1 at 100 μM hydrogen peroxide, the half-life for inactivation during catalytic cycling with reductant was ∼2 min. The inactivation is due to overoxidation of the CP side chain that occurs when a second peroxide substrate molecule attacks the Cys-SPOH, forming a dead-end sulfinic acid (Cys-SPO2H) (step 4 in Figure 1a). This reaction is in competition with the resolution step (step 2 in Figure 1a) of the normal catalytic cycle.
In contrast to these “sensitive” Prxs, the activity of a number of members of the typical 2-Cys Prx family from bacteria such as S. typhimurium AhpC, are robust, requiring about 100-fold higher hydrogen peroxide to be inactivated. Wood et al. [27] followed up this observation to draw two conclusions: first, that the sensitivity to inactivation by hydrogen peroxide correlated with two amino-acid sequence motifs – a GGLG-containing motif in the middle of the protein, and a YF-containing C-terminal extension (Figure 2a); and second, that the Prxs that conserved these two motifs (i.e. were putative sensitive Prxs) were from eukaryotic organisms. These or similar motifs are largely absent from bacterial Prx sequences, although potentially interesting exceptions exist3.
As described by Wood et al. [27], the YF motif is not part of the peroxidatic active site itself, but forms a helix that packs just above the active site in the fully folded form of the protein (Figure 2a). In contrast, this feature is missing in robust Prxs so that the peroxidatic active site region is much more open. The explanation for sensitivity runs as follows: the YF-containing C-terminal helix packs above the active site region like a cork in a bottle, limiting the active site dynamics and hindering it from unfolding; because of this, the local unfolding of the active site required for the resolution reaction (step 2 in Figure 1a) is a much rarer event, causing the Cys-SPOH containing active site to be longer-lived and thus more susceptible to attack by a second molecule of peroxide [27]. That the presence of the C-terminal helix is responsible for sensitivity has been confirmed by mutagenesis [30].
The reason for conservation of the GGLG motif is less clear, but it is speculated to be required for rescuing the overoxidized (sulfinic acid, Cys-SPO2H) form of the protein [31]. Although Cys-SPO2H formation was originally thought to be biologically irreversible, sulfiredoxins (Srxs) and possibly sestrins are able to reduce the Cys-SPO2H to Cys-SPOH in an ATP-dependent reaction [31]. The existence of the “resurrection” activity supports a physiological role for the overoxidized form of the protein. Recent structural studies of a Prx-Srx complex revealed a surprising C-terminal tail “embrace” [32] (Figure 2b). In this structure, the GGLG motif forms part of the ATP binding site and thus may have been selected for due to the reduction reaction with Srx [32].
That some Prxs are sensitive to overoxidation by their own substrates, making them worse peroxidases, raises the question of why a worse peroxidase would be maintained. In theory, the selective pressure to maintain sensitivity could be directly due to the importance of the sensitivity or it could be a pressure directed at conserving the C-terminal extension for another reason and the sensitivity is an unwanted byproduct. The existence of robust Prxs that otherwise conserve the active site and mechanistic details proves that the sensitivity is not an obligatory limitation related to the enzyme mechanism. Furthermore, because the sensitivity is not only avoidable, but could be very easily lost during evolution (simply through the mutation or loss of one or a few C-terminal residues) [28], its conservation throughout eukarya implies that there must be a very strong selective pressure to conserve it. Wood et al. proposed that the built-in sensitivity is important for facilitating non-stress related hydrogen peroxide signaling in eukaryotes [27], but this remains to be proven.
Stress and non-stress related peroxide signaling
Much evidence has accumulated that implicates hydrogen peroxide as an important and widespread signaling molecule [5, 9-11], both as an indicator of oxidative stress and as a part of normal cellular development. Whereas both of these processes involve signaling, some authors use “H2O2 signaling” to refer to just the second process, leading to some confusion. In this text, hydrogen peroxide signaling includes both and will be referred to as either stress (exogenous peroxide induced) or non-stress (endogenous peroxide) related. It is necessary to differentiate the two because the supporting evidence and pathways of the two processes are quite different.
All of the well-characterized pathways for hydrogen peroxide signaling describe stress related signaling, and the response triggered is generally the protective activation of a broad antioxidant response involving increased transcription of antioxidants and repair proteins. Well-characterized examples of stress related signaling include the OxyR and OhrR transcriptional regulators in prokaryotes, which act both as molecular sensors and as transducers of the H2O2 signal, and the Yap1/Gpx3 system in Saccharomyces cerevisiae [9, 10, 33]. In the latter system, yeast transcriptional responses to elevate protective antioxidant enzyme levels rely on communication of the H2O2 signal from a thiol-based peroxidase, the glutathione peroxidase-like Gpx3 (also known as Orp1), to a transcriptional regulator, Yap1, through thiol-disulfide interchange [34]. There is no controversy about the relevance of these events.
In contrast, the role of hydrogen peroxide in non-stress related signaling associated with endogenously generated hydrogen peroxide is still controversial. These mechanisms require that the hydrogen peroxide signal is generated in a regulated manner without a global change in the redox state of the cell. Long standing evidence for such signaling comes from studies showing that the exposure of cells to low levels of hydrogen peroxide stimulates proliferation. More recent evidence has shown that at least in mammals, tightly regulated NADPH oxidases (NOXs) become activated by hormones and produce superoxide that gets converted to hydrogen peroxide which in turn oxidizes specific cysteine residues in target proteins (such as protein tyrosine phosphatases) to influence the fate of the cell. These pathways can be blocked by increased catalase expression, supporting a role for hydrogen peroxide as a signaling molecule [9-11].
Despite the growing body of evidence for such non-stress related peroxide signaling, there is active debate about the physiological relevance of these putative signaling pathways. One of the major concerns is that many of the implicated target proteins, such as Prxs and phosphatases, appear to require peroxide concentrations in the 10 - 300 μM range in order to become (over)oxidized whereas in healthy cells the peroxide levels are not thought to exceed 700 nM [9]. As Stone and Yang summarize, alternative hypotheses to explain the discrepancy include (1) that there are yet unidentified, much more sensitive peroxide sensor proteins that transduce the signals, (2) that hydrogen peroxide itself is not the key signaling molecule but perhaps it is superoxide, peroxynitrite or a nitrosothiol, or (3) that the hydrogen peroxide buildup is highly localized [9].
Among these possibilities, new evidence suggests that subcellular localization (possibility 3) is indeed a key component of certain non-stress related peroxide signaling pathways. Early evidence supporting localization was published by Choi et al. [35] who demonstrated that Prx2 from mouse embryonic fibroblasts is recruited to the platelet-derived growth factor (PDGF) receptor in response to PDGF stimulation. This site specific recruitment of Prx2 was associated with the suppression of protein tyrosine phosphatase inactivation. Later, Li et al. [36] showed that specific recruitment of Nox2 to the endosome was required for redox dependent recruitment of TRAF6 to the active interleukin-1 (IL-1) receptor complex, ultimately leading to IL-1β-dependent NF-κB activation. Similarly, Nox localization has been implicated in vascular endothelial growth factor (VEGF) signaling in angiogenesis [37]. Most recently, Chen et al. [38] showed that for epidermal growth factor signaling, activated Nox4 is localized to the endoplasmic reticulum (ER) and is able to oxidatively inactivate ER-localized protein tyrosine phosphatase 1B (PTP1B), but not cytosolic PTP1B. Furthermore, ER-localized antioxidant enzymes were able to block the signal whereas untargeted counterparts were not. This last report provides powerful evidence that localization allows for levels of reactive oxygen species that can oxidize a less reactive target such as PTP1B.
What is the role of Prx sensitivity in peroxide signaling?
The use of hydrogen peroxide as a signaling molecule requires very tight regulation due to the damaging nature of peroxides. The high expression level (0.1 – 1% of total soluble protein) and ubiquitous distribution [1] would make Prxs one of the first proteins that a hydrogen peroxide molecule would encounter. This combined with their fast reactivity (∼107 M−1s−1) implies that in mammalian cells, 10,000 times more hydrogen peroxide would react with Prx than with glutathione [11]. In characterized systems, the most highly expressed Prxs are sensitive: mammalian mRNA levels suggest that this is generally Prx1 [39], and in yeast it is Tpx1 [40]4. The importance of Prx1 expression for controlled growth in mammals is demonstrated by the high rate of malignant cancers in the Prx1 knockout mouse [41].
So what is the role of sensitivity? In principle, there are two possibilities – overoxidation could cause a gain in function and/or overoxidation could cause a loss of function. In either case, the Prx could act as a molecular switch, influenced by a change in peroxide level whether for stress or non-stress related signaling. It is worth noting that phosphorylation, nitrosylation and C-terminal cleavage can also modulate Prx activity and sensitivity and contribute to the regulation of cell signaling [5]. Three models describing a role for sensitivity in signal transduction have been proposed. Two models rely on a gain of function mechanism: (1) disulfide exchange with other downstream sensor proteins and (2) chaperone activity. Both of these models are supported by evidence derived from stress related signaling pathways. The only loss of function paradigm is (3) the floodgate model, proposed to be involved in non-stress related signaling. Figure 3 summarizes these three models and the role of sensitivity in each as is described in the following three paragraphs.
Figure 3.
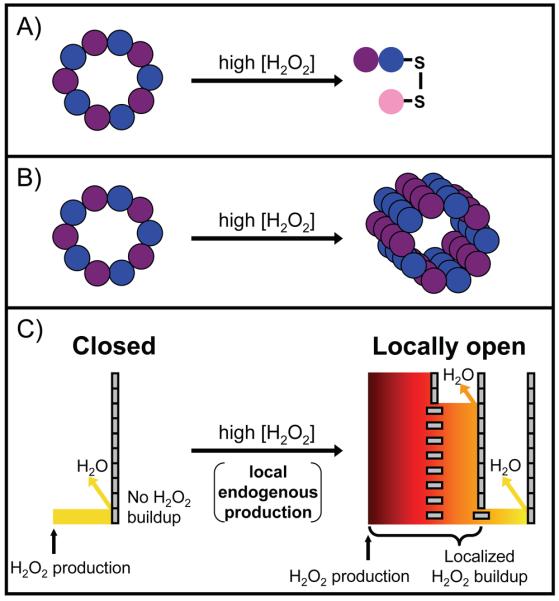
Three proposed roles for Prxs in peroxide signaling. In each case, different high levels of hydrogen peroxide cause a shift in function from peroxidase activity. (A) Disulfide exchange, represented by an interprotein disulfide bond between Prx and a downstream protein (pink). In the one case studied, signaling is not stress related and does not require sensitivity. (B) The chaperone model, represented by the formation of higher order oligomers of overoxidized Prxs. This is involved in stress related signaling and requires sensitivity. In (A) and (B), the Prxs are represented as a purple and blue decamer under normal cellular conditions. (C) The floodgate model is an unproven mechanism. The Prxs are represented as tall barriers made up of gray rectangles – vertical for active, horizontal for overoxidized and inactive. The multiple barriers on the right reflect the cell-wide Prx distribution; Prxs that are close to the peroxide generation site (marked by an arrow) are overwhelmed and inactivated, whereas those at increasing distances away are not. This creates a steep peroxide gradient and allows for localized peroxide buildup after endogenous peroxide generation. The level of hydrogen peroxide is represented by both color gradient and height. This may be involved in both stress and non-stress related signaling and requires sensitivity.
In the disulfide exchange model for peroxide signaling (Figure 3a), Prx acts as a specific transducer of the peroxide signal by forming an intermolecular disulfide bond with a partner protein, much like the Gpx3/Yap1 system mentioned earlier. Although in theory the target protein may itself continue to transmit the signal through continued disulfide exchange with downstream proteins, this has not been seen in the two examples of intermolecular disulfide bond formation observed in stress related signaling. Vivancos et al. [42] showed that at low levels of hydrogen peroxide, the Schizosaccharomyces pombe Prx, Tpx1, activates the transcription factor Pap1 through intermolecular disulfide bond formation. At higher hydrogen peroxide levels, Pap1 is not activated but instead the transcriptional factor Sty1 is activated through an intermolecular disulfide bond with Tpx1 [25]. Activation of Sty1 leads to the transcription of antioxidant defense proteins and Srx. While early evidence suggested that the regulation of these two pathways required sensitivity, mutants with C-terminal truncations that render Tpx1 insensitive to overoxidation were fully functional in these two pathways [43].
In the second gain of function model, Prxs have been shown to act as chaperones or cell cycle regulators when the overoxidized enzyme aggregates into larger assemblies [44, 45] (Figure 3b). Higher order molecular weight oligomers have been shown to have chaperone activity in Helicobacter pylori, Saccharomyces cerevisiae, and Homo sapiens [29, 46, 47], an activity which could protect cells from oxidation induced protein unfolding. Interestingly, chaperone activity appears to be higher for H. sapiens Prx1 than Prx2 because an additional cysteine present in Prx1 forms a disulfide to stabilize the higher order aggregates [48]. Consistent with the proposed requirement for sensitivity, C-terminal truncation mutants of H. sapiens Prx2 do not respond to oxidative stress with increasing levels of chaperone activity [47]. While sensitivity is important for Prx chaperone activity in S. cerevisiae and H. sapiens, the case for H. pylori AhpC is unclear as studies documenting the sensitivity of H. pylori AhpC have not been published.
While direct evidence exists supporting both of the gain of function models for stress related signaling, the loss of function model for Prxs in non-stress signaling is still largely speculative. This mechanism requires sensitivity. The “floodgate model” [27] predicts that under normal conditions Prxs act as a barrier hindering peroxide encounters with sensitive cellular components (Figure 3c). In the presence of a high peroxide pulse, such as could be produced by hormone-triggered activation of cellular NOXs, the rapid production of hydrogen peroxide causes high local peroxide concentrations which would inactivate the proximal floodgate Prxs, allowing peroxide to locally build up to concentrations that can oxidize specific downstream target proteins. The recent demonstrations of localized NOX signaling are consistent with this model but do not prove that Prxs play this role. As seen in Figure 3c, the high Prx concentration throughout the cell implies that the term floodgate is somewhat of a misnomer as sensitive Prxs will actually not allow peroxide to spread throughout the cell but will instead localize the peroxide buildup. Thus Prxs are more like an adjustable buffer than a floodgate. The recent evidence that non-stress related growth factor signaling involves localized peroxide buildup of a sufficient concentration to oxidize PTP1B implies that the nearby sensitive Prxs would also be overoxidized [38]. Operation of a Prx floodgate in such systems may be challenging to discern due to the difficulty in detecting a small overoxidized population located close to the peroxide source within the large cellular pool of Prxs [10].
Hormone-triggered apoptosis is, in contrast, a signaling process that appears to involve more global Prx overoxidation. This was shown years ago for signaling by tumor necrosis factor [49]. More recently, a report of apoptotic signaling in dopaminergic neurons (used as an experimental model of Parkinson's Disease) shows that 6-hydroxydopamine activation of p38 MAP kinase and caspase-3 was associated with significant overoxidation of Prx1 and other Prxs [50]. The cells were protected against elevated levels of reactive oxygen species and apoptotic death by overexpression of Prx1 or addition of other antioxidants, and displayed enhanced apoptosis when Prx1 expression was knocked down. Whether or not these two cases can truly be considered as “non-stress related redox signaling” is debatable because the extensive overoxidation of Prxs implies that apoptotic signaling is accompanied by rises in reactive oxygen species that are much higher than in other types of signaling.
With the increasing attention given to Prxs, we are learning that they are efficient catalysts and that their catalytic repertoire is broader than that of a simple peroxidase. The high level of expression of sensitive Prxs in eukaryotes allows them to carry out functions under conditions of oxidative stress, such as that of a chaperone, that depend on their high concentration rather than their peroxidase activity. Nevertheless, why the sensitivity of the highly expressed eukaryotic Prxs is so strongly conserved and why repair systems have evolved for recovering activity in the overoxidized Prxs are still open questions. Some answers may come from the study of a Prx1 knockout mouse with a robust Prx gene knockin that mimics the expression pattern of the missing sensitive Prxs. The local nature of non-stress related redox signaling has made it challenging to ferret out the mechanisms involved and to discern whether the overoxidized Prxs act primarily as a passive floodgate/buffer or as an active positive signal or both.
Acknowledgements
The authors thank Todd Lowther, Kim Nelson and Derek Parsonage for their editorial suggestions. This publication was made possible in part by a grant from the National Institute of General Medical Sciences to L.B.P. with a subcontract to P.A.K. (RO1 GM50389), and by a grant from the National Institute of Environmental Health Sciences (P30 ES00210).
Footnotes
Abbreviations: Prx, peroxidredoxin; CP, peroxidatic cysteine; CR, resolving cysteine; 1-Cys, Prx with CR coming from another molecule; 2-Cys, Prx with CR coming from a Prx; Cys-SPOH, peroxidatic cysteine sulfenic acid; Cys-CPO2H, peroxidatic cysteine sulfinic acid; Srx, sulfiredoxin; NOXs, NADPH oxidases; VEGF, vascular endothelial growth factor; PDGF, platelet-derived growth factor; PTP1B, protein tyrosine phosphatase 1B.
In addition to decamers, octamers and dodecamers have also been observed. The role of oligomerization is thought to be the same in all three cases [6].
Among the exceptions to the distribution of Prxs with the GGLG and YF motifs within eukaryotes only, some parasitic bacteria (from H. pylori, Y. pestis, C. pneumoniae) have Prxs with a GGIG motif and a C-terminal extension containing a YL motif. The sensitivity of Prxs to overoxidation with these and other variations on the GGLG and YF motifs found within both prokayotes and eukaryotes has not been fully characterized, although it is clear that point mutations within the C-terminus of sensitive Prxs are sufficient to disrupt the C-terminal helix packing [28]. The presence of these potentially sensitive Prxs in parasitic bacteria may be due to horizontal gene transfers [29], and so does not necessarily break from the expected limited distribution of sensitive Prxs to eukaryotes.
Of the five Prxs expressed in Saccharomyces cerevisiae, three are present in relatively low abundance (cTpxII, mTpx and nTpx, each at less than 5,000 molecules per cell), while cTpxI and cTpxIII are present at 378,000 and 162,000 molecules per cell, respectively. cTpxI and cTpxII are sensitive Prxs. Expression levels were estimated using GFP-fusion proteins [40].
References
- 1.Wood ZA, Schröder E, Harris JR, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 2.Copley SD, Novak WR, Babbitt PC. Divergence of function in the thioredoxin fold suprafamily: evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry. 2004;43:13981–13995. doi: 10.1021/bi048947r. [DOI] [PubMed] [Google Scholar]
- 3.Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J Biol Chem. 1994;269:27670–27678. [PubMed] [Google Scholar]
- 5.Fourquet S, Huang ME, D'Autreaux B, Toledano MB. The dual functions of thiol-based peroxidases in H2O2 scavenging and signaling. Antioxid Redox Signal. 2008;10:1565–1576. doi: 10.1089/ars.2008.2049. [DOI] [PubMed] [Google Scholar]
- 6.Karplus PA, Hall A. Structural Survey of the Peroxiredoxins. In: Flohé L, Harris JR, editors. Peroxiredoxin Systems. Springer; New York: 2007. pp. 41–60. [DOI] [PubMed] [Google Scholar]
- 7.Poole LB. The Catalytic Mechanism of Peroxiredoxins. In: Flohé L, Harris JR, editors. Peroxiredoxin Systems. Springer; New York: 2007. pp. 61–81. [Google Scholar]
- 8.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 9.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 10.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 12.Ogusucu R, Rettori D, Munhoz DC, Soares Netto LE, Augusto O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic Biol Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 13.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 14.Nelson KJ, Parsonage D, Hall A, Karplus PA, Poole LB. Cysteine pKa Values for the Bacterial Peroxiredoxin AhpC. Biochemistry. 2008;47:12860–12868. doi: 10.1021/bi801718d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 16.Barranco-Medina S, Kakorin S, Lazaro JJ, Dietz KJ. Thermodynamics of the dimer-decamer transition of reduced human and plant 2-cys peroxiredoxin. Biochemistry. 2008;47:7196–7204. doi: 10.1021/bi8002956. [DOI] [PubMed] [Google Scholar]
- 17.Cao Z, Bhella D, Lindsay JG. Reconstitution of the mitochondrial PrxIII antioxidant defence pathway: general properties and factors affecting PrxIII activity and oligomeric state. J Mol Biol. 2007;372:1022–1033. doi: 10.1016/j.jmb.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 18.Matsumura T, Okamoto K, Iwahara S, Hori H, Takahashi Y, Nishino T, Abe Y. Dimer-oligomer interconversion of wild-type and mutant rat 2-Cys peroxiredoxin: disulfide formation at dimer-dimer interfaces is not essential for decamerization. J Biol Chem. 2008;283:284–293. doi: 10.1074/jbc.M705753200. [DOI] [PubMed] [Google Scholar]
- 19.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schröder E, Littlechild JA, Lebedev AA, Errington N, Vagin AA, Isupov MN. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure. 2000;8:605–615. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- 21.Cha MK, Yun CH, Kim IH. Interaction of human thiol-specific antioxidant protein 1 with erythrocyte plasma membrane. Biochemistry. 2000;39:6944–6950. doi: 10.1021/bi000034j. [DOI] [PubMed] [Google Scholar]
- 22.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci U S A. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dietz KJ, Jacob S, Oelze ML, Laxa M, Tognetti V, de Miranda SM, Baier M, Finkemeier I. The function of peroxiredoxins in plant organelle redox metabolism. J Exp Bot. 2006;57:1697–1709. doi: 10.1093/jxb/erj160. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto Y, Ritz D, Planson AG, Jonsson TJ, Faulkner MJ, Boyd D, Beckwith J, Poole LB. Mutant AhpC peroxiredoxins suppress thiol-disulfide redox deficiencies and acquire deglutathionylating activity. Mol Cell. 2008;29:36–45. doi: 10.1016/j.molcel.2007.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veal EA, Findlay VJ, Day AM, Bozonet SM, Evans JM, Quinn J, Morgan BA. A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol Cell. 2004;15:129–139. doi: 10.1016/j.molcel.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 26.Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, Rhee SG. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem. 2002;277:38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 27.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 28.Koo KH, Lee S, Jeong SY, Kim ET, Kim HJ, Song K, Chae H-Z. Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch Biochem Biophys. 2002;397:312–318. doi: 10.1006/abbi.2001.2700. [DOI] [PubMed] [Google Scholar]
- 29.Chuang MH, Wu MS, Lo WL, Lin JT, Wong CH, Chiou SH. The antioxidant protein alkylhydroperoxide reductase of Helicobacter pylori switches from a peroxide reductase to a molecular chaperone function. Proc Natl Acad Sci U S A. 2006;103:2552–2557. doi: 10.1073/pnas.0510770103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sayed AA, Williams DL. Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J Biol Chem. 2004;279:26159–26166. doi: 10.1074/jbc.M401748200. [DOI] [PubMed] [Google Scholar]
- 31.Jönsson TJ, Lowther WT. The peroxiredoxin repair proteins. In: Flohé L, Harris JR, editors. Peroxiredoxin Systems. Springer; New York: 2007. pp. 115–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jönsson TJ, Johnson LC, Lowther WT. Structure of the sulphiredoxin-peroxiredoxin complex reveals an essential repair embrace. Nature. 2008;451:98–101. doi: 10.1038/nature06415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 34.Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 35.Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 36.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 38.Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF., Jr. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leyens G, Donnay I, Knoops B. Cloning of bovine peroxiredoxins-gene expression in bovine tissues and amino acid sequence comparison with rat, mouse and primate peroxiredoxins. Comp Biochem Physiol B Biochem Mol Biol. 2003;136:943–955. doi: 10.1016/s1096-4959(03)00290-2. [DOI] [PubMed] [Google Scholar]
- 40.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 41.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 42.Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci U S A. 2005;102:8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jara M, Vivancos AP, Hidalgo E. C-terminal truncation of the peroxiredoxin Tpx1 decreases its sensitivity for hydrogen peroxide without compromising its role in signal transduction. Genes Cells. 2008;13:171–179. doi: 10.1111/j.1365-2443.2007.01160.x. [DOI] [PubMed] [Google Scholar]
- 44.Phalen TJ, Weirather K, Deming PB, Anathy V, Howe AK, van der Vliet A, Jönsson TJ, Poole LB, Heintz NH. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trotter EW, Rand JD, Vickerstaff J, Grant CM. The yeast Tsa1 peroxiredoxin is a ribosome-associated antioxidant. Biochem J. 2008;412:73–80. doi: 10.1042/BJ20071634. [DOI] [PubMed] [Google Scholar]
- 46.Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW, Choi YO, Kim WY, Kang JS, Cheong GW, Yun DJ, Rhee SG, Cho MJ, Lee SY. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Moon JC, Hah YS, Kim WY, Jung BG, Jang HH, Lee JR, Kim SY, Lee YM, Jeon MG, Kim CW, Cho MJ, Lee SY. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem. 2005;280:28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- 48.Lee W, Choi KS, Riddell J, Ip C, Ghosh D, Park JH, Park YM. Human peroxiredoxin 1 and 2 are not duplicate proteins: the unique presence of CYS83 in Prx1 underscores the structural and functional differences between Prx1 and Prx2. J Biol Chem. 2007;282:22011–22022. doi: 10.1074/jbc.M610330200. [DOI] [PubMed] [Google Scholar]
- 49.Rabilloud T, Heller M, Gasnier F, Luche S, Rey C, Aebersold R, Benahmed M, Louisot P, Lunardi J. Proteomics analysis of cellular response to oxidative stress. Evidence for in vivo overoxidation of peroxiredoxins at their active site. J Biol Chem. 2002;277:19396–19401. doi: 10.1074/jbc.M106585200. [DOI] [PubMed] [Google Scholar]
- 50.Lee YM, Park SH, Shin DI, Hwang JY, Park B, Park YJ, Lee TH, Chae HZ, Jin BK, Oh TH, Oh YJ. Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J Biol Chem. 2008;283:9986–9998. doi: 10.1074/jbc.M800426200. [DOI] [PubMed] [Google Scholar]