

Abstract
Using 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-one and 4,5-dihydroxy-1H-isoindole-1,3(2H)-dione based HIV-1 integrase inhibitors as display platforms, we undertook a thorough examination of the effects of modifying the halogen substituents on a key benzyl ring that is hypothesized to bind in a hydrophobic pocket of the integrase•DNA complex. Data from this study suggest that in general dihalo – substituted analogues have higher potency than monohalo – substituted compounds, but that further addition of halogens is not beneficial.
Integrase (IN) is a key enzyme in the life cycle of human immunodeficiency virus type 1 (HIV-1), the causative agent of acquired immunodeficiency syndrome (AIDS). Approximately 30 drugs that have been approved by the FDA for treatment of HIV-1 infection,1 Raltegravir [Merck & Co., Inc. (MK-0518)]2 is the most recently approved drug (October 2007) and the only integrase inhibitor. Another IN inhibitor, Elvitegravir, [Gilead Sciences, Inc. (GS-9137)]3 is currently undergoing phase III clinical trials in HIV-1-infected patients. These two inhibitors show high potency against IN – catalyzed “strand transfer” (ST) reactions, while being less effective against the IN 3′ –processing (3′-P) step.4 This combination of characteristics is characteristic of a broad range of IN inhibitors, many of which contain the elements typified by General Structure I (Figure 1). A key component of these inhibitors is an array of heteroatoms that are hypothesized to chelate two divalent metal ions associated with catalytically essential IN residues Asp64, Asp116 and Glu152 (“DDE” motif).5 Aromatic functionality, frequently in the form of a benzyl group linked to the chelating portion of the inhibitor, can make a significant contribution to overall binding affinity. The empirical observation that halogen substituents on this aryl ring can enhance potency has led to the hypothesis that this aryl ring may bind in one or more hydrophobic pockets of the IN•DNA complex.6,7
Figure 1.

Hypothetical IN binding of a metal chelating inhibitor showing halogen substituent patterns on a key benzylamide group.
A diversity of halogen-substituted benzyl groups has been introduced into IN inhibitors (Figure 1).8–16 We recently reported a series of 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones (general structure A, Table 1) that exhibit potent ST inhibition in extracellular assays and antiviral efficacies in HIV-1 infected cells.14 The closely related 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (general structure B, Table 1) represent phthalimide –based analogues that can be prepared by the chemistry described in our earlier report.14 For series A we had observed an enhancement of up to two orders of magnitude in potency depending on the halogen substitution of a key benzyl ring.14 Numerous reports of enhancing effects of halogens across a wide range of IN inhibitors prompted us to undertake a thorough investigation of the effects of halogen substituents on IN inhibitory potency. Here we compare the effects of varying halogen substitutions on an isolated benzyl ring in related compounds with similar scaffolds. The structural simplicity of A and B, suggested that they could serve as useful platforms for this study. During the preparation of this manuscript a report was published describing the effects of phenyl ring fluorine substituents on the inhibitory potencies of 1H-benzylindole IN inhibitors.17
Table 1.
General structures of inhibitors and in vitro IN inhibitory potencies for mono-halogen substituted analogues.
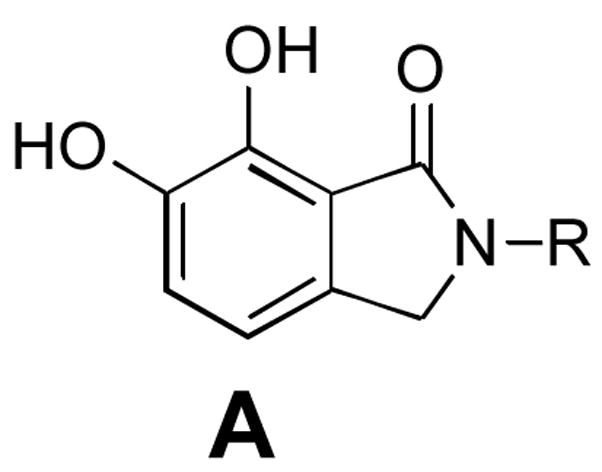 |
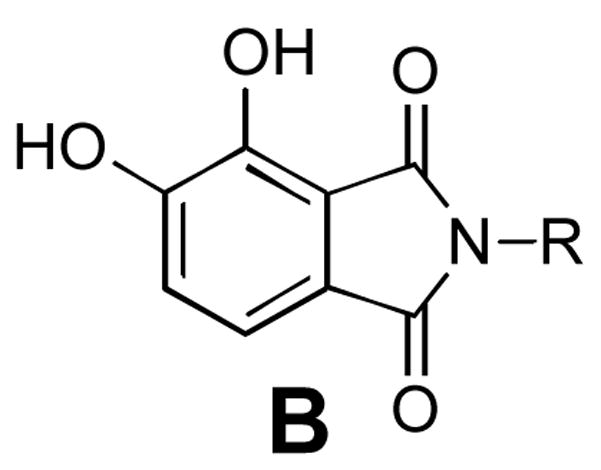 |
||||
|---|---|---|---|---|---|
| No. | R | IC50 (μM) | IC50 (μM) | ||
| 3′-P | ST | 3′-P | ST | ||
| 1 | 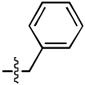 |
>333a | 12 ± 6a | 72 ± 23 | 0.4 ± 0.2 |
| 2 | 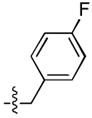 |
282 ± 41a | 10 ± 4a | 8 ± 3 | 5 ± 2 |
| 3 | 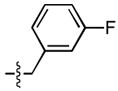 |
238 ± 1 | 21 ± 2 | 11 ± 3 | 0.3 ± 0.2 |
| 4 | 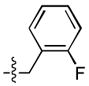 |
>333 | 25 ± 5 | 68 ± 3 | 2 ± 1 |
| 5 | 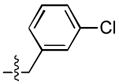 |
72 ± 34 | 4 ± 1 | 14 ± 3 | 0.4 ± 0.1 |
| 6 | 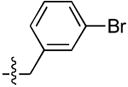 |
264 ± 98 | 3 ± 1 | 16 ± 1 | 0.4 ± 0.1 |
| 7 | 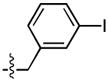 |
169 ± 68 | 4 ± 3 | 85 ± 35 | 1 ± 0.4 |
Reported in reference 14.
We prepared analogues by procedures similar to those previously reported.14 Adding a second carbonyl to the lactam ring of series A to yield the phthalimide – based series B enhanced inhibitory potency against both 3′–P and ST reactions. In vitro IN assays were performed in the presence of Mg+2 as described.14 Introduction of a 4-F substitutent (2) had little effect on the potencies of the series A compounds, while in series B this resulted in an order of magnitude change in inhibitory potencies. There was an enhancement in the inhibition of 3′ – P and loss of efficacy against ST. Moving the fluorine from the 4 – to the 3 – position (3) gave a slight decrease in ST inhibitory potency in series A and an order of magnitude enhancement of ST inhibition in series B. Finally, going from the 3 – F to the 2 – F isomer (4) had essentially no effect in potency on series A, while a significant drop in both 3′ – P and ST inhibitory potencies was observed in series B. In series A Cl (5), Br (6) or I (7) substituents at the 3 – position gave slightly higher ST inhibitory potencies relative to the 3 – F analogue (3), while in series B the potencies of the 3 – Cl and 3 – Br analogues were unchanged relative to the 3 – F inhibitor, and the 3 – I compound had reduced inhibitory potency (Table 1).
Results of an exhaustive examination of difluoro substituents are shown in Table 2. The 3,4 – difluoro and 2,4 – difluoro analogues (8 and 12, respectively) were significantly more potent ST inhibitors than the mono – substituted 4 – F compounds. The 3,5 – isomer (9A) showed the greatest ST – selectivity (700 – fold) among all fluoro compounds tested. The 2,6 – difluoro isomers (13) were among the least potent of both series A and B, with only the 2,5 – difluoro analogue (10) in series A showing lower ST inhibitory potency.
Table 2.
Examination of difluoro analogues in an in vitro IN assay.
| No. | R | IC50(μM) | IC50(μM) | ||
|---|---|---|---|---|---|
| 3′-P | ST | 3′-P | ST | ||
| Aa | Ba | ||||
| 8 | 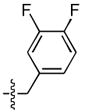 |
72 ± 3 | 0.9 ± 0.4 | 27 ± 1 | 0.1 ± 0.03 |
| 9 | 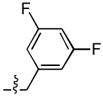 |
233 ± 12 | 0.3 ± 0.1 | 48 ± 4 | 0.4 ± 0.1 |
| 10 | 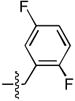 |
>333 | 27 ± 4 | 60 ± 7 | 0.23 ± 0.18 |
| 11 | 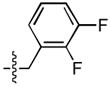 |
72 ± 5 | 0.7 ± 0.2 | 24 ± 8 | 0.2 ± 0.1 |
| 12 | 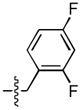 |
37 ± 2 | 0.5 ± 0.1 | 16 ± 4 | 0.3 ± 0.1 |
| 13 | 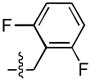 |
71 ± 2 | 20 ± 2 | 103 ± 1 | 2 ± 1 |
Structures A and B are as shown in Table 1.
The effect of replacing one or both fluorines with chloro substituents was examined (Table 3). In series A, the 3 – Cl, 4 – F analogue (14) was approximately four fold more potent than the 3,4 – difluoro containing compound (8), while in series B there was little difference between the two inhibitors. The corresponding 3 – Br, 4 – F and 3 – methyl, 4 – F compounds (15 and 16, respectively) had reduced ST inhibitory potencies in both series A and B. Reversing the substitution pattern to 3 – F, 4 – Cl (17) resulted in a loss of inhibitory potency, while replacing the 4 – F with a chloro to give the 3,4 – dichloro analogues (18) provided ST inhibitory potencies that were equivalent to the 3 – Cl, 4 – F analogues and gave the highest ST –selectivity in the A series (800 – fold). This indicated the importance of having a chloro substituent at the 3 –position, with the choice of halogen at the 4 – position being less critical. Maintaining a chloro substituent at the 3 – position, while placing the fluoro group at the 2 – or 6 – position (21 and 20 respectively), resulted in reduced ST inhibitory potency. With the chloro substituent at the 2 – position, placement of a fluoro group at the 4 – or 6 – position (19 and 22, respectively) reduced ST inhibitory potency.
Table 3.
Examination of disubstituted analogues in an in vitro IN assay.
| No. | R | IC50 (μM) | IC50 (μM) | ||
|---|---|---|---|---|---|
| 3′-P | ST | 3′-P | ST | ||
| Aa | Ba | ||||
| 14 | 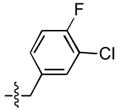 |
13 ± 3b | 0.16 ± 0.08b | 14 ± 3 | 0.17 ± 0.06 |
| 15 | 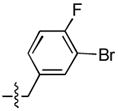 |
34 ± 5 | 1 ± 0.1 | 11 ± 1 | 0.9 ± 0.3 |
| 16 | 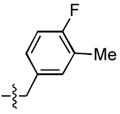 |
17 ± 2 | 0.9 ± 0.1 | 11 ± 1 | 0.8 ± 0.2 |
| 17 | 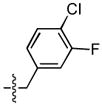 |
97 ± 26 | 6 ± 3 | 58 ± 20 | 0.9 ± 0.4 |
| 18 | 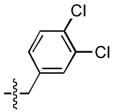 |
89 ± 15 | 0.1 ± 0.02 | 18 ± 4 | 0.12 ± 0.02 |
| 19 | 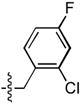 |
>333 | 2.5 ± 1.2 | 90 ± 26 | 1.3 ± 0.2 |
| 20 | 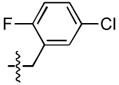 |
18 ± 1 | 1.2 ± 0.4 | 9 ± 1 | 0.31 ± 0.06 |
| 21 | 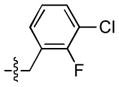 |
36 ± 17 | 2 ± 1 | 25 ± 16 | 2 ± 1 |
| 22 | 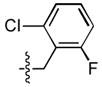 |
>333 | 18 ± 1 | 54 ± 9 | 3.5 ± 1.4 |
Finally, a series of tri – halo substituted analogues (23–28) and one pentafluoro analogue (29) were prepared (Table 4). In general, the additional halogens did not enhance inhibitory potency. Rather, as in the case of 28, the addition of a 6 – F to the 3 – Cl, 4 – F substituted analogue (14) resulted a 27 – fold reduction of ST inhibitory potency in series A series, whereas the ST inhibitory potency was only marginally reduced in series B. On the other hand, the addition of a 4 – F to the 3,5 – difluoro analogue (9) to give 23 was accompanied by an order of magnitude reduction in ST inhibitory potency as well as by a 30 – fold decrease in ST – selectivity in the A series. The same compound in sereis B was almost unaffected by this modification.
Table 4.
Examination of polyhalogen substituted analogues in an in vitro IN assay.
| No. | R | IC50 (μM) | IC50 (μM) | ||
|---|---|---|---|---|---|
| 3′-P | ST | 3′-P | ST | ||
| Aa | Ba | ||||
| 23 | 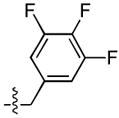 |
99 ± 8 | 4 ± 1 | 10 ± 2 | 0.6 ± 0.2 |
| 24 | 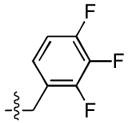 |
153 ± 4 | 2.7 ± 0.9 | 18 ± 2 | 0.8 ± 0.2 |
| 25 | 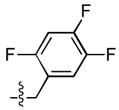 |
>333 | 3.7 ± 1.2 | 54 ± 4 | 0.5 ± 0.1 |
| 26 | 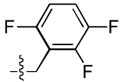 |
>333 | 25 ± 6 | 158 ± 16 | 1.8 ± 0.4 |
| 27 | 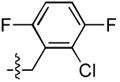 |
>333 | 8.9 ± 2.5 | 34 ± 2 | 4 ± 0.9 |
| 28 | 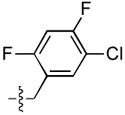 |
>333 | 4.4 ± 2.1 | 79 ± 31 | 0.4 ± 0.2 |
| 29 | 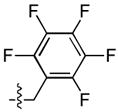 |
230 ± 19 | 8 ± 3 | 41 ± 7 | 1.3 ± 0.5 |
Structures A and B are as shown in Table 1.
The antiviral efficacy of a subset of these inhibitors was examined using HIV-1 vectors in cultured cells as previously reported (Table 5).14 Antiviral potencies for series A compounds were better than for series B compounds. Submicromolar antiviral efficacies were observed for the 3 – Cl and 3 – Br substituted analogues (5A and 6A, respectively); the 3 – Cl, 4 – F (14A) and 2 – F, 3 – Cl (21A) and the 3 – Cl, 3,6 – difluoro (28A) analogues. A common feature of these inhibitors is the presence of a chloro or bromo substituent at the 3 –position. However, the relatively high cytotoxicity of these compounds means that they have relatively unfavorable therapeutic indexes as indicated by the ratio of the half-maximal cytotoxic concentration (CC50) to the half maximal effective concentration (EC50)
Table 5.
Antiviral potencies of selected inhibitors in HIV-1 infected cells.a
| No | EC50(μM) | CC50(μM) | No | EC50(μM) | CC50(μM) |
|---|---|---|---|---|---|
| 1A | 0.68b | ND | 1B | 11.2 | ND |
| 2A | 0.77b | ND | 2B | 0.86 | ND |
| 3A | 1.06 | 8.60 | 3B | – | – |
| 5A | 0.54 | 9.02 | 5B | 4.20 | 13.29 |
| 6A | 0.59 | 7.24 | 6B | 3.78 | 11.36 |
| 7A | 1.07 | 6.92 | 7B | 7.77 | 11.99 |
| 8A | 1.08 | 9.01 | 8B | 5.88 | 7.94 |
| 9A | 2.42 | 17.15 | 9B | – | – |
| 10A | 1.24 | 13.44 | 10B | – | – |
| 14A | 0.48 | 6.87 | 14B | 2.83 | 9.54 |
| 18A | 3.30 | 5.68 | 18B | 2.33 | 4.43 |
| 21A | 0.62 | 9.43 | 21B | 2.74 | 11.49 |
| 28A | 0.65 | 8.38 | 28B | 4.43 | 13.74 |
Our results are consistent with selected previous reports. For example, Gilead’s clinical candidate (GS-9137)3 contains a 3 – Cl, 2 – F substituted benzyl group. However, in our series, maintaining a chloro substituent at the 3 – position, while placing the fluoro group at the 2 – or 6 – position resulted in reduced inhibitory potency for the ST reaction. The recent report that for fluorobenzyl-1H-benzylindoles, ortho - fluoro substitution enhanced the potency,17 suggests that subtle differences in the binding orientation of the benzyl ring may be important. Despite progress in development of potent IN inhibitors, cytotoxicity remains a problem that might be addressed by increasing target specificity through appropriate choice of peripheral substituents.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Intstitute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.See: http://www.fda.gov/oashi/aids/virals.html for a list of FDA approved drugs for the treatment of HIV infection.
- 2.Evering TH, Markowitz M. Expert Opin Invest Drugs. 2008;17:413. doi: 10.1517/13543784.17.3.413. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K, Ikeda S, Kodama E, Matsuoka M, Shinkai H. J Med Chem. 2006;49:1506. doi: 10.1021/jm0600139. [DOI] [PubMed] [Google Scholar]
- 4.Marinello J, Marchand C, Mott BT, Bain A, Thomas CJ, Pommier Y. Biochemistry. 2008;47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johns BA, Svolto AC. Expert Opin Ther Pat. 2008;18:1225. [Google Scholar]
- 6.Jin H, Cai RZ, Schacherer L, Jabri S, Tsiang M, Fardis M, Chen X, Chen JM, Kim CU. Bioorg Med Chem Lett. 2006;16:3989. doi: 10.1016/j.bmcl.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Tsiang M, Yu F, Hung M, Jones GS, Zeynalzadegan A, Qi X, Jin H, Kim CU, Swaminathan S, Chen JM. J Mol Biol. 2008;380:504. doi: 10.1016/j.jmb.2008.04.054. [DOI] [PubMed] [Google Scholar]
- 8.Walker MA, Johnson T, Naidu BN, Banville J, Remillard R, Plamondon S, Martel A, Li C, Torri A, Samanta H, Lin Z, Dicker I, Krystal M, Meanwell NA. Bioorg Med Chem Lett. 2007;17:4886. doi: 10.1016/j.bmcl.2007.06.042. [DOI] [PubMed] [Google Scholar]
- 9.Petrocchi A, Koch U, Matassa VG, Pacini B, Stillmock KA, Summa V. Bioorg Med Chem Lett. 2007;17:350. doi: 10.1016/j.bmcl.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 10.Gardelli C, Nizi E, Muraglia E, Crescenzi B, Ferrara M, Orvieto F, Pace P, Pescatore G, Poma M, del Rosario Rico Ferreira M, Scarpelli R, Homnick CF, Ikemoto N, Alfieri A, Verdirame M, Bonelli F, Gonzalez Paz O, Taliani M, Monteagudo E, Pesci S, Laufer R, Felock P, Stillmock KA, Hazuda D, Rowley M, Summa V. J Med Chem. 2007;50:4953. doi: 10.1021/jm0704705. [DOI] [PubMed] [Google Scholar]
- 11.Wai JS, Kim B, Fisher TE, Zhuang L, Embrey MW, Williams PD, Staas DD, Culberson C, Lyle TA, Vacca JP, Hazuda DJ, Felock PJ, Schleif WA, Gabryelski LJ, Jin L, Chen IW, Ellis JD, Mallai R, Young SD. Bioorg Med Chem Lett. 2007;17:5595. doi: 10.1016/j.bmcl.2007.07.092. [DOI] [PubMed] [Google Scholar]
- 12.Verschueren WG, Dierynck I, Amssoms KIE, Hu L, Boonants PMJG, Pille GME, Daeyaert FFD, Hertogs K, Surleraux DLNG, Wigerinck PBTP. J Med Chem. 2005;48:1930. doi: 10.1021/jm049559q. [DOI] [PubMed] [Google Scholar]
- 13.Fisher TE, Kim B, Staas DD, Lyle TA, Young SD, Vacca JP, Zrada MM, Hazuda DJ, Felock PJ, Schleif WA, Gabryelski LJ, Anari MR, Kochansky CJ, Wai JS. Bioorg Med Chem Lett. 2007;17:6511. doi: 10.1016/j.bmcl.2007.09.086. [DOI] [PubMed] [Google Scholar]
- 14.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr J Med Chem. 2008;51:251. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 15.Di Francesco ME, Pace P, Fiore F, Naimo F, Bonelli F, Rowley M, Summa V. Bioorg Med Chem Lett. 2008;18:2709. doi: 10.1016/j.bmcl.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. J Med Chem. 2008;51:5843. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 17.Ferro S, De Luca L, Barreca Maria L, Iraci N, De Grazia S, Christ F, Witvrouw M, Debyser Z, Chimirri A. J Med Chem. 2009;52:569. doi: 10.1021/jm8009266. [DOI] [PubMed] [Google Scholar]