

Abstract
Using our recently disclosed 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-one and 4,5-dihydroxy-1H-isoindole-1,3(2H)-dione integrase inhibitors, we report differential effects on inhibitory potency induced by introduction of an α-chiral center into a key aryl substitutent. We show that introduction of the chiral center is uniformly deleterious to binding, with the (R)-enantiomer being more deleterious than the (S)-enantiomer. A greater enantiomeric difference in potency is shown by inhibitors that have restricted rotation of the aryl ring, with the larger difference being due to poorer potency of the (R)-enantiomer rather than higher potency of the (S)-enantiomer. The potency difference for enantiomers based on the isoindoline-1,3-dione ring system is less than for those derived from the isoindol-1-one ring system. Our findings provide useful information that should aid in understanding molecular binding interactions of DKA – derived IN inhibitors.
Keywords: HIV-1 integrase, inhibit, enantiomer, arylamide, diketoacid
1. Introduction
Integrase (IN) is a key enzyme encoded within the genome of the human immunodeficiency virus type 1 (HIV-1) that is required for productive infection. The recent FDA approval of the first IN inhibitor, raltegravir (Isentress®), validates IN as an antiretroviral target for the treatment of AIDS.1-3 IN catalyzes the incorporation of viral cDNA into host DNA in a process involving two distinct steps, termed 3′-processing (3′-P) and strand transfer (ST).4 These reactions are catalyzed by three conserved acidic residues (Asp64, Asp116 and Glu152, the “DDE motif”) with the assistance of divalent metal cofactors.5 Numerous IN inhibitors that trace their conceptual ancestry to diketoacid (DKA) progenitors6 contain heteroatom functionality that has been hypothesized to chelate the catalytically-essential divalent metal ions (Figure 1).7, 8 These inhibitors often include aryl-containing amine or amide groups that are thought to bind in a hydrophobic pocket(s) of the IN•DNA complex.9, 10 Recent studies examining structural variation of the aryl group highlight the importance of binding interactions originating from this portion of the inhibitor.11-13 Previous reports have shown that introduction of a substituent at the prochiral aryl α-carbon provides preferentially higher potency for the (S)-enantiomer relative to the (R)-enantiomer.14-17 For example, enantiomeric potency differences of from 2 – fold to more than 10 – fold have been observed for the α-methylbenzylamides 1 – 3 shown in Table 1.
Figure 1.

Table 1. Examples of differences in HIV-1 IN inhibitory potencies for enantiomeric α-methylbenzyl amides.
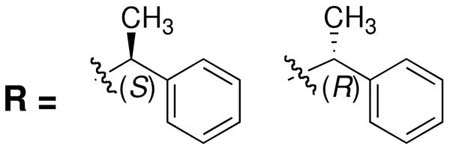
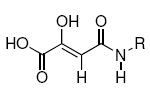
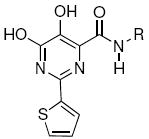
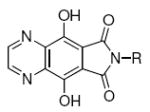
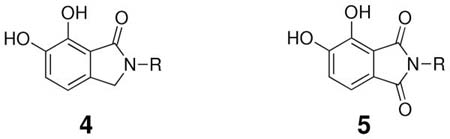
Although these are potentially interesting observations, there have been no systematic investigations examining enantiomeric effects of the α-methylbenzylamide moiety within IN inhibitors. Our recently reported 2,3-dihydro-6,7-dihydroxy-1H-isoindol-1-ones and 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (4 and 5, Figure 1) represent structurally simple IN inhibitors that exhibit good potency and ST selectivity in the presence of Mg2+ cofactor.13, 18 In our current report we use 4 and 5 as platforms for a detailed examination centered on enantiomeric effects of the arylamide group.
2. Results and Discussion
2.1 Chemical synthesis
Analogues 4 were prepared by reaction of the appropriately substituted amines with methyl 2-chloromethyl-3,4-dimethoxybenzoate 618-20 followed by demethylation of the resulting lactams (7) with boron tribromide (Scheme 1).
Scheme 1.

Analogues 5 were formed by refluxing the appropriately substituted amines with triethylamine in toluene in the presence of 3,4-dimethoxyphthalic anhydride 8 (obtained in three steps from 2,3-dimethoxytoluene21) followed by demethylation of the resulting phthalimides (9) with boron tribromide (Scheme 2).
Scheme 2.

2.2 Inhibition of integrase in in vitro assays
In the current study achiral arylamides were first examined in the context of platforms 4 and 5 by measuring 3′-P and ST inhibitory potencies in the presence of Mg2+ cofactor using previously reported procedures (Table 2).18 With 4 as a platform, at least 10 – fold selectivity for ST inhibition was observed for all analogues tested (4a – 4f). Similar selectivity was also shown in the series 5 derivatives, with the exception of the 4-F analogue 5b, which exhibited little ST selectivity. For this latter compound the lack of selectivity resulted from a nearly 10 – fold increase in 3′-P inhibitory potency and a 10 – fold decrease in ST inhibitory potency relative to the unsubstituted benzyl analogue 5a. A very minor difference in potency was observed for 4a relative to 4b. The 1-naphthylmethyl and 2-naphthylmethyl analogues (4c and 4d, respectively) were among the most potent inhibitors in both the series 4 and the series 5 compounds. This indicated a moderate degree of steric tolerance within the IN binding pocket to allow accommodation of the larger aryl group. For both series 4 and series 5 inhibitors the potential importance of alignment of the aryl rings within the pocket was indicated through the loss of binding affinity shown by the phenylethyl (4e and 5e) and 2-indanyl (4f and 5f) analogues. It can be postulated that these analogues are disadvantageous either due to greater flexibility of the aryl substituent (for 4e and 5e) or unfavorable conformational restriction of the aryl ring.
Table 2. Inhibition data from an in vitro HIV-1 IN assay.
 | |||||
|---|---|---|---|---|---|
| No. | R | IC50 (μM) | IC50(μM) | ||
| 3′-P | ST | 3′-P | ST | ||
| a | 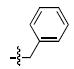 |
>333a | 12.3 ± 5.6a | 72 ± 23 | 0.39 ± 0.21 |
| b | 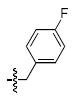 |
282 ± 41a | 10 ± 3a | 8 ± 3b | 5 ± 2b |
| c | 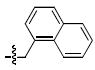 |
61 ± 29a | 0.74 ± 0.16a | 150 ± 36 | 0.72 ± 0.25 |
| d | 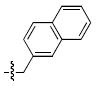 |
24 ± 4 | 0.8 ± 0.2 | 18 ± 3 | 0.96 ± 0.19 |
| e | 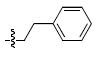 |
> 333a | 28 ± 10a | 157 ± 28 | 6.8 ± 2.5 |
| f | 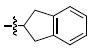 |
> 333 | 23 ± 6 | 37 ± 4 | 4 ± 1 |
Data as previously reported in reference 18.
Data as previously reported in reference 13.
Within the series 4 compounds the effects were next examined of introducing a chiral center into the arylamide moiety through addition of an α-methyl group (compounds 4g – 4l, Table 3). Consistent with previous reports,14-17 the ST inhibitory potency for all analogues tested was greater for the (S)-enantiomer than for the respective (R)-enantiomer. The entiomeric difference in potencies ranged from approximately 2 – fold for the benzyl analogues 4g and 4h to more than 10 – fold for 1-naphthylmethyl, 1-indanyl and 1-methyl(tetrahydronaphthyl) compounds (4i, 4k and 4l, respectively). For the benzyl and naphthylmethyl derivatives, the ST inhibitory potencies of the more potent (S)-methyl enantiomers were approximately 4 – fold lower then the corresponding unsubstituted achiral analogues shown in Table 2. Therefore, while addition of an α-methyl group was uniformly deleterious to binding, the (S)-enantiomer was less deleterious than the (R)-enantiomer. A greater enantiomeric difference in potency (more than 10 – fold) was shown for analogues 4i, 4k and 4l. These inhibitors exhibit restricted rotation of the aryl ring either through steric hindrance (for 4i) or through ring structures (4k and 4l). In each case the larger enantiomeric difference was due to poorer potency of the (R)-enantiomer rather than higher potency of the (S)-enantiomer. This could indicate that conformations for the aryl rings for the (R)-enantiomers are particularly unfavorable.
Table 3. Inhibition data from an in vitro HIV-1 IN assay.
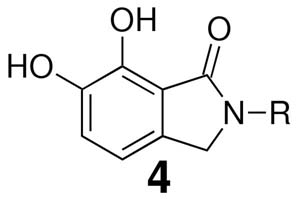 | |||||
|---|---|---|---|---|---|
| No. | R | IC50(μM) | IC50(μM) | ||
| 3′-P | ST | 3′-P | ST | ||
| (S)-Chirality | (R)-Chirality | ||||
| g | 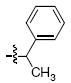 |
>333 | 49.0 ± 1.1 | >333 | 100 ± 2.5 |
| h | 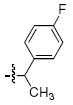 |
>111 | 12.1 ± 1.1 | >333 | 24.4 ± 1.5 |
| i | 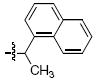 |
>111 | 4.0 ± 0.42 | >111 | 48.3 ± 1.2 |
| j | 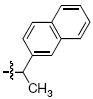 |
>111 | 4.6 ± 0.86 | >111 | 24.0 ± 1.3 |
| k | 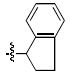 |
> 333 | 6 ± 1 | > 333 | 99 ± 7 |
| l | 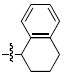 |
> 333 | 9 ± 1 | > 333 | 121 ± 17 |
While the chiral analogues above dealt with series 4 compounds, it was also of interest to examine enantiomeric effects in series 5 compounds, which contain an additional carbonyl group that could influence binding orientation. For this purpose the (S)- and (R)-enantiomers of the 1-indanyl (5k) and 1-methyl(tetrahydronaphthyl) (5l) compounds were prepared (Table 4). While the corresponding series 4 analogues (4k and 4l) showed an approximate 15 – fold greater ST inhibitory potency for the (S)-enantiomer relative to the (R)-enantiomer, the preference in the series 5 compounds was approximately one half as great (approximately 7 – fold to 8 – fold). It was of note that the previously reported analogue 3, which also contains a second carbonyl group situated similarly to the series 5 compounds, also exhibits a relatively low degree of enantiomeric preference.14
Table 4. Inhibition data from an in vitro HIV-1 IN assay.
 | |||||
|---|---|---|---|---|---|
| No. | R | IC50(μM) | IC50(μM) | ||
| 3′-P | ST | 3′-P | ST | ||
| (S)-Chirality | (R)-Chirality | ||||
| k | 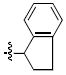 |
128 ±11 | 1.8 ± 0.3 | 290 ± 18 | 12 ± 2 |
| l | 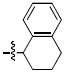 |
111 ± 13 | 10 ± 1 | > 333 | 81 ± 2 |
3. Conclusion
In conclusion, understanding the molecular basis for the binding of DKA – derived inhibitors to IN•DNA complexes has been hampered by the lack of X-ray crystal data leading to the development of computationally derived binding models.22, 23 Correlation of hypothetical inhibitor binding interactions with known patterns of resistance IN mutants has provided an indirect means of validating these models.24 An emerging feature in the understanding of how DKA – derived inhibitors bind to IN is the concept that hydrophobic interactions are critical. This has led to the postulation of a hypothetical hydrophobic binding pocket(s) that accommodates specific aryl components of the inhibitor structure.9, 10 Recent studies have focused on modulating interactions with this hydrophobic pocket by variation of aryl substituents.11-13 Previous reports have also cursorily indicated the existence of differential binding effects induced by introduction of an α-chiral center to the key aryl substitutent. Our current study examines this latter phenomenon in detail. We show that addition of an α-methyl group is uniformly deleterious to binding, with the (S)-configuration being less deleterious than the (R)-configuration. A greater enantiomeric difference in potency is shown by inhibitors that have restricted rotation of the aryl ring, with this being due to poorer potency of the (R)-enantiomer rather than higher potency of the (S)-enantiomer. This potentially indicates that conformations of the aryl rings for the (R)-enantiomers are particularly unfavorable. Addition of a second carbonyl group to the isoindoline-1-one platform (series 4) to give the isoindoline-1,3-dione ring system (series 5) reduces the potency difference by increasing the potency (decreasing unfavorable binding interactions) of the (R)-enantiomer relative to the (S)-enantiomer. Taken in total, findings of our current study contribute to understanding the molecular interactions of DKA – derived IN inhibitors.
4. Experimental
4.1 Integrase Catalytic Assay
Expression of the recombinant IN in Escherichia coli and subsequent purification of the protein were performed as previously reported25,26 with addition of 10% glycerol to all buffers. Preparation of oligonucleotide substrates has been described.27 Integrase reactions were performed in 10 μL with 400 nM of recombinant IN, 20 nM of 5′-end [32P]-labeled oligonucleotide substrate and inhibitors at various concentrations. Solutions of 10% DMSO without inhibitors were used as controls. Reactions were incubated at 37 °C (30 minutes) in buffer containing at a final concentration of 50 mM MOPS, pH 7.2 and 7.5 mM of divalent cations (MgCl2 unless MnCl2 is otherwise indicated). Reactions were stopped by addition of 20 μL of loading dye (10 mM EDTA, 98% deionized formamide, 0.025% xylene cyanol and 0.025% bromophenol blue). Reactions were heated at 95 °C (1 minute) then subjected to electrophoresis in 20% polyacrylamide–7 M urea gels. Gels were dried and reaction products were visualized and quantitated with a PhosphorImager (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric analyses were performed using ImageQuant from Molecular Dynamics Inc. The concentrations at which enzyme activity was reduced by 50% (IC50) were determined using “Prism” software (GraphPad Software, San Diego, CA) for nonlinear regression to fit dose-response data to logistic curve models.
4.2 General Synthetic
1H and 13C NMR data were obtained on a Varian 400 MHz spectrometer and are reported in ppm relative to TMS and referenced to the solvent in which the spectra were collected. Solvent was removed by rotary evaporation under reduced pressure and anhydrous solvents were obtained commercially and used without further drying. Purification by silica gel chromatography was performed using EtOAc – hexanes sovent systems. Preparative high pressure liquid chromatography (HPLC) was conducted using a Waters Prep LC4000 system having photodiode array detection and Phenomenex C18 columns (250 mm × 21.2 mm 5 μm particle size, 110 Å pore) [or YMC J'sphere ODS-H80 columns (250 mm × 20 mm 4 μm particle size, 80 Å pore) as indicated] at a flow rate of 10 mL/min. A binary solvent systems consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in acetonitrile was employed with the gradients as indicated. Products were obtained as amorphous solids following lyophilization. High-resolution mass spectra (HRMS) were obtained from the UCR Mass Spectrometry Facility, University of California at Riverside and fast-atom bombardment mass spectra (FABMS) were acquired with a VG Analytical 7070E mass spectrometer under the control of a VG 2035 data system.
4.3 General Procedure for the Synthesis of Lactams (7d – 7l)
Triethylamine (2.0 mmol) was added to a solution of methyl 2-chloromethyl-3,4-dimethoxybenzoate (6)18-20 (1.0 mmol) and an appropriate amine (1.0 mmol) in anhydrous acetonitrile (3.0 mL) was added. The mixture was stirred at 110° C until the starting material was consumed as indicated by silica gel TLC. The solvent was evaporated and the residue was partitioned between chloroform and brine. The combined organic phase was dried (Na2SO4), evaporated and the residue was purified by silica gel column chromatography. (Note: Lactams 7a – 7c and 7e have been previously reported.18)
4.3.1 6,7-Dimethoxy-2-(naphthalen-2-ylmethyl)isoindolin-1-one (7d)
Prepared in 37% yield. 1H NMR (CDCl3): δ 7.72-7.69 (m, 3H), 7.66 (s, 1H), 7.39-7.34 (m, 3H), 6.95 (d, 1H, J = 8.4 Hz), 6.87 (d, 1H, J = 8.4 Hz), 4.80 (s, 2H), 4.08 (s, 3H), 4.04 (s, 2H), 3.78 (s, 3H). 13C NMR (CDCl3): δ 166.7, 152.3, 147.2, 134.6, 134.5, 133.3, 132.8, 128.6, 127.7, 127.6, 126.9, 126.3, 126.2, 126.0, 124.7, 117.9, 116.4, 62.5, 56.7, 48.5, 46.4. APCI-MS m/z: 334.1 (MH+).
4.3.2 2-(2,3-Dihydro-1H-inden-2-yl)-6,7-dimethoxyisoindolin-1-one (7f)
Prepared in 54% yield. 1H NMR (CDCl3): δ 7.21-7.18 (m, 2H), 7.16-7.13 (m, 2H), 6.99 (d, 1H, J = 8.4 Hz), 6.92 (d, 1H, J = 8.4 Hz), 5.29-5.25 (m, 1H), 4.03 (s, 3H), 3.98 (s, 2H), 3.81 (s, 3H), 3.27 (dd, 2H, J = 7.6 Hz, 16.4 Hz), 2.94 (dd, 2H, J = 5.2 Hz, 16.4 Hz). 13C NMR (CDCl3): δ 166.4, 152.3, 147.1, 140.9 (2C), 134.3, 126.8 (2C), 124.9, 124.4 (2C), 117.8, 116.4, 62.5 (d, 1C, J = 1.5 Hz), 56.7 (d, 1C, J = 2.2 Hz), 51.4, 45.5, 37.7 (2C). APCI-MS m/z: 310.1 (MH+).
4.3.3 (S)- and (R)-6,7-Dimethoxy-2-(1-phenylethyl)isoindolin-1-one [(S)-7g] and [(R) – 7g]
(S) – 7g Prepared in 47% yield; prepared in (R) – 7g 45% yield. 1H NMR (CDCl3): δ 7.29-7.21 (m, 4H), 7.18-7.14 (m, 1H), 6.96 (d, 1H, J = 8.2 Hz), 6.90 (d, 1H, J = 8.2 Hz), 5.68 (dd, 1H, J = 7.2 Hz, 14.0 Hz), 4.141 (d, 1H, J = 16.8 Hz), 4.03 (s, 3H), 3.78 (d, 1H, J = 16.8 Hz), 3.77 (s, 3H), 1.57 (d, 3H, J = 7.2 Hz). 13C NMR (CDCl3): δ 166.2, 152.2, 147.1, 140.7, 134.6, 128.5 (2C), 127.4, 127.1 (2C), 125.0, 117.8, 116.4, 62.4, 56.7, 48.9, 44.6, 17.1. APCI-MS m/z: 298.1 (MH+).
4.3.4 (S)- and (R)-2-(1-(4-Fluorophenyl)ethyl)-6,7-dimethoxyisoindolin-1-one [(S) – 7h] and [(R) – 7h]
(S) – 7h Prepared in 64% yield; [(R) – 7h] prepared in 60% yield. 1H NMR (CDCl3): δ 7.23-7.19 (m, 2H), 6.95-6.84 (m, 4H), 5.60 (dd, 1H, J = 7.2 Hz, 14.0 Hz), 4.11 (d, 1H, J = 16.4 Hz), 3.98 (s, 3H), 3.76 (d, 1H, J = 16.4 Hz), 3.73 (s, 3H), 1.51 (d, 3H, J = 7.2 Hz). 13C NMR (CDCl3): δ 166.2, 161.9 (d, 1C, J = 244.9 Hz), 152.2, 147.1, 136.6 (d, 1C, J = 3.0 Hz), 134.4, 128.7 (d, 2C, J = 8.4 Hz), 124.9, 117.9, 116.4, 115.2 (d, 2C, J = 20.6 Hz), 62.3, 56.6, 48.3, 44.4, 17.2. APCI-MS m/z: 316.1 (MH+).
4.3.5 (S)- and (R)-6,7-Dimethoxy-2-(1-(naphthalen-1-yl)ethyl)isoindolin-1-one [(S) – 7i] and [(R) – 7i]
(S) – 7i Prepared in 74% yield; (R) – 7i prepared in 73% yield. 1H NMR (CDCl3): δ 8.17 (d, 1H, J = 8.4 Hz), 7.79 (d, 1H, J = 8.0 Hz), 7.59 (d, 1H, J = 7.2 Hz), 7.48-7.39 (m, 3H), 6.92 (d, 1H, J = 8.4 Hz), 6.77 (d, 1H, J = 8.0 Hz), 6.39 (dd, 1H, J = 6.4 Hz, 14.0 Hz), 4.11 (s, 3H), 4.07 (d, 1H, J = 16.8 Hz), 3.79 (s, 3H), 3.32 (d, 1H, J = 16.8 Hz), 1.747 (d, 3H, J = 6.8 Hz). 13C NMR (CDCl3): δ 165.7, 152.2, 147.2, 135.7, 134.7, 133.8, 131.6, 128.8, 128.5, 127.0, 126.0, 125.0, 124.8, 123.9, 123.6, 117.7, 116.4, 62.5, 56.8, 45.4, 44.6, 17.1. APCI-MS m/z: 348.1 (MH+).
4.3.6 (S)- and (R)-6,7-Dimethoxy-2-(1-(naphthalen-2-yl)ethyl)isoindolin-1-one [(S) – 7j] and [(R) – 7j]
(S) – 7j Prepared in 56% yield; (R) – 7j prepared in 65% yield. 1H NMR (CDCl3): δ 7.73-7.65 (m, 4H), 7.37-7.31 (m, 3H), 6.89 (d, 1H, J = 8.0 Hz), 6.80 (d, 1H, J = 8.0 Hz), 5.83 (dd, 1H, J = 6.8 Hz, 14.0 Hz), 4.06(s, 3H), 3.72 (s, 3H), 1.63 (d, 3H, J = 7.2 Hz). 13C NMR (CDCl3): δ 166.4, 152.2, 147.1, 138.2, 134.5, 133.2, 132.7, 128.4, 127.9, 127.5, 126.2, 126.0, 125.8, 125.3, 124.9, 117.9, 116.4, 62.4, 56.6, 48.9, 44.6, 17.0. APCI-MS m/z: 348.1 (MH+).
4.3.7 (S)- and (R)-2-(2,3-Dihydro-1H-inden-1-yl)-6,7-dimethoxyisoindolin-1-one [(S) – 7k] and [(R) – 7k]
(S) – 7k Prepared in 55% yield, [α]D23 (CHCl3) = -98.0.; (R) – 7k prepared in 57% yield, [α]D23 (CHCl3) = 91.5. 1H NMR (CDCl3): δ 7.24-7.19 (m, 2H), 7.13-7.10 (m, 2H), 7.02 (d, 1H, J = 8.0 Hz), 6.94 (d, 1H, J = 8.0 Hz), 6.02 (t, 1H, J = 7.6 Hz), 4.08 (s, 3H), 4.06 (d, 1H, J = 16.8 Hz), 3.90 (d, 1H, J = 16.8 Hz), 3.84 (s, 3H), 3.07-3.00 (m, 1H), 2.96-2.88 (m, 1H), 2.54-2.45 (m, 1H), 2.06-1.97 (m, 2H). 13C NMR (CDCl3): δ 166.8, 152.3, 147.2, 143.5, 141.4, 134.6, 128.0, 126.8, 125.0, 124.9, 124.4, 117.8, 116.5, 62.5 (d, 1C, J = 3.8 Hz), 56.8 (d, 1C, J = 6.1 Hz), 56.1 (d, 1C, J = 5.4 Hz), 44.9 (t, 1C, J = 5.0 Hz), 30.6, 30.4. APCI-MS m/z: 310.1 (MH+).
4.3.8 (S)- and (R)-6,7-Dimethoxy-2-(1,2,3,4-tetrahydronaphthalen-1-yl)isoindolin-1-one [(S) – 7l] and [(R) – 7l]
(S) – 7l Prepared in 51% yield, [α]D22 (MeOH) = -109.7; (R) – 7l prepared in 56% yield, [α]D22 (MeOH) = 131.8. 1H NMR (CDCl3): δ 7.11-7.07 (m, 2H), 7.05-7.00 (m, 2H), 6.95-6.93 (m, 2H), 5.64 (t, 1H, J = 7.4 Hz), 4.11 (d, 1H, J = 16.8 Hz), 4.08 (s, 3H), 3.90 (d, 1H, J = 16.8 Hz), 3.83 (s, 3H), 2.80-2.75 (m, 2H), 2.10-2.07 (m, 1H), 1.99-1.95 (m, 1H), 1.88-1.83 (m, 2H). 13C NMR (CDCl3): δ 167.2, 152.3, 147.2, 138.0, 135.1, 134.7, 129.2, 127.5, 127.0, 126.2, 124.8, 117.8, 116.5, 62.5 (d, 1C, J = 3.0 Hz), 56.8 (d, 1C, J = 4.6 Hz), 49.6, 45.3, 29.4, 28.7, 21.7.
4.4 General Procedure for the Demethylation of Methyl Ethers (Conversion of 7 to 4 and 9 to 5)
Boron tribromide (1.0 M in dichloromethane, 8.5 mmol) was added carefully to a solution of appropriate methyl ether (1.0 mmol in 1.0 mL anhydrous dichloromethane) and the mixture was stirred at room temperature (overnight). The reaction was quenched by the careful addition of ice water (1.0 mL) then the mixture was stirred at room temperature (overnight). The resulting suspension was filtered and the collected solid was purified by preparative HPLC. (Note: Products 4a – 4c and 4e have been previously reported18 as well as 5b.13)
4.4.1 6,7-Dihydroxy-2-(naphthalen-2-yl)isoindolin-1-one (4d)
HPLC conditions: Linear gradient of 40% B to 60% B over 30 minutes; retention time = 22.1 minutes. 1H NMR (DMSO): δ 7.84-7.81 (m, 3H), 7.22 (s, 1H), 7.46-7.41 (m, 2H), 7.35 (dd, 1H, J = 1.6 Hz, 8.0 Hz), 6.92 (d, 1H, J = 8.0 Hz), 6.69 (d, 1H, J = 8.0 Hz), 4.76 (s, 2H), 4.17 (s, 2H). 13C NMR (DMSO): δ 168.5, 144.8, 143.5, 135.6, 133.4, 132.7 (2C), 128.8, 128.0 (2C), 126.7, 126.5 (2C), 126.3, 120.0, 118.3, 114.2, 49.2, 45.7. APCI-MS m/z: 306.1 (MH+). HRMS calcd for C19H16NO3 [MH+]: 306.1130. Found: 306.1132.
4.4.2 2-(2,3-Dihydro-1H-inden-2-yl)-6,7-dihydroxyisoindolin-1-one (4f)
HPLC conditions: linear gradient of 40% B to 55% B over 30 minutes; retention time = 20.6 minutes. 1H NMR (DMSO): δ 7.21-7.19 (m, 2H), 7.14-7.11 (m, 2H), 6.90 (d, 1H, J = 7.6 Hz), 6.69 (d, 1H, J = 7.6 Hz), 4.98-4.95 (m, 1H), 4.07 (s, 2H), 3.14 (dd, 2H, J = 8.0 Hz, 16.0 Hz), 2.99 (dd, 2H, J = 8.0 Hz, 16.0 Hz). 13C NMR (DMSO): δ 168.4, 144.6, 143.2 (2C), 141.3 (2C), 132.4, 127.0 (2C), 124.7 (2C), 120.0, 118.5, 114.2, 51.6, 46.2, 37.2 (2C). APCI-MS m/z: 282.0 (MH+).
4.4.3 (S)- and (R)-6,7-Dihydroxy-2-(1-phenylethyl)isoindolin-1-one [(S) – 4g] and [(R) – 4g]
HPLC conditions: linear gradient of 40% B to 45% B over 30 minutes; retention time = 20.1 minutes. 1H NMR (CD3OD): δ 7.31 (d, 4H, J = 4.8 Hz), 7.24-7.21 (m, 1H), 6.92 (d, 1H, J = 8.0 Hz), 6.73-6.70 (m, 1H), 5.54 (dd, 1H, J = 6.8 Hz, 14.4 Hz), 4.31 (d, 1H, J = 17.2 Hz), 3.88 (d, 1H, J = 17.2 Hz), 1.63 (d, 3H, J = 7.2 Hz). 13C NMR (CD3OD): δ 169.3, 144.1, 143.0, 140.6, 132.5, 128.3 (2C), 127.2, 126.5 (2C), 119.6, 117.6, 113.6, 49.4, 45.3, 16.5. APCI-MS m/z: 270.1 (MH+).
4.4.4 (S)- and (R)-2-(1-(4-Fluorophenyl)ethyl)-6,7-dihydroxyisoindolin-1-one [(S) – 4h] and [(R) – 4h]
HPLC conditions: linear gradient of 40% B to 50% B over 30 minutes; retention time = 21.6 minutes. 1H NMR (DMSO): δ 7.33-7.29 (m, 2H), 7.14-7.09 (m, 2H), 6.90 (d, 1H, J = 8.0 Hz), 6.71 (d, 1H, J = 8.0 Hz), 5.37 (dd, 1H, J = 7.2 Hz, 14.4 Hz), 4.30 (d, 1H, J = 17.2 Hz), 3.89 (d, 1H, J = 17.2 Hz), 1.54 (d, 3H, J = 7.2 Hz). 13C NMR (DMSO): δ 168.1, 161.7 (d, 1C, J = 242.6 Hz), 144.7, 143.3, 138.0 (d, 1C, J = 3.1 Hz), 132.5, 129.2 (d, 2C, J = 7.6 Hz), 120.0, 118.5, 115.7 (d, 2C, J = 20.5 Hz), 114.2, 48.8, 45.5, 18.3. APCI-MS m/z: 288.1 (MH+).
4.4.5 (S)- and (R)-6,7-Dihydroxy-2-(1-(naphthalen-1-yl)ethyl)isoindolin-1-one [(S) – 4i] and [(R) – 4i]
HPLC conditions: linear gradient of 40% B to 60% B over 30 minutes; retention time = 25.5 minutes. 1H NMR (DMSO): δ 8.07-8.05 (m, 1H), 7.92-7.86 (m, 2H), 7.64 (d, 1H, J = 7.2 Hz), 7.54-7.49 (m, 1H), 7.48-7.43 (m, 2H), 6.82 (d, 1H, J = 8.0 Hz), 6.58 (d, 1H, J = 8.0 Hz), 6.11 (dd, 1H, J = 7.2 Hz, 13.6 Hz), 4.24 (d, 1H, J = 17.6 Hz), 3.29 (d, 1H, J = 17.6 Hz), 1.67 (d, 3H, J = 7.2 Hz). 13C NMR (DMSO): δ 167.3, 144.8, 143.3, 136.3, 133.8, 132.5, 131.3, 129.2, 128.8, 127.1, 126.3, 125.7, 124.6, 123.2, 119.8, 118.4, 114.2, 45.3, 45.2, 17.8. APCI-MS m/z: 320.1 (MH+).
4.4.6 (S)- and (R)-6,7-Dihydroxy-2-(1-(naphthalen-2-yl)ethyl)isoindolin-1-one [(S) – 4j] and [(R) – 4j]
HPLC conditions: linear gradient of 40% B to 54% B over 30 minutes; retention time = 24.0 minutes. 1H NMR (CD3OD): δ 7.83-7.76 (m, 4H), 7.45-7.38 (m, 3H), 6.91 (d, 1H, J = 8.0 Hz), 6.70 (d, 1H, J = 8.0 Hz), 5.70 (dd, 1H, J = 7.2 Hz, 14.4 Hz), 4.33 (d, 1H, J = 16.8 Hz), 3.88 (d, 1H, J = 16.8 Hz), 1.74 (d, 3H, J = 7.2 Hz). 13C NMR (CD3OD): δ 169.4, 144.1, 143.0, 138.0, 133.3, 132.8, 132.5, 128.1, 127.6, 125.9, 125.7, 125.0, 124.9, 119.6, 117.6, 113.7, 49.4, 45.4, 16.4. APCI-MS m/z: 320.1 (MH+).
4.4.7 (S)- and (R)-2-(2,3-Dihydro-1H-inden-1-yl)-6,7-dihydroxyisoindolin-1-one [(S) – 4k] and [(R) – 4k]
HPLC conditions: linear gradient of 40% B to 55% B over 30 minutes; retention time = 20.2 minutes. 1H NMR (DMSO): δ 7.26 (d, 1H, J = 7.6 Hz), 7.19 (t, 1H, J = 7.2 Hz), 7.15 (t, 1H, J = 7.6 Hz), 7.02 (d, 1H, J = 7.2 Hz), 6.90 (d, 1H, J = 7.6 Hz), 6.68 (d, 1H, J = 7.6 Hz), 5.73 (t, 1H, J = 8.0 Hz), 4.16 (d, 1H, J = 17.2 Hz), 3.75 (d, 1H, J = 17.2 Hz), 3.03-2.96 (m, 1H), 2.89-2.81 (m, 1H), 2.38-2.30 (m, 1H), 2.08-1.99 (m, 1H). 13C NMR (DMSO): δ 168.6, 144.7, 143.7, 143.3, 141.8, 132.5, 128.3, 127.1, 125.3, 124.3, 120.0, 118.4, 114.3, 55.9, 45.4, 30.5, 30.3. [(S) – 4k] HRMS calcd for C17H15NO3Na [MNa+]: 304.0950. Found: 304.0955. [(R) – 4k] HRMS calcd for C17H15NO3Na [MNa+]: 304.0950. Found: 304.0951.
4.4.8 (S)- and (R)-6,7-Dihydroxy-2-(1,2,3,4-tetrahydronaphthalen-1-yl)isoindolin-1-one [(S) – 4l] and [(R) – 4l]
HPLC conditions: linear gradient of 40% B to 50% B over 30 minutes; retention time = 24.7 minutes. 1H NMR (DMSO): δ 7.12-7.07 (m, 2H), 7.06-7.02 (m, 2H), 6.91 (d, 1H, J = 8.0 Hz), 6.80 (d, 1H, J = 7.6 Hz), 6.69 (d, 1H, J = 7.6 Hz), 5.37-5.33 (m, 1H), 4.18 (d, 1H, J = 17.2 Hz), 3.75 (d, 1H, J = 17.2 Hz), 2.78-2.66 (m, 2H), 1.95-1.85 (m, 3H), 1.82-1.74 (m, 1H). 13C NMR (DMSO): δ 169.0, 144.8, 143.4, 138.2, 135.6, 132.5, 129.6, 127.3, 127.1, 126.6, 120.0, 118.3, 114.3, 49.4, 45.8, 29.2, 28.5, 21.9. ESI-MS m/z: 294.1 (M-H). [(S) – 4l] HRMS calcd for C18H18NO3 [MH+]: 296.1281. Found: 296.1278. [(R) – 4l] HRMS calcd for C18H18NO3 [MH+]: 296.1281. Found: 296.1282.
4.5 General Procedure for the Synthesis of Phthalimides (9a – 9f, 9k, 9l)
Triethylamine (2.0 mmol) was added dropwise to a solution of 3,4-dimethoxyphthalic anhydride) (8)22 (1.0 mmol) and an appropriate amine (1.0 mmol) in toluene (5.0 mL) and the mixture was stirred at 80° C (overnight). The solvent was evaporated and the residue was taken up in chloroform, dried (Na2SO4) and evaporated. The product was obtained following purification by silica gel column chromatography. (Note: Phthalimide 9b has been previously reported.13)
4.5.1 2-Benzyl-4,5-dimethoxyisoindoline-1,3-dione (9a)
Prepared in 16% yield. 1H NMR (CDCl3): δ 7.49 (dd, 1H, J = 1.2 Hz, 8.0 Hz), 7.39 (dd, 2H, J = 1.2 Hz, 8.0 Hz), 7.29-7.23 (m, 3H), 7.05 (d, 1H, J = 8.0 Hz), 4.76 (s, 2H), 4.10 (d, 3H, J = 1.2 Hz), 3.90 (d, 3H, J = 1.2 Hz). 13C NMR (CDCl3): δ 167.3, 166.0, 157.7, 147.2, 136.5, 128.6 (4C), 127.7, 124.6, 121.9, 119.4, 115.7, 62.5, 56.6, 41.6. FAB-MS m/z: 298.2 (MH+).
4.5.2 4,5-Dimethoxy-2-(naphthalen-1-ylmethyl)isoindoline-1,3-dione (9c)
Prepared in 18% yield. 1H NMR (CDCl3): δ 8.34 (d, 1H, J = 8.8 Hz), 7.79 (dd, 2H, J = 8.0 Hz, 24.4 Hz), 7.58-7.52 (m, 1H), 7.50 (d, 1H, J = 8.0 Hz), 7.48-7.44 (m, 2H), 7.39 (t, 1H, J = 8.0 Hz), 7.04 (d, 1H, J = 8.0 Hz), 5.24 (s, 2H), 4.09 (s, 3H), 3.89 (s, 3H). 13C NMR (CDCl3): δ 167.5, 166.2, 157.7, 147.2, 133.7, 131.5, 131.2, 128.6, 128.5, 127.4, 126.4, 125.7, 125.3, 124.5, 123.5, 121.9, 119.5, 115.8, 62.5, 56.6, 39.5. FAB-MS m/z: 348.1 (MH+).
4.5.3 4,5-Dimethoxy-2-(naphthalen-2-ylmethyl)isoindoline-1,3-dione (9d)
Prepared in 39% yield. 1H NMR (CDCl3): δ 7.84 (s, 1H), 7.76-7.70 (m, 3H), 7.51 (dd, 1H, J = 1.6 Hz, 8.4 Hz), 7.42 (d, 1H, J = 8.4 Hz), 7.38-7.35 (m, 2H), 6.92 (d, 1H, J = 8.4 Hz), 4.90 (s, 2H), 4.08 (s, 3H), 3.79 (s, 3H). 13C NMR (CDCl3): δ 167.3, 166.0, 157.6, 147.1, 134.0, 133.2, 132.8, 128.4, 127.9, 127.6, 127.5, 126.5, 126.1, 126.0, 124.5, 121.8, 119.4, 115.7, 62.5, 56.5, 41.7. APCI-MS m/z: 348.1 (MH+).
4.5.4 4,5-dimethoxy-2-phenethylisoindoline-1,3-dione (9e)
Prepared in 61% yield. 1H NMR (CDCl3): δ 7.47 (d, 1H, J = 8.0 Hz), 7.24-7.22 (m, 5H), 7.05 (d, 1H, J = 8.0 Hz), 4.08 (s, 3H), 3.90 (s, 3H), 3.84 (t, 2H, J = 7.6 Hz), 2.93 (t, 2H, J = 7.6 Hz). 13C NMR (CDCl3): δ 167.4, 166.1, 157.6, 147.1, 138.1, 128.8 (2C), 128.5 (2C), 126.5, 124.6, 121.8, 119.3, 115.6, 62.5, 56.6, 39.2, 34.5. FAB-MS m/z: 312.1 (MH+).
4.5.5 2-(2,3-Dihydro-1H-inden-2-yl)-4,5-dimethoxyisoindoline-1,3-dione (9f)
Prepared in 23% yield. 1H NMR (CDCl3): δ 7.50 (d, 1H, J = 8.0 Hz), 7.19-7.13 (m, 4H), 7.07 (d, 1H, J = 8.0 Hz), 5.11-5.06 (m, 1H), 4.10 (s, 3H), 3.92 (s, 3H), 3.59 (dd, 2H, J = 9.2 Hz, 15.2 Hz), 3.12 (dd, 2H, J = 8.8 Hz, 15.2 Hz). 13C NMR (CDCl3): δ 167.7, 166.3, 157.7, 147.1, 140.9, 124.8, 126.6 (2C), 134.6, 124.3 (2C), 121.9, 119.3, 115.8, 62.5 (d, 1C, J = 3.1 Hz), 56.6 (d, 1C, J = 4.6 Hz), 49.9, 36.0 (2C). APCI-MS m/z: 324.0 (MH+).
4.5.6 (S)- and (R)-2-(2,3-Dihydro-1H-inden-1-yl)-4,5-dimethoxyisoindoline-1,3-dione [(S) – 9k] and [(R) – 9k]
(S) – 9k Prepared in 14% yield, [α]D23 (CHCl3) = -127.7; (R) – 9k prepared in 15% yield, [α]D23 (CHCl3) = -119.4. 1H NMR (CDCl3): δ 7.46 (d, 1H, J = 8.0 Hz), 7.24 (d, 1H, J = 7.6 Hz), 7.20-7.16 (m, 1H), 7.12-7.06 (m, 2H), 7.05 (d, 1H, J = 8.0 Hz), 5.80 (dd, 1H, J = 6.8, 8.4 Hz), 4.07 (s, 3H), 3.89 (s, 3H), 3.34-3.29 (m, 1H), 2.98-2.90 (m, 1H), 2.53-2.40 (m, 2H), 13C NMR (CDCl3): δ 167.3, 165.9, 157.7, 147.1, 143.8, 140.6, 127.9, 126.4, 124.8, 124.6, 123.3, 121.9, 119.3, 115.8, 62.5, 56.6, 54.7, 31.1, 29.5. APCI-MS m/z: 324.1 (MH+).
4.5.7 (S)- and (R)-4,5-Dimethoxy-2-(1,2,3,4-tetrahydronaphthalen-1-yl)isoindoline-1,3-dione [(S) – 9l] and [(R) – 9l]
(S) – 9l Prepared in 8% yield, [α]D22 (MeOH) = -81.3; (R) – 9l prepared in 14% yield, [α]D22 (MeOH) = 78.2. 1H NMR (CDCl3): δ 7.50 (d, 1H, J = 8.4 Hz), 7.10-7.07 (m, 3H), 7.04-7.00 (m, 1H), 6.92-6.90 (m, 1H), 5.47 (dd, 1H, J = 6.0, 16.8 Hz), 4.09 (s, 3H), 3.92 (s, 3H), 3.02-2.94 (m, 1H), 2.80-2.76 (m, 1H), 2.42-2.33 (m, 1H), 2.11-2.03 (m, 2H), 1.86-1.75 (m, 1H). 13C NMR (CDCl3): δ 167.4, 166.0, 157.7, 147.2, 137.8, 134.8, 129.2, 126.8, 126.0, 125.8, 124.6, 121.9, 119.3, 115.8, 62.5, 56.6 (d, 1C, J = 3.8 Hz), 49.2, 29.4, 27.9, 22.4.
4.5.8 2-Benzyl-4,5-dihydroxyisoindoline-1,3-dione (5a)
HPLC conditions: linear gradient of 30% B to 55% B over 30 minutes; retention time = 24.2 minutes. 1H NMR (DMSO): δ 7.26-7.16 (m, 5H), 7.14 (d, 1H, J = 8.0 Hz), 6.97 (d, 1H, J = 8.0 Hz), 4.81 (s, 2H), 4.68 (s, 2H). 13C NMR (DMSO): δ 168.1, 167.6, 152.3, 144.0, 137.0 (2C), 128.1 (2C), 127.5 (2C), 127.1, 122.4, 118.5, 115.9, 40.5. FAB-MS m/z: 268.1 (M-H). HRMS calcd for C15H10NO4 [M-H]: 268.0610. Found: 268.0613.
4.5.9 4,5-Dihydroxy-2-(naphthalen-1-ylmethyl)isoindoline-1,3-dione (5c)
HPLC condisitons: linear gradient of 40% B to 60% B with YMC HPLC column over 30 minutes; retention time = 22.3 minutes. 1H NMR (DMSO): δ 8.21 (d, 1H, J = 8.0 Hz), 7.91 (d, 1H, J = 8.0 Hz), 7.81 (d, 1H, J = 8.0 Hz), 7.56-7.51 (m, 3H), 7.39 (t, 1H, J = 8.0 Hz), 7.26 (d, 1H, J = 6.8 Hz), 7.17 (d, 1H, J = 7.6 Hz), 7.04 (d, 1H, J = 7.6 Hz), 5.10 (s, 2H). 13C NMR (DMSO): δ 167.9, 167.0, 153.0, 144.7, 133.7, 132.5, 130.8, 129.0, 128.2, 126.9, 126.4, 125.8, 125.2, 123.5, 122.6, 119.2, 116.3, 116.1, 38.9. FAB-MS m/z: 320.1 (MH+). HRMS calcd for C19H14NO4 [M-H]: 320.0923. Found: 320.0917.
4.5.10 4,5-Dihydroxy-2-(naphthalen-2-ylmethyl)isoindoline-1,3-dione (5d)
HPLC conditions: linear gradient of 40% B to 60% B over 30 minutes; retention time = 23.1 minutes. 1H NMR (DMSO): δ 7.83-7.81 (m, 3H), 7.72 (s, 1H), 7.44-7.38 (m, 3H), 7.17 (d, 1H, J = 8.0 Hz), 7.04 (d, 1H, J = 8.0 Hz), 4.80 (s, 2H). 13C NMR (DMSO): δ 167.8, 166.9, 152.9, 144.7, 135.2, 133.2, 132.6, 128.6, 128.0, 127.9, 126.7, 126.3, 126.1 (2C), 126.0, 122.7, 119.2, 116.2, 41.1. APCI-MS m/z: 318.1 (M-H). HRMS calcd for C19H12NO4 [M-H]: 318.0766. Found: 318.0759.
4.5.11 4,5-Dihydroxy-2-phenethylisoindoline-1,3-dione (5e)
HPLC conditions: linear gradient of 40% B to 50% B with YMC HPLC column over 30 minutes; retention time = 18.3 minutes. 1H NMR (CD3OD): δ 7.22-7.11 (m, 6H), 6.96 (d, 1H, J = 7.6 Hz), 6.70 (d, 1H, J = 8.0 Hz), 3.76 (dd, 2H, J = 7.6 Hz, 8.4 Hz), 2.89 (t, 2H, J = 7.6 Hz). 13C NMR (CD3OD): δ 168.2, 167.8, 152.2, 143.8, 138.3, 128.4 (2C), 128.0 (2C), 126.1, 122.4, 118.4, 115.7, 115.5, 38.5, 34.0. FAB-MS m/z: 284.1 (MH+). HRMS calcd for C16H14NO4 [MH+]: 284.0923. Found: 284.0914.
4.5.12 2-(2,3-Dihydro-1H-inden-2-yl)-4,5-dihydroxyisoindoline-1,3-dione (5f)
HPLC conditions: linear gradient of 30% B to 55% B over 30 minutes; retention time = 29.5 minutes. 1H NMR (DMSO): δ 7.18-7.15 (m, 2H), 7.13-7.11 (m, 2H), 7.11 (d, 1H, J = 8.0 Hz), 7.01 (d, 1H, J = 8.0 Hz), 4.91-4.87 (m, 1H), 3.36 (dd, 2H, J = 9.2 Hz, 15.6 Hz), 3.08 (dd, 2H, J = 9.2 Hz, 15.6 Hz). 13C NMR (DMSO): δ 167.9, 167.1, 152.8, 144.5, 141.4 (2C), 126.9 (2C), 124.7 (2C), 122.7, 119.1, 116.1, 115.9, 49.0, 36.1 (2C). MALDI-MS m/z: 295.54 (MH+). HRMS calcd for C17H12NO4 [M-H]: 294.0766. Found: 294.0769.
4.5.13 (S)- and (R)-2-(2,3-Dihydro-1H-inden-1-yl)-4,5-dihydroxyisoindoline-1,3-dione [(S) – 5k] and [(R) – 5k]
HPLC conditions: linear gradient of 40% B to 55% B over 30 minutes; retention time = 20.8 minutes. 1H NMR (DMSO): δ 7.21 (d, 1H, J = 7.6 Hz), 7.15 (t, 1H, J = 7.2 Hz), 7.11-7.04 (m, 2H), 7.02-6.99 (m, 2H), 5.61 (t, 1H, J = 8.0 Hz), 3.14-3.07 (m, 1H), 2.90-2.82 (m, 1H), 2.38-2.28 (m, 2H). 13C NMR (DMSO): δ 167.5, 166.7, 152.8, 144.5, 143.7, 141.6, 127.9, 126.8, 125.0, 123.4, 122.6, 119.2, 116.1, 116.0, 54.0, 30.9, 29.6. ESI-MS m/z: 294.1 (M-H). (S) – 5k HRMS calcd for C17H12NO4 [M-H]: 294.0766. Found: 294.0767. (R) – 5k HRMS calcd for C17H12NO4 [M-H]: 294.0766. Found: 294.0762.
4.5.14 (S)- and (R)-4,5-Dihydroxy-2-(1,2,3,4-tetrahydronaphthalen-1-yl)isoindoline-1,3-dione [(S) – 5l] and [(R) – 5l]
HPLC conditions: linear gradient of 40% B to 55% B over 30 minutes; retention time = 23.5 minutes. 1H NMR (CD3OD): δ 7.17 (d, 1H, J = 8.0 Hz), 7.07-7.06 (m, 2H), 7.01 (d, 1H, J = 8.0 Hz), 7.01-6.98 (m, 2H), 6.85 (d, 1H, J = 7.6 Hz), 5.37 (dd, 1H, J = 6.4, 10.8 Hz), 2.96-2.88 (m, 1H), 2.82-2.76 (m, 1H), 2.41-2.32 (m, 1H), 2.10-2.00 (m, 2H), 1.85-1.76 (m, 1H). 13C NMR (CD3OD): δ 168.2, 167.7, 152.3, 137.5, 135.0, 128.7, 126.3, 125.7, 125.2, 122.4, 121.8, 118.6, 115.8, 115.5, 48.8, 29.0, 27.6, 22.3. ESI-MS m/z: 308.1 (M-H). (S) – 5l HRMS calcd for C18H14NO4 [M-H]: 308.0928. Found: 308.0928. (R) – 5l HRMS calcd for C18H14NO4 [M-H]: 308.0928. Found: 308.0921.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Evering TH, Markowitz M. Expert Opin Invest Drugs. 2008;17:413. doi: 10.1517/13543784.17.3.413. [DOI] [PubMed] [Google Scholar]
- 2.Anker M, Corales RB. Expert Opin Invest Drugs. 2008;17:97. doi: 10.1517/13543784.17.1.97. [DOI] [PubMed] [Google Scholar]
- 3.Croxtall JD, Lyseng-Williamson KA, Perry CM. Drugs. 2008;68:131. doi: 10.2165/00003495-200868010-00009. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y, Johnson AA, Marchand C. Nat Rev Drug Discovery. 2005;4:236. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 5.Johns BA, Svolto AC. Expert Opin Ther Pat. 2008;18:1225. [Google Scholar]
- 6.Grobler Jay A, Stillmock K, Hu B, Witmer M, Felock P, Espeseth Amy S, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai John S, Young S, Vacca J, Hazuda Daria J. Proc Natl Acad Sci U S A. 2002;99:6661. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayam R, Gundla R, Al-Mawsawi LQ, Neamati N. Med Res Rev. 2008;28:118. doi: 10.1002/med.20116. [DOI] [PubMed] [Google Scholar]
- 8.Bacchi A, Biemmi M, Carcelli M, Carta F, Compari C, Fisicaro E, Rogolino D, Sechi M, Sippel M, Sotriffer Christoph A, Sanchez Tino W, Neamati N. J Med Chem. 2008;51:7253. doi: 10.1021/jm800893q. [DOI] [PubMed] [Google Scholar]
- 9.Jin H, Cai RZ, Schacherer L, Jabri S, Tsiang M, Fardis M, Chen X, Chen JM, Kim CU. Bioorg Med Chem Lett. 2006;16:3989. doi: 10.1016/j.bmcl.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Tsiang M, Yu F, Hung M, Jones GS, Zeynalzadegan A, Qi X, Jin H, Kim CU, Swaminathan S, Chen JM. J Mol Biol. 2008;380:504. doi: 10.1016/j.jmb.2008.04.054. [DOI] [PubMed] [Google Scholar]
- 11.Ferro S, De Luca L, Barreca Maria L, Iraci N, De Grazia S, Christ F, Witvrouw M, Debyser Z, Chimirri A. J Med Chem. 2009;52:569. doi: 10.1021/jm8009266. [DOI] [PubMed] [Google Scholar]
- 12.Jin H, Metobo S, Jabri S, Mish M, Lansdown R, Chen X, Tsiang M, Wright M, Kim CU. Bioor Med Chem Lett. 2009;18:2263. doi: 10.1016/j.bmcl.2009.02.092. [DOI] [PubMed] [Google Scholar]
- 13.Zhao XZ, Maddali K, Vu BC, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr Bioorg Med Chem Lett. 2009 doi: 10.1016/j.bmcl.2009.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verschueren WG, Dierynck I, Amssoms KIE, Hu L, Boonants PMJG, Pille GME, Daeyaert FFD, Hertogs K, Surleraux DLNG, Wigerinck PBTP. J Med Chem. 2005;48:1930. doi: 10.1021/jm049559q. [DOI] [PubMed] [Google Scholar]
- 15.Walker MA, Johnson T, Naidu BN, Banville J, Remillard R, Plamondon S, Martel A, Li C, Torri A, Samanta H, Lin Z, Dicker I, Krystal M, Meanwell NA. Bioor Med Chem Lett. 2007;17:4886. doi: 10.1016/j.bmcl.2007.06.042. [DOI] [PubMed] [Google Scholar]
- 16.Petrocchi A, Koch U, Matassa VG, Pacini B, Stillmock KA, Summa V. Bioorg Med Chem Lett. 2007;17:350. doi: 10.1016/j.bmcl.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 17.Pace P, Di Francesco ME, Gardelli C, Harper S, Muraglia E, Nizi E, Orvieto F, Petrocchi A, Poma M, Rowley M, Scarpelli R, Laufer R, Gonzalez Paz O, Monteagudo E, Bonelli F, Hazuda D, Stillmock KA, Summa V. J Med Chem. 2007;50:2225. doi: 10.1021/jm070027u. [DOI] [PubMed] [Google Scholar]
- 18.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr J Med Chem. 2008;51:251. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 19.Naplolitano E, Giannone E, Fiaschi R, Marsili A. J Org Chem. 1983;48:3653. [Google Scholar]
- 20.Naplitano E, Spinelli G, Fiaschi R, Marsili A. J Chem Soc, Perkin Trans. 1986;1:785. [Google Scholar]
- 21.Baudart MG, Hennequin LF. J Antibiot. 1993;46:1458. doi: 10.7164/antibiotics.46.1458. [DOI] [PubMed] [Google Scholar]
- 22.Barreca ML, De Luca L, Iraci N, Chimirri A. J Med Chem. 2006;49:3994. doi: 10.1021/jm060323r. [DOI] [PubMed] [Google Scholar]
- 23.Barreca ML, Ortuso F, Iraci N, De Luca L, Alcaro S, Chimirri A. Biochem Biophys Res Commun. 2007;363:554. doi: 10.1016/j.bbrc.2007.08.199. [DOI] [PubMed] [Google Scholar]
- 24.Serrao E, Odde S, Ramkumar K, Neamati N. Retrovirol. 2009 doi: 10.1186/1742-4690-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leh H, Brodin P, Bischerour J, Deprez E, Tauc P, Brochon JC, LeCam E, Coulaud D, Auclair C, Mouscadet JF. Biochemistry. 2000;39:9285. doi: 10.1021/bi000398b. [DOI] [PubMed] [Google Scholar]
- 26.Marchand C, Neamati N, Pommier Y. Methods Enzymol (Drug-Nucleic Acid Interactions) 2001;340:624. doi: 10.1016/s0076-6879(01)40446-0. [DOI] [PubMed] [Google Scholar]
- 27.Semenova EA, Johnson AA, Marchand C, Davis DA, Yarchoan R, Pommier Y. Mol Pharmacol. 2006;69:1454. doi: 10.1124/mol.105.020321. [DOI] [PubMed] [Google Scholar]