

Abstract
The RNA-editing adenosine deaminases (ADARs) catalyze deamination of adenosine to inosine in double stranded structure found in various RNA substrates, including mRNAs. Here we describe the synthesis of a phosphoramidite of 2’-deoxy-2'-mercaptoadenosine and its incorporation into an ADAR substrate. Surprisingly, no deamination product was observed with this substrate indicating replacing the 2’-OH with a 2’-SH at the editing site is highly inhibitory. Modeling of nucleotide binding into the active site suggests the side chain of T375 of human ADAR2 to be in proximity of the 2’-substituent. Mutation of this residue to cysteine caused a greater that 100-fold reduction in deamination rate with the 2’-OH substrate.
INTRODUCTION
The RNA editing adenosine deaminase enzymes (ADARs) convert adenosines to inosines in various RNAs, including mRNAs. When this reaction takes place within a coding sequence, the meaning of codons can be altered.1–3 Thus, ADARs play a pivotal role in the basic process of information transfer that takes place during protein expression. A role for ADARs in nervous system function is implied by the observation that deletion of ADAR genes leads to significant behavioral defects in model organisms.4–6 Interestingly, in addition to the neurological phenotypes observed, both ADAR1 and ADAR2 knockout mice die prematurely.4,7 Thus, the precise role of ADAR activity in adult mammals remains to be fully defined.
Structural and biochemical studies have shed light on how ADARs recognize substrates and catalyze adenosine deamination in RNA.8–14 In general, members of the ADAR family contain multiple RNA-binding motifs that bind double-stranded RNA substrates in addition to a zinc-containing catalytic domain where deamination occurs.8,15,16 Although no structures of ADAR/RNA complexes have been reported, the structure of the human ADAR2 deaminase domain alone has been solved by x-ray crystallography.8 Furthermore, modeling of AMP into the zinc-containing active site suggests residues that may be involved in substrate recognition (Figure 1).8 In addition to the residues that bind zinc and the catalytic water molecule (H394, C451, C516 and E396), R455 and T375 are present in the ADAR2 active site. The nucleotide binding model suggests R455 is positioned near the purine N7 and T375 is near the 2’-hydroxyl group of the reactive nucleotide (Figure 1). Studies in our laboratory with 7-substituted-8-aza-7-deazaadenosine derivatives in RNA and residue 455 mutants of human ADAR2 support this proposed model (Maydanovych, Jayalath, Pokharel, Wang, Tantillo and Beal, unpublished results). Furthermore, we hypothesized that the close approach of the 2’ substituent and the side chain of the 375 residue in human ADAR2 might be used to create a covalent linkage between the enzyme and substrate. For instance, the combination of an A2’-SH-containing RNA and the T375C mutant of the enzyme might lead to the formation of a disulfide bond between enzyme and substrate. Such a stabilization of the enzyme/substrate complex would be useful in its biophysical characterization. Making and testing these modifications independently would also contribute to our understanding of structure/activity relationships in the ADAR/RNA complex. Here we report the synthesis of a phosphoramidite o f 2’-deoxy-2’-mercaptoadenosine and its use in the generation and evaluation of an A2’-SH-containing ADAR substrate. We also analyzed the effect of the T375C mutation on ADAR2 activity.
Figure 1.

A) A model for nucleotide recognition in the human ADAR2 active site.8 B) Structural changes made for this study: 2' OH → 2' SH, T375 → 375C
RESULTS AND DISCUSSION
Synthesis of the phosphoramidite of 2’-deoxy-2’-mercaptoadenosine proceeded from the disiloxyl-protected arabinofuranosyladenine derivative 1 in analogy to published reports for related nucleosides (Scheme 1).17,18 The 2’-triflate was prepared and used without purification in an SN2 displacement with the sodium salt of tritylmercaptan generating S-trityl compound 2 in 78% yield.19,20 Removal of the disiloxyl group with TBAF in THF provided intermediate 3. Proton NMR analysis of 3 revealed a 1'H-2'H J = 9.6 Hz, consistent with ribose configuration at the 2' position as expected for the SN2 displacement.18 Protection of the exocyclic amino group as the dimethylacetamidine and the 5’-hydroxyl as the DMT ether proceeded with good yields. Phosphitylation with 2-cyanoethyl-(N,N-diisopropylamino)chlorophosphite gave the fully protected phosphoramidite 6 in 74% yield. This phosphoramidite was used in the solid phase synthesis of oligoribonucleotides corresponding in sequence to that found in the pre-mRNA encoding the B subunit of the glutamate receptor near a known editing site (the R/G site).21,22 Successful incorporation of the modified nucleotide into RNA was confirmed by electrospray ionization mass spectrometry (see Experimental for details). Figure 2 shows the different sequences of the oligonucleotides prepared for this study. Oligonucleotide 9 was prepared for analysis of covalent complex formation using gel mobility shift assays. Oligonucleotide 12 was generated for analysis of the effect of 2'-SH modification on the adenosine deamination rate, which is more readily assayed when the targeted nucleotide is at the 5' end.22
Scheme 1.

Figure 2.

Oligonucleotides used in this study. Abbreviations: A2’STr = 2'-deoxy-2'-mercaptoadenosine protected as S-trityl; A2’SH = 2'-deoxy-2'-mercaptoadenosine.
Trityl was chosen as the protecting group for the 2’-mercaptan because of its ability to survive the conditions of RNA synthesis and deprotection of the common nucleotides and its lability in the presence of AgNO3, a reagent known to be compatible with RNA.17 Once the A2'-STr-containing oligonucleotide was purified, removal of the S-trityl group was achieved using AgNO3 essentially as described by Hamm and Piccirilli for cytidine-2’-S-trityl-containing RNAs.17 We confirmed these conditions were effective at protecting group removal by monitoring the mobility of 32P-labeled oligonucleotides in a polyacrylamide gel (Figure 3). After treatment with AgNO3 followed by DTT, the rate of migration of the oligonucleotide through the gel increased consistent with the resulting decrease in molecular weight. This is apparent when comparing the mobility of oligonucleotide 8 (no AgNO3, -STr 2' substituent) to oligonucleotide 9 (+ AgNO3, -SH 2' substituent) (Figure 3).
Figure 3.
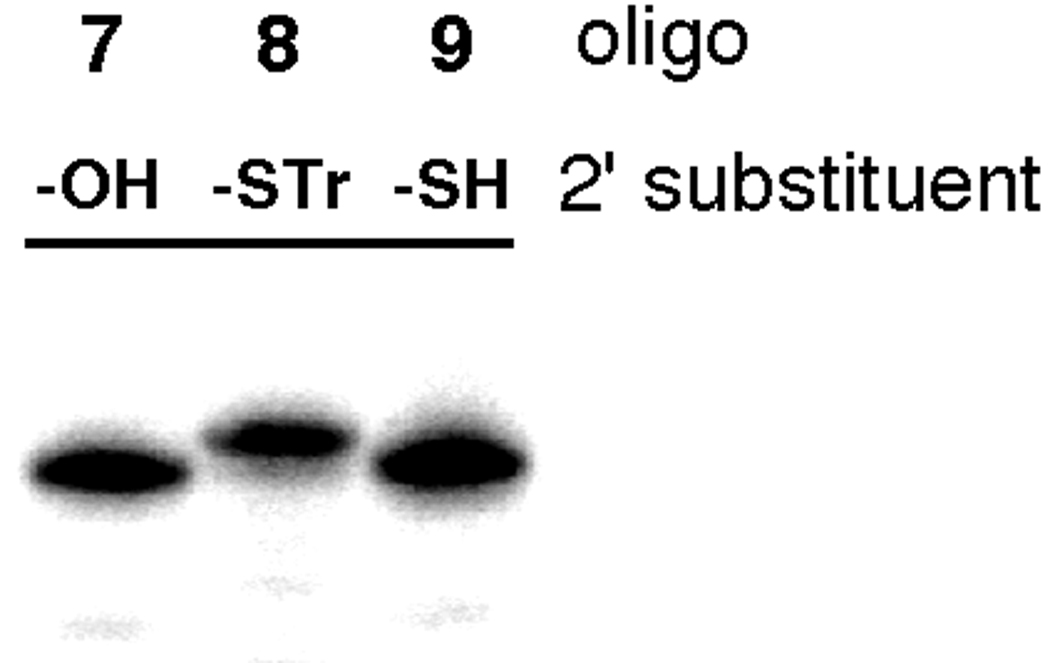
Gel mobility assay to monitor the AgNO3-promoted S-trityl deprotection of an A2’STr-containing RNA (See Experimental section for conditions).
We evaluated the effect of replacing the editing site 2'-hydroxyl group with a 2'-mercaptan using duplex structure designed to mimic the R/G editing site of GluR B pre-mRNA formed by hybridization of either oligonucleotides 10 or 12 with 13 (Figure 2, Figure 4). Similar duplexes have been shown previously to support editing by human ADAR2 in vitro. Indeed, the rate of deamination for the 2'-OH containing substrate (10 + 13) is similar to previous reports with the edited base at the duplex 5' end (Table 1).21,22 However, no product was observed with the 2'-SH substrate under these conditions (12 + 13), even after prolonged incubation with the enzyme. Previously, we had shown that although 2'-deoxyadenosine and 2'-deoxy-2'-fluoroadenosine in RNA were deaminated by ADAR2 with moderately reduced rates, 2'-O-methyl modification inhibited deamination by more than two orders of magnitude.23 Together these results indicate that ADAR2 is highly sensitive to modifications at the 2'-position that are more sterically demanding than the 2'-hydroxyl group, an effect that likely arises from a clash with the T375 residue.
Figure 4.

Duplex RNA substrate with varying nucleotide X at a known editing site (GluR-B pre-mRNA R/G site) prepared via hybridization of oligonucleotides 10 + 13 (X = A) or 12 + 13 (X = A2'-SH).
Table 1.
Rate constants for the deamination of duplex substrates a
| X | ADAR2 | ADAR2 T375C |
|---|---|---|
| A | 0.08 ± 0.002 min−1 | 6.8 ± 0.2 × 10−4 min−1 |
| A-2'SH | NDb | ND |
Reactions were carried out with 250 nM enzyme, 25 nM RNA substrate.
ND = no product detected.
Our interpretation of the low reactivity of the 2'-SH substrate suggested the ADAR2 reaction might also be sensitive to mutation at position 375. Therefore, we evaluated the effect of the T375C mutation on the ADAR2 deamination rate measured in vitro. Indeed, a > 100-fold reduced deamination rate was observed with this mutant compared to wild type ADAR2 using the 2'-OH substrate (10 + 13) (Table 1). It is interesting to note that the loop of ADAR2 containing T375 is not found in the evolutionarily related cytidine deaminases and it has been suggested that the adenosine selectivity of the ADARs arises from the positioning of this loop to prevent cytidine in RNA from entering the ADAR active site.8 Thus, this part of the enzyme appears to have evolved to sense the shape of its cognate substrate. As might be expected from the results described above, no stable covalent complex could be observed using electrophoretic mobility shift assays when duplexes containing the 2'-SH substrate oligonucleotide 9 were incubated with the ADAR2 T375C mutant under a variety of conditions.
In summary, we prepared a new phopshoramidite for incorporation of 2'-deoxy-2'-mercaptoadenosine into RNA. A duplex RNA mimic of a known editing site containing 2'-deoxy-2'-mercaptoadenosine was evaluated as a substrate for human ADAR2 in vitro with no deamination product detected. This result, along with our previous observation that 2'-O-methlyladenosine is an extremely poor ADAR2 substrate, suggests the ADAR reaction is highly sensitive to 2' modifications more sterically demanding than a hydroxyl group. Furthermore, we show here the ADAR2 reaction is also sensitive to cysteine mutation at position 375, a residue likely in proximity to the 2'-substituent in the enzyme/substrate complex.
EXPERIMENTAL
General Synthetic Procedures
Glassware for all reactions was oven dried at 125 °C overnight and cooled in a desiccator prior to use. Reactions were carried out under an atmosphere of dry nitrogen when anhydrous conditions were necessary. All reagents were purchased from commercial sources (Sigman/Aldrich or Fischer Scientific) and were used without further purification unless noted otherwise. Liquid reagents are introduced by oven-dried microsyringes. Tetrahydrofuran was passed through a column of activated aluminum.24 Thin layer chromatography (TLC) was performed with Merck silica gel 60 F254 precoated TLC plates, eluting with the solvents indicated. Short and long wave visualization was performed with a Mineralight multiband ultraviolet lamp at 254 and 365 nm, respectively. Flash column chromatography was performed on Mallinckrodt Baker silica gel 150 (60–200 mesh) or using an ISCO™ CombiFlash Companion with Teledyne RediSep Rx columns. 1H, 13C, and 31P Nuclear Magnetic Resonance spectra of pure compounds were acquired at 600, 125, and 121 MHz, respectively. Chemical shifts reported in parts per million (ppm) in the reference to a solvent peak. The abbreviations such as s, t, m, bs, dd, d stand for singlet, triplet, multiplet, broad singlet, doublet of doublets and doublet. High-resolution mass spectra were obtained at Department of Chemistry, University of Utah or at University of California, Davis Mass spectroscopy facility.
3´,5´-O-(1,1,3,3-Tetraisopropyldisiloxane-1,3-diyl)-2´-deoxy-2´-(triphenylmethylthio)adenosine (2)
Trifluoromethanesulfonyl chloride (0.232 mL, 2.17 mmol) was added to a cold (0 °C ) stirred solution of 9-[3´,5´-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-β-D-arabinofuranosyl]adenine 1 (1.01 g, 1.98 mmol) and DMAP (0.29 g, 2.37 mmol) in anhydrous CH2Cl2 (15 mL). The mixture was stirred at 0 ° C for 3 h and partitioned between ice-cold AcOH/H2O (1:99) and CH2Cl2 (2 × 100 mL). The combined organic phases were washed with ice-cold 5% (w/v) aqueous NaHCO3 (100 mL), brine (150 mL) and dried over anhydrous Na2SO4. The solvents were evaporated and the residue was dried under reduced pressure for 6 h. The above residue was dissolved in DMF (10 mL) and added dropwise during 30 min to a stirred solution of triphenylmethyl thiol (2.73 g, 9.9 mol) and NaH (0.2 g, 7.92 mmol) in DMF (15 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 3 h and allowed to reach room temperature overnight. The reaction mixture was poured into ice-cold water (150 mL) and extracted into hexane/ethyl acetate (3:7). The combined organic layers were washed with brine and dried over anhydrous Na2SO4 and evaporated under reduced pressure. Silica gel chromatography (3% MeOH in CH2Cl2 ) of the residue gave compound (2) as a white solid (1.87 g, 78%). 1H NMR (CDCl3 600 MHz): δ (ppm) 8.07 (s, 1H), 7.41-7.39 (m, 6H), 7.11-7.05 (m, 9H), 5.68 (bs, 2H), 5.50 (d, J = 3.0 Hz, 1H), 5.22 (t, J = 7.2 Hz, 1H), 4.14 (dd, J = 2.4, 7.2 Hz, 1H), 3.97-3.94 (m, 1H), 3.90 (d, J = 3.0, 12.6 Hz, 1H), 3.81 (d, J = 5.4, 12.6 Hz, 1H). 13C NMR (CDCl3 125 MHz): δ (ppm) 155.5, 152.7, 148.9, 144.7, 140.8, 129.6, 128.0, 126.9, 120.2, 90.3, 84.2, 71.8, 67.1, 61.9, 51.5, 17.6, 17.5, 17.49, 17.40, 17.39, 17.36, 17.3, 13.5, 13.3, 13.2, 12.9. ESIHRMS cald for C42H57N5O4SSi2 [M+H]+ 768.3435, obsd 768.3457
2´-Deoxy-2´-triphenylmethylthioadenosine (3)
Compound 2 (0.73 g, 0.95 mmol) was dissolved in anhydrous THF (25 mL) and treated with 1M n-Bu4NF (1.5 mL, 1.5 mmol) in THF. The mixture was stirred at 0 ° C for 3 h under an argon atmosphere and concentrated. The crude product was purified by column chromatography using silica gel and CH2Cl2/MeOH (20:1) to give compound 3 as white solid (0.44 g, 90%). 1H NMR (CDCl3 600 MHz): δ (ppm) 7.95 (s, 1H), 7.89 (s, 1H), 7.12-7.11 (m, 6H), 7.01-6.97 (m, 9H), 5.71 (d, J = 9.6 Hz, 1H), 4.04 (s, 1H), 3.77 (dd, J = 4.2, 10.2 Hz, 1H), 3.68 (dd, J = 1.8, 13.2 Hz, 1H), 3.3 (d, J = 12.6 Hz, 1H). 13C NMR (CDCl3 125 MHz): δ (ppm) 156.1, 152.2, 148.3, 143.9, 141.0, 128.9, 128.1, 127.1, 90.3, 88.4, 88.3, 72.0, 68.0, 63.2, 53.3, 53.2. ESIHRMS cald for C29H27N5O3S [M+H]+ 526.1913, obsd 526.1912.
N-(Dimethylacetamidine)-2'-deoxy-2'-triphenylmethylthioadenosine (4)
To a stirred solution of 3 (0.26 g, 0.49 mmol) in 7 mL of CH2Cl2/MeOH (4:3) was added N,N-dimethylacetamide dimethyl acetal (0.24 mL, 1.45 mmol) and stirring was continued overnight at room temperature. The solvents were evaporated under reduced pressure and the resulting residue was purified on silica gel column chromatography (CH2Cl2/MeOH (30:1) to give compound 4 (0.24 g) in 84% yield. 1H NMR (CDCl3 600 MHz): δ (ppm) 8.33 (s, 1H), 7.98 (s, 1H), 7.18-7.17 (m, 6H), 7.09-7.02 (m, 9H), 6.50 (dd, J = 1.2, 12.0 Hz, 1H), 5.76 (d, J = 10.2 Hz, 1H), 4.10 (s, 1H), 4.03 (d, J = 4.2, 10.2 Hz, 1H), 3.79 (d, J = 13.2 Hz, 1H), 3.40 (t, J = 12.6 Hz, 1H), 3.29 (bs, 3H), 3.16 (bs, 3H), 2.74 (d, J = 4.2 Hz, 1H). 13C NMR (CDCl3 125 MHz): δ (ppm) 162.2, 161.0, 152.5, 149.9, 143.9, 141.9, 129.05, 129.0, 128.3, 128.0, 127.2, 90.2, 88.1, 71.9, 67.9, 63.6, 53.6, 46.4, 38.8, 38.7, 17.8, 11.7. ESIHRMS calcd for C33H35N6O3S [M+H]+ 595.2491, obsd 595.2484.
N-(Dimethylacetamidine)-5'-O-(4,4'-dimethoxytriphenylmethyl)-2'-deoxy-2'-triphenylmethylthioadenosine (5)
A suspension of 4 (155 mg, 0.26 mmol), and N,N-diisopropylethylamine (50 µL, 313 mmol) in anhydrous pyridine (1 mL) is treated with 4,4'-dimethoxytriphenylmethyl chloride (112 mg, 313 mmol). After stirring at room temperature overnight the reaction mixture was diluted with EtOAc (25 mL) and washed with saturated aqueous NaHCO3. The organic layer was removed and dried over anhydrous Na2SO4 and evaporated under reduced pressure. Silica gel chromatography (EtOAc ) of the residue gave compound (5) as a white solid (168 mg, 72%). 1H NMR (CD2Cl3 600 MHz): δ (ppm) 8.36 (s, 1H), 8.10 (s, 1H), 7.27-7.09 (m, 24H), 6.70-6.68 (m, 4H), 6.11 (d, J = 9.6 Hz, 1H), 4.07 (dd, J = 4.8, 9.6 Hz, 1H), 4.02 (t, J = 4.2 Hz, 1H), 4.17-4.13 (m, 1H), 3.77 (s, 3H), 3.76 (s, 3H), 3.27 (bs, 3H), 3.22 (dd, J = 4.2, 10.8 Hz, 1H), 3.19 (bs, 3H), 3.03 (dd, J = 3.0, 10.2 Hz, 1H), 2.58 (d, J = 4.8 Hz, 1H), 2.23 (s, 3H). 13C NMR (CD2Cl2 125 MHz): δ (ppm) 158.8, 153.1, 151.8, 144.7, 144.1, 140.8, 135.99, 135.98, 130.2, 130.19, 129.2, 128.5, 128.4, 127.9, 127.3, 127.0, 126.7, 113.2, 86.6, 86.5, 85.4, 70.9, 67.5, 63.9, 55.5, 54.8, 45.0, 17.5. ESIHRMS calcd for C33H35N6O3S [M+H]+ 897.3798, obsd 897.3816.
N-(Dimethylacetamidine)-5'-O-(4,4'-dimethoxytriphenylmethyl)-3'-O-[(2-cyanoethoxy)(N,N-diisopropylamino)phosphino]-2'-deoxy-2'-triphenylmethylthioadenosine (6)
A solution of 5 (92 mg, 0.1 mmol), DMAP (13 mg, 0.11 mmol) and N,N-diisopropylethylamine (39 µL, 0.23 mmol) in anhydrous THF (0.8 mL) was treated dropwise with 2-cyanoethyl-(N,N-diisopropylamino) chlorophosphite (53 µL, 0.23 mmol). After stirring at room temperature for 4 h the reaction mixture was diluted with EtOAc (25 mL) and washed with 5% (w/v) aqueous NaHCO3. The organic layer was removed and dried over anhydrous Na2SO4 and evaporated under reduced pressure. Silica gel chromatography (CH2Cl2/TEA; 99:1) of the residue gave compound (6) as a white foam (82 mg, 74%). 31P NMR (CD2Cl2, 121 MHz); δ (ppm) 153.10, 150.55. ESIHRMS calcd for C63H69N8O6PS [M+H]+ 1097.3187, obsd. 1097.3418.
Synthesis, purification and mass spectrometric analysis of RNA
RNA oligonucleotides were synthesized on a ABI 394 synthesizer (DNA/Peptide Core Facility, University of Utah, Salt Lake City) using 5'-DMT protected β-cyanoethyl phosphoramidites on a 1 µmol scale with coupling times of 25 min for more efficient coupling. The RNA oligonucleotides after polyacrylamide gel electrophoresis purification were analyzed by mass spectrometry at the Mass Spectrometry and Proteomics Core Facility, University of Utah. Oligonucleotides were analyzed in an acetonitrile solution at pH = 11 by electrospray ionization on a Quattro-II mass spectrometer (Micromass, Inc.). The multiply-charged molecular ions were deconvoluted into a molecular mass spectrum using MaxEnt (Micromass, Inc.) software. The Quattro-II instrument was operated using Masslynx software version 3.4 (Micromass, Inc.). ESI-MS analysis results: 2'-deoxy-2'-triphenylmethylthioadenosine 26 mer RNA oligonucleotide 8 calcd. (M-H)− 9003.7, obsd 9003.2; 2'-deoxy-2'-triphenylmethylthioadenosine 22 mer RNA oligonucleotide 11 calcd. (M-H)− 7427.7; obsd 7427.5.
General Biochemical Procedures
Distilled, deionized water was used for all aqueous reactions and dilutions. Biochemical reagents were purchased from Sigma/Aldrich unless otherwise noted. Common enzymes were purchased from Roche, Promega, or New England Biolab. ADAR2 overexpression and purification were carried out as previously described.25 5’-End labeling of RNA oligonucleotides was accomplished using T4 polynucleotide kinase and [γ-32P]ATP, 6000 Ci/mmol (Perkin-Elmer Life Sciences) as previously described.22
Removal of S-trityl group to generate oligonucleotides 9 and 12
To remove the S-trityl protecting group from the RNA oligonucleotide, 750 pmol of RNA dissolved in 7.3 µL of 100 mM TEAA, pH= 6.5 was treated with 1 µL of 1M aqueous silver nitrate solution. This reaction was mixed at room temperature by shaking in orbital shaker for 45 min. Dithiothreitol (2 µL of 1M aqueous solution) was added to the reaction and further mixed by shaking an additional five min. TEAA, pH = 6.5 (50 µL of 100 mM) was added, mixed and centrifuged to remove silver/DTT precipitate. The supernatant containing the deprotected oligonucleotide was transferred to a new Eppendorf tube. The precipitate was washed with 50 µL of 100 mM TEAA, pH=6.5 buffer. The supernatants containing the oligonucleotide were combined. Prior to end labeling, the buffer was exchanged for 50 mM BisTris, pH = 7.0, 100 mM NaCl, 10 mM MgCl2 with 100 µM TCEP using a Microcon-3 concentrator. For monitoring the deprotection reaction, end labeled RNAs 7, 8 and 9 were resolved on a 19% PAGE gel and visualized using storage phosphor autoradiography (Figure 3).
Preparation of duplex RNA ADAR substrate and deaminase assay
5'-End 32P-labeled duplex RNA substrates were prepared as previously described with a minor modification as follows. End labeled A2’-SH-containing RNA oligonucleotide (12) was mixed with a two-fold excess of unlabeled 12 and 10-fold excess of unlabeled complement strand (13) in presence of 5 mM DTT. ADAR activity assays were carried out as previously described.22
ACKNOWLEDGMENTS
P.A.B. acknowledges the National Institutes of Health for financial support in the form of grant GM061115.
REFERENCES
- 1.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 2.Grosjean H, Benne R. Modification and editing of RNA. Washington, D.C: ASM Press; 1998. [Google Scholar]
- 3.Higuchi M, Single FN, Koehler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron-exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 4.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 5.Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. A-to-I Pre-mRNA Editing in Drosophila Is Primarily Involved in Adult Nervous System Function and Integrity. Cell. 2000;102(4):437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 6.Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002;21(22):6025–6035. doi: 10.1093/emboj/cdf607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA Editing Deaminase ADAR1 Gene for Embryonic Erythropoiesis. Science. 2000;290(5497):1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 8.MacBeth MR, Schubert HL, VanDemark AP, Lingam AT, Hill CP, Bass BL. Inositol Hexakisphosphate Is Bound in the ADAR2 Core and Required for RNA Editing. Science. 2005;309(5740):1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacBeth MR, Lingam AT, Bass BL. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA. 2004;10(10):1563–1571. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho D-SC, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of Dimerization for RNA Editing Activity of Adenosine Deaminases Acting on RNA. J. Biol. Chem. 2003;278(19):17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- 11.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH-T. Structure and Specific RNA Binding of ADAR2 Double-Stranded RNA Binding Motifs p345. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Dawson TR, Sansam CL, Emeson RB. Structure and Sequence Determinants Required for the RNA Editing of ADAR2 Substrates. J. Biol. Chem. 2004;279:4941–4951. doi: 10.1074/jbc.M310068200. [DOI] [PubMed] [Google Scholar]
- 13.Maydanovych O, Easterwood LM, Cui T, Veliz EA, Pokharel S, Beal PA. Probing Adenosine-to-Inosine Editing Reactions Using RNA-Containing Nucleoside Analogs. Methods Enzymol. 2007;424:369–386. doi: 10.1016/S0076-6879(07)24017-0. [DOI] [PubMed] [Google Scholar]
- 14.Stephens OM, Haudenschild BL, Beal PA. The Binding Selectivity of ADAR2's dsRBMs Contributes to RNA-editing Site Selectivity. Chem. Biol. 2004;11(9):1239–1250. doi: 10.1016/j.chembiol.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Bass BL, Weintraub H. A developmentally regulated activity that unwinds RNA duplexes p607. Cell. 1987;48(4):607–613. doi: 10.1016/0092-8674(87)90239-x. [DOI] [PubMed] [Google Scholar]
- 16.Doyle M, Jantsch MF. Distinct in vivo roles for double-stranded RNA-binding domains of the Xenopus RNA-editing enzyme ADAR1 in chromosomal targeting. J. Cell Biol. 2003;161(2):309–319. doi: 10.1083/jcb.200301034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamm ML, Piccirilli JA. Incorporation of 2'-Deoxy-2'-mercaptocytidine into Oligonucleotides via Phosphoramidite Chemistry. J. Org. Chem. 1997;62:3415–3420. doi: 10.1021/jo970096o. [DOI] [PubMed] [Google Scholar]
- 18.Marriott JH, Mottahedeh M, Reese CB. Synthesis of 2'-thioadenosine. Carbohydr. Res. 1991;216:257–269. [Google Scholar]
- 19.Robbins MJ, Hawrelak SD, Hernandez AE, Wnuk SF. Nucleic Acid Related Compounds. 71. Efficient general synthesis of purine (amino, azido, and triflate)-sugar nucleoside. Nucleosides Nucleotides. 1992;11(2–4):821–834. [Google Scholar]
- 20.Gruen M, Becker C, Beste A. Synthesis of 2'-Iodo- and 2'-Bromo-ATP and GTP Analogues as Potential Phasing Tools for X-ray rystallography. Nucleosides Nucleotides. 1999;18(1):137–151. [Google Scholar]
- 21.Easterwood LM, Veliz EA, Beal PA. Demethylation of 6-O-Methylinosine by an RNA-editing Adenosine Deaminase. J. Am. Chem. Soc. 2000;122:11537–11538. [Google Scholar]
- 22.Stephens OM, Yi-Brunozzi HY, Beal PA. Analysis of the RNA-editing Reaction of ADAR2 with Structural and Fluorescent Analogs of the GluR-B R/G Editing Site. Biochemistry. 2000;39(40):12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 23.Yi-Brunozzi HY, Easterwood LM, Kamilar GM, Beal PA. Synthetic Substrate Analogs for the RNA-Editing Adenosine Deaminase ADAR-2. Nucleic Acids Res. 1999;27(14):2912–2917. doi: 10.1093/nar/27.14.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15(5):1518–1520. [Google Scholar]
- 25.MacBeth MR, Bass BL. Large-Scale Overexpression and Purification of ADARs from Saccharomyces cerevisiae for Biophysical and Biochemical Studies. Methods Enzymol. 2007;424:319–331. doi: 10.1016/S0076-6879(07)24015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]