

Abstract
Traditional crosslinked polyester elastomers are inherently weak, and the strategy of increasing crosslink density to improve their mechanical properties makes them brittle materials. Biodegradable polyurethanes, although strong and elastic, do not fare well in dynamic environments due to the onset of permanent deformation. The design and development of a soft, strong and completely elastic (100% recovery from deformation) material for tissue engineering still remains a challenge. Herein, we report the synthesis and evaluation of a new class of biodegradable elastomers, crosslinked urethane-doped polyesters (CUPEs), which is able to satisfy the need for soft, strong, and elastic biomaterials. Tensile strength of CUPE was as high as 41.07 ± 6.85 MPa with corresponding elongation at break of 222.66 ± 27.84%. The initial modulus ranged from 4.14 ± 1.71 MPa to 38.35 ± 4.5 MPa. Mechanical properties and degradation rates of CUPE could be controlled by varying the choice of diol used for synthesis, the polymerization conditions, as well as the concentration of urethane bonds in the polymer. The polymers demonstrated good in vitro and in vivo biocompatibilities. Preliminary hemocompatibility evaluation indicated that CUPE adhered and activated lesser number of platelets compared to PLLA. Good mechanical properties and easy processability make these materials well suited for soft tissue engineering applications. The introduction of CUPEs provides new avenues to meet the versatile requirements of tissue engineering and other biomedical applications.
Keywords: Biodegradation, Elastomer, Scaffold, Biocompatibility, Tissue engineering
1. Introduction
The healthy growth and development of native soft tissues such as vasculature, cardiac tissue, bladder, and cartilage is predominantly dependent on biochemical and mechanical interactions between cells and the underlying extracellular matrix (ECM) [1–3]. For cells seeded on a synthetic scaffold used for soft tissue engineering, the scaffold must provide cues similar to those between the cells and the native ECM. One strategy aimed at fulfilling this requirement involves the use of scaffolds, which are analogous to the ECM of the target tissue in terms of its mechanical, physical, and chemical properties. To better mimic the ECM and transfer mechanical stimulation from the tissue surroundings to the seeded cells, the scaffold has to be soft and elastic, so that they are able to sustain and recover from cyclic deformations which are characteristic of these mechanically active tissues.
In the search for an ideal scaffold material for soft tissue engineering, numerous synthetic biodegradable elastomeric polymers have been synthesized [4–11]. Elastomers such as poly(diol citrates) and poly(glycerol sebacate) (PGS) have received much attention recently [4,12]. These elastomers are polymer networks comprising aliphatic polyester chains bonded to each other by ester crosslinks. They are soft and elastic (100% recovery from deformation) with excellent cell/tissue compatibility. Although these polyester elastomer scaffolds have been proposed for engineering tissues like nerve guides [13–15] and small diameter blood vessels [16], their success in in vivo tissue engineering is still not proven. They are weak and hence, unsuitable for engineering tissues like ligaments, which are characterized by high tensile strength and load bearing abilities. For example, the human anterior cruciate ligament has a ultimate tensile strength of at least 38 MPa [17], which is much stronger that that of poly(diol citrates) (up to 11.15 ± 2.62 MPa). In addition to matching the properties of the target tissue, an ideal tissue engineering scaffold should also possess sufficient mechanical strength to support surgical handling soon after initial cell seeding. These stringent requirements on mechanical properties must be taken into consideration when designing scaffolds for tissue engineering. The tensile strength of poly(diol citrates) and PGS films varies from 11.15 ± 2.62 MPa to 0.5 ± 0.2 MPa, respectively. However, when these materials are fabricated into porous scaffolds, they lose mechanical strength significantly. For example, poly(1,8-octanediol citrate) (POC), one of the members of the poly(diol citrates) family, underwent a significant loss in mechanical properties, with the peak stress of the material dropping from 2.93 ± 0.09 MPa (film) to 0.3 ± 0.1 MPa (scaffold) upon scaffold fabrication [16]. Although the strength and stability of these network polyesters like poly(diol citrates) and PGS can be improved substantially by increasing the crosslinking density, such an approach would also compromise its elasticity and make the material more brittle. As one can expect a significant loss of mechanical strength when polymers are fabricated into porous scaffolds, it is important that the polymers used for scaffold fabrication should be strong enough to allow room for potential reduction of mechanical strength. Currently, no biodegradable polyester elastomeric scaffold can mechanically function for immediate implantation. The development of a soft, strong and totally elastic biodegradable elastomer, therefore, remains a challenge in the field of biomaterial science.
Biodegradable polyurethanes (BPUs) are another class of elastic biomaterials that have been developed for soft tissue engineering applications [18–22]. These polymers have good mechanical strength (up to 29 MPa tensile strength) and elasticity (up to 895% elongation). However, due to their aliphatic nature BPUs are susceptible to permanent creep under cyclic mechanical loading. Therefore, the potential long-term success of polyurethanes as scaffold materials for dynamic tissues like blood vessels and ligaments is still questionable.
In this work, we report the synthesis and characterization of a new class of crosslinked polyester elastomer networks. This class of polymers termed crosslinked urethane-doped polyesters (CUPEs) combines the advantages of fully elastic (100% recovery) and biocompatible/hemocompatible crosslinked polyester networks with mechanically strong polyurethanes. The previously developed poly(diol citrates) have excellent elasticity with 100% recovery from deformation due to the crosslinking nature of the polymers. The high mechanical strength of polyurethanes primarily stems from the strong hydrogen bonding between inter-chain urethane bonds. Taken together, the rationale behind CUPE synthesis is: (1) crosslinking confers excellent elasticity to CUPEs; (2) ester bonds confer degradability to CUPEs, all the crosslinks of the polymer’s network consist of ester bonds to ensure a degradable crosslinked polymer network; (3) introduction of urethane bonds into the polyester chains between ester crosslinks enhances the hydrogen bonding within the polyester network thus significantly improving mechanical strength of the CUPE network. In this work, we describe the synthesis and characterization of the new CUPE polymers. Both in vitro and in vivo models were used to assess the biocompatibility of CUPEs. The hemocompatibility of CUPE was studied with respect to its interactions with human blood.
2. Materials and methods
All chemicals, cell culture medium and supplements were purchased from Sigma—Aldrich (St. Louis, MO), except where mentioned otherwise. LIVE/DEAD Viability/Cytotoxicity Kit was purchased from Invitrogen (Carlsbad, CA). All chemicals were used as-received.
2.1. CUPE synthesis
CUPE synthesis was carried out in three distinct steps (Fig. 1). Step 1 involved the synthesis of the POC pre-polymer. POC was synthesized as per previously published methods with slight modifications [23]. Briefly, citric acid and 1,8-octanediol, with a monomer ratio of 1:1.1, were bulk polymerized in a three-necked reaction flask, fitted with an inlet and outlet adapter, at 160–165 °C. Once the mixture had melted, the temperature was lowered to 140 °C, and the reaction mixture was stirred for another 60 min to create the POC pre-polymer. The pre-polymer was purified by drop wise precipitation in deionized water. Undissolved pre-polymer was collected and lyophilized to obtain the purified POC pre-polymer. The average molecular weight of pre-POC was characterized as 850 Da by Matrix assisted laser desorption/ionization mass spectroscopy (MALDI-MS) which was done using an Autoflex MALDI-TOF Mass Spectrometer (Bruker Daltonics, Manning Park, MA). Chain extension of the POC pre-polymer to obtain pre-CUPE was done in Step 2. Purified pre-POC was dissolved in 1,4-dioxane to form a 3% (wt/wt) solution. The polymer solution was reacted with 1,6-hexamethyl diisocyanate (HDI) in a clean reaction flask under constant stirring, with stannous octoate as catalyst (0.1% wt). Different pre-CUPE polymers were synthesized with different feeding ratios of pre-POC:HDI (1:0.6, 1:0.9 and 1:1.2, molar ratio). The system was maintained at 55 °C throughout the course of the reaction. Small amounts of the reaction mixture were removed at 6-h intervals and subjected to Fourier transform infrared (FT-IR) analysis. The reaction was terminated when the isocyanate peak at 2267 cm−1 disappeared. In step 3, the pre-CUPE solution was cast into a Teflon mold and allowed to dry in a chemical hood equipped with a laminar airflow until all the solvents had evaporated. The resulting pre-CUPE film was moved into an oven maintained at 80 °C for pre-determined time periods to obtain crosslinked urethane-doped polyester (CUPE).
Fig. 1.

Synthesis schematic of CUPE polymers. The monomers citric acid and 1,8-octanediol underwent condensation polymerization to produce hydroxyl group capped pre-poly(1,8-octanediol citrate) in step 1. In step 2, 1,6-hexamethyl diisocyanate was used to extend the pre-POC chain. In step 3, the pre-CUPE was post-polymerized to obtain CUPE network.
The synthesis procedure was repeated with poly(ethylene glycol) (PEG) (Mw = 200) by replacing all or part of 1,8-octanediol as the diol portion. This class of CUPEs was abbreviated as CA-PEGUX/Y, where CA-PEGU refers to citric acid-PEG-urethane, X/Y denotes the molar ratio of 1,8-octanediol to PEG used in the synthesis. For example, CA-PEGU8/3 indicates that 0.08 mol of 1,8-octanediol and 0.03 mol of PEG were reacted with 0.1 mol of citric acid. The molar ratio of the CA-PEGU pre-polymer:HDI was maintained at 1:0.9 for all the different variants.
2.2. Polymer characterization
Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet 6700 FT-IR spectrometer (Thermo Fisher Scientific) at room temperature. A dilute solution of CUPE and POC pre-polymers in 1,4-dioxane (3% wt/wt) was cast on KBr crystals and allowed to dry for 12 h under vacuum before being used to obtain the spectra. Attenuated total reflectance spectroscopic analysis was performed using an IRAffinity-1 ATR spectrometer (Shimadzu Corp, Japan) on solvent cast CUPE and POC films which had been polymerized in an 80 °C oven for 2 days.
Differential Scanning Calorimetry (DSC) using a DSC2010 Differential Scanning Calorimeter (TA Instruments, New Castle, DE) and Thermogravimetric Analysis (TGA) using an SDT2960 Simultaneous DSC—TGA (TA Instruments, New Castle, DE) were used to characterize the thermal properties of different CUPE polymers. For DSC, the polymer samples were scanned to 150 °C under nitrogen with a step size of 10 °C/min, followed by cooling at a rate of −40 °C/min, until a temperature of −60 °C was achieved. Readings were then taken during a second heating scan from −60 °C to 230 °C, with a constant heating rate of 10 °C/min. The glass transition temperature Tg was determined from the middle of the recorded step change in heat capacity from the second heating run. For TGA, the polymer samples were heated at a rate of 10 °C/min from 50 °C to 600 °C, in an atmosphere of nitrogen gas. The temperature, at which 10% loss of the initial weight occurred, was recorded as the decomposition temperature Td.
Mechanical testing was conducted with an MTS Insight 2 machine fitted with a 500 N load cell. The CUPE films were cut into a dog bone shape, as per ASTM D412a (25 × 6 × 1.5 mm, length × width × thickness). The deflection rate was adjusted to 500 mm/min. Samples were elongated to failure. The initial modulus was calculated by measuring the gradient at 10% of elongation of the stress—strain curve. Five specimens per sample were tested and averaged.
The density of the different polymers was measured by the fluid displacement method using a density measurement kit (Mettler Toledo, Columbus, OH). The auxiliary liquid used was deionized water.
Crosslink density and molecular weight between crosslinks were evaluated according to the theory of rubber elasticity [4,5].
| (1) |
where N is the number of moles of active network chains per unit volume, E0 is the initial modulus of the polymer, R stands for the universal gas constant, T represents the absolute temperature, ρ is the density of the polymer and Mc is the relative molar mass between crosslinks.
Water in air contact angle measurements were made on CUPE thin films by sessile drop method, using a KSV101 Optical Contact Angle and Surface Tension Meter (KSV Instruments Inc. Helsinki, Finland). Readings were collected within 10 s after dropping. Eight readings were collected from different regions on the CUPE films.
2.3. In vitro degradation
Degradation studies were conducted in both phosphate buffered saline (PBS) (pH 7.4) and NaOH solutions (0.01 M and 0.05 M). NaOH degradation was used to screen the polymer degradation in a relatively short period of time [5]. The polymer films were cut into 7 mm discs using a cork borer. The initial weights of the samples were noted, and they were placed in test tubes containing 10 ml of either PBS or NaOH and incubated at 37 °C for the period of the study. At pre-set time points, the samples were removed and washed thoroughly with deionized water (3 times) to remove any residual salt. The samples were then lyophilized for 3 days to remove traces of water and weighed. Degradation was measured by determining the mass loss of the sample over the period of the study, as shown in Eq. (2), where W0 represents the initial weight of the specimen and Wt represents its weight after it has been degraded.
| (2) |
2.4. Scaffold fabrication
Both particulate leaching and thermal induced phase separation (TIPS) methods were used to fabricate CUPE scaffolds. Sodium chloride salt (99% purity) with an average size in the range of 150–250 μm was used as the porogen in the particulate leaching technique. Briefly, 3% (wt/wt) solution of CUPE1.2 in 1,4-dioxane was mixed with sodium chloride salt (150–250 μm) in a 1:9 ratio of CUPE to salt (by weight). The slurry obtained was mixed thoroughly, until it became a viscous paste, and was then cast into Teflon molds. The Teflon molds were kept overnight in a vacuum oven, to aid solvent evaporation. The dried scaffolds were polymerized in an oven maintained at 80 °C for 4 days. The salt in the scaffold was leached out by immersion in DI water for 48 h. Finally, the scaffolds were freeze dried for three days to remove any traces of water. These scaffolds were used for subsequent in vivo foreign response studies.
Thin CUPE scaffold sheets were fabricated by a thermal induced phase separation (TIPS) method with minor modifications [24–27]. Briefly, 3% solutions of pre-CUPE (wt/wt) in 1,4-dioxane were cast into Teflon molds. The molds with the polymer solution were immediately transferred to a −80 °C freezer. The frozen polymer solution was freeze dried for 4 days to obtain porous pre-CUPE scaffolds. The resulting pre-CUPE scaffolds were further polymerized in an oven (80 °C, 2 days) to obtain CUPE scaffolds. Scanning electron microscopy (SEM) was utilized to study the morphology and pore structure of the obtained scaffolds.
2.5. In vitro cell culture
Cytotoxicity of CUPE was evaluated in vitro using both qualitative and quantitative methods. CUPE1.2 was used for all cytotoxicity evaluation studies. The CUPE films and TIPS fabricated scaffolds were cut into discs (7 mm) and sterilized by treatment with 70% ethanol for 30 min, followed by another 30 min of UV light exposure. NIH 3T3 fibroblasts and human aortic smooth muscle cells (HASMCs) were used as model cells for cytotoxicity evaluation. The cells were cultured in 75 cm2 tissue culture flasks with Dulbecco’s modified eagle’s medium (DMEM), which had been supplemented with 10% fetal bovine serum (FBS). The culture flasks were kept in an incubator maintained at 37 °C, 5% CO2 and 95% humidity. The cells were trypsinized, centrifuged and suspended in media to obtain a seeding density of 3 × 105 cell/ml for the films. A seeding density of 8 × 105 cell/ml was used for seeding on the scaffolds. 200 μl of the cell suspension was added on top of the polymer specimens in the culture dishes. After 1 h of incubation at 37 °C, 5 ml of media was added to each culture dish. After 3 days and 6 days in culture for the films and scaffolds, respectively, the cells were fixed by addition of 5 ml of 2.5% (wt/v) glutaraldehyde—PBS solution. The fixed film and scaffold specimens were sequentially dehydrated by treatment with a graded series of ethanol (50%, 75%, 95%, and 100%), freeze dried and then sputter coated with silver. SEM examination was carried out on a Hitachi 3000N SEM to observe the morphology of the cells on the CUPE surface. Cell seeded scaffolds were also stained using a LIVE/DEAD assay kit (LIVE/DEAD Viability/Cytotoxicity Kit, Invitrogen, Carlsbad, CA) and observed under a fluorescence microscope.
Quantitative assessment of cell growth and proliferation on CUPE1.2 films was done using the Methylthiazoletetrazolium (MTT) cell proliferation and viability assay. CUPE films were cut into discs (7 mm diameter) and placed in a 96-well plate. PLLA films were used as a relative control. HASMCs were seeded on the CUPE and PLLA films as per the aforementioned seeding protocol. Three time points were used for the MTT Assay analysis (1, 3, and 5 days). At the end of each time point, the assay was performed as per the manufacturer’s protocol. Briefly, the old media was aspirated, and each specimen was thoroughly washed 3 times with PBS to remove any loosely attached cells and dead cells. 100 μl of incomplete media (DMEM without serum) was added to these specimens. 10 μl of 3-(4,5-dimethylthiazol-2yl)-diphenyltetrazolium bromide solution was then added to the wells containing the cells and the blanks, and they were incubated at 37 °C for 3 h. At the end of incubation period, the mixture of the MTT solution and incomplete media was aspirated and replaced with 100 μl of MTT solvent. Dissolution of formazan crystals was facilitated by constantly agitating the well plate on an orbital shaker for 15 min. Absorbance was analyzed with an Infinite200 microplate reader (Tecan Group Ltd., Switzerland) at 570 nm, with a reference wavelength of 690 nm, within 30 min of MTT solvent addition.
2.6. Foreign body response
CUPE1.2 was used in order to evaluate the in vivo response to the polymer. Both films and salt-leached scaffold forms of CUPE and PLLA samples (as relative control) were used. The films and scaffolds were cut into discs (10 mm diameter, and 1 mm thickness), and then implanted subcutaneously in the back of healthy, female Sprague Dawley (SD) rats (Harlan Sprague Dawley, Inc., Indianapolis, IN). Animals were cared for in compliance with regulations of the animal care and use committee of University of Texas at Arlington. Briefly, the discs were first sterilized by treatment with 75% ethanol for 30 min followed by treatment under UV light for another 30 min. 24 SD rats were divided into groups of 4 rats each for the different time points of the study. The rats were anesthetized in a chamber through which an isofluorane—oxygen mixture was passed. Test samples were randomly implanted in the upper or lower back of the rats by blunt dissection. At the end of each time point, the rats were sacrificed and the implant and surrounding tissue were frozen in OCT embedding media (Polysciences Inc., Warrington, PA) at −80 °C for histological analyses. To assess the tissue responses to film and scaffold implants, tissue sections were hematoxylin & eosin (H&E) stained. Images of stained sections were taken at 10 × magnifications using a Leica DMLP microscope fitted with a Nikon E500 camera (Nikon Corp. Japan). Three images per section were collected from different parts of the section for analysis. A total of three sections per animal were examined in this manner. Fibrous capsule thickness was measured in each of the images using the Image J analysis software. At least 25 readings from different parts of the images were collected and averaged to determine the capsule thickness.
2.6.1. Platelet adhesion
All methods related to collection and handling of whole blood and blood components like plasma and platelets were approved by the Institutional Review Board at the University of Texas at Arlington. Acid citrate dextrose (ACD) anticoagulant containing tubes were used to collect blood drawn from healthy individuals by venipuncture. Platelet-rich-plasma (PRP) was prepared using methods previously described in literature [12]. Briefly, whole blood was centrifuged at 250g for 15 min to separate the blood components. Using a sterile transfer pipette, the clear supernatant containing the PRP was collected for platelet adhesion studies. All hemocompatibility studies were conducted on CUPE1.2.
Platelet adhesion to the different polymer surfaces was examined by SEM. 200 μl of PRP was added on top of the PLLA and CUPE discs (7 mm diameter). After 1 h of incubation at 37 °C, PRP was aspirated and the samples were rinsed thoroughly with phosphate buffered saline (PBS) 3 times to remove any loosely adhered and unattached platelets. The adhered platelets were fixed using 2.5% glutaraldehyde—PBS solution, sequentially dehydrated in a graded series of alcohol (50%, 75%, 90% and 100%) and lyophilized. The samples were sputter coated with silver and examined using SEM. Three specimen discs of each polymer were prepared for SEM observation.
Quantitative assessment of adherent platelets was done using the lactate dehydrogenase (LDH) assay as previously described [12,28]. Polymer samples were incubated and washed as per the aforementioned procedure for SEM sample preparation. The samples were then treated with 2% Triton—PBS buffer for 30 min at 37 °C to lyse the adhered platelets. 100 μl of LDH assay substrate was added to the platelet lysate and incubated for 30 min at 37 °C in darkness. 0.1 N of HCl was added to stop the reaction at the end of the incubation period. Optical density was measured at 490 nm with a reference wavelength of 650 nm.
2.6.2. Platelet activation
Platelet activation was assessed by measuring the expression of P-selectin on platelet plasma membrane [29,30]. Briefly, CUPE, PLLA, and TCP samples were treated with 200 μl of PRP for 1 h at 37 °C. 5 μl of the material exposed PRP was incubated with 20 μl phycoerythrin (PE)-conjugated mouse anti-human CD42a monoclonal antibodies and 20 μl allophycocyanin (APC)-conjugated mouse antihuman CD62p monoclonal antibodies at room temperature for 30 min in darkness. CD42a, known as platelet glycoprotein IX (GP9), is a small membrane glycoprotein found on the platelet plasma membrane, and is used as a platelet marker. CD62p, a member of the P-selectin family, is used as an indicator of activated platelets. After staining, the cells were fixed with 1% paraformaldehyde and measured for the antibodies bound to platelet membranes using FACS array bioanalyzer (Becton Dickinson FACSArray, San Jose, CA). 10,000 platelets were acquired and the mean values of red fluorescence intensity (APC-CD62p), which corresponded to the activated platelets, were measured. These values were compared with the fluorescence intensity obtained from the PLLA group, which was set to 100% as the control.
2.7. Statistical methods
Data are expressed as mean ± standard deviation. The statistical significance between two sets of data was calculated using two-tail Student’s t-test. Data were taken to be significant, when p < 0.05 was obtained.
3. Results
3.1. Polymer characterization
Thermal properties of the different CUPE polymers were characterized by DSC and TGA. DSC curves (Fig. 2a) showed that, under similar polymerization conditions (4 days, 80 °C), the glass transition temperature of CUPEs increased with increasing isocyanate content. CUPE1.2 had the highest glass transition temperature (Sample C, Tg = 5.20 °C) and CUPE0.6 had the lowest value (Sample A, Tg = 0.64 °C). No crystallization or melt peaks were observed for any of the polymers examined. Post-polymerization conditions were also observed to affect the Tg. CUPE1.2 pre-polymer had a lower value of Tg (Sample D, −6.31 °C) compared to the CUPE1.2 sample which had been post-polymerized (crosslinked) at 80 °C for four days (sample C). TGA was used to determine the effect of the isocyanate content on the thermal stability of the CUPE polymers. Results from TGA curves (Fig. 2b) indicate that the thermal stability also increased with increasing the amount of isocyanate in the polymer synthesis. CUPE1.2 was most thermally stable (Sample C, Td = 322.93 °C), while CUPE0.6 degraded fastest under applied heat (Sample A, Td = 292.18 °C). Thermal stability of the CUPEs increased with post-polymerization. The CUPE1.2 pre-polymer (Sample D, Td = 270 °C) had a smaller value of Td compared to post-polymerized CUPE1.2 (Sample C, Td 322.93 °C).
Fig. 2.

DSC thermograms of CUPEs (a); TGA curves of CUPEs (b). Samples A, B and C correspond to crosslinked CUPEs (80 °C, 4 days) while sample D is a pre-polymer without crosslinking.
The FT-IR spectra of both POC and CUPE pre-polymers showed sharp peaks at 1733 cm−1, which are characteristic of carbonyl groups on the ester bond. The carbonyl groups of the carboxylic acid side chain of citric acid also contributed to this absorbance. Methylene groups in the polymer chain were represented by the peaks between 2931 cm−1 and 2919 cm−1 in the pre-CUPE and pre-POC spectra [31]. Absorbance from the urethane bonds was observed in the form of a narrow shoulder peak at 3350 cm−1 in CUPE, while it was absent in POC spectra. Additionally, amide I and amide II vibrations were also observed at 1670 cm−1 and 1560 cm−1, respectively [19] in CUPE pre-polymers and were absent in the POC pre-polymer spectrum. Absence of a peak at 2267 cm−1 in the CUPE spectra indicated that all the isocyanate groups had been incorporated into the polymer chains (Fig. 3).
Fig. 3.

FT-IR spectra of a representative crosslinked urethane-doped polyester pre-polymer: (A) poly (1,8-octanediol citrate) pre-polymer before chain extension; (B) CUPE0.9 pre-polymer after incorporation of urethane bonds.
Initial contact angles of the CUPE pre-polymer films increased with increase in the hydrophobicity of the diol used in the synthesis (Fig. 4). CA-PEGU0/1 had the smallest contact angle (42.78 ± 1.51°) while the CUPE with 1,8-octanediol was most hydrophobic (94.20 ± 2.87°).
Fig. 4.
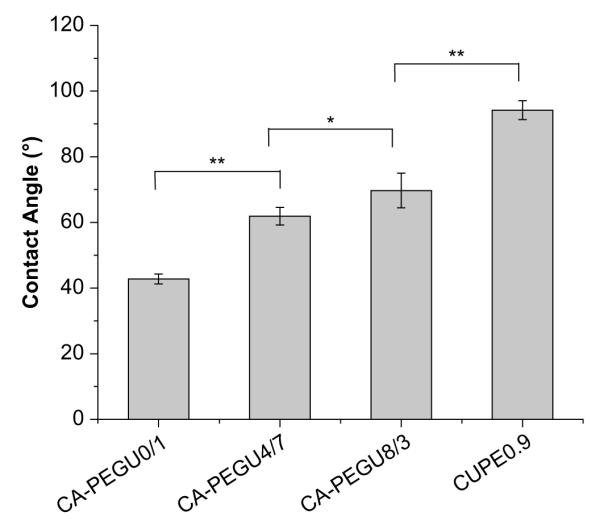
Initial contact angles of the different CUPE pre-polymers. Readings were taken for each specimen and averaged (N = 8). Contact angle was observed to decrease with increasing the ratio of hydrophilic PEG in the polymer backbone. *p < 0.05; **p < 0.01.
3.2. Mechanical properties
The stress—strain curves of CUPE were characteristic of elastomers (Fig. 5). The tensile strength was observed to increase with increasing the isocyanate composition during synthesis. CUPE1.2 had the highest break stress (41.07 ± 6.85 MPa), while CUPE0.6 had the least tensile stress at break (14.6 ± 1.004 MPa). There was no significant difference in the break elongation of the different CUPE polymers. Initial modulus of the different polymers ranged from 4.14 ± 1.71 MPa to 38.35 ± 4.49 MPa. The effect of polymerization conditions on the mechanical properties was also investigated. Increasing the post-polymerization time and temperature was observed to increase polymer tensile strength with a corresponding decrease in polymer elongation at break. Crosslink density between polymer chains varied in the order, CUPE1.2 > CUPE0.9 > CUPE0.6. On the other hand, both the density and the molecular weight between crosslinks varied as CUPE0.6 > CUPE0.9 > CUPE1.2 (Table 1).
Fig. 5.

Tensile stress-strain curves of the different CUPE polymers and crosslinked POC polymer. All the polymers shown were polymerized at 80 °C for 2 days (a). Effect of post-polymerization conditions on the peak stress (b), and elongation at break (c) are shown. **p < 0.01; *p < 0.05; N = 5.
Table 1.
Density measurements, mechanical properties and crosslink densities of different CUPE polymers
| Sample | Density (g/cm3) | Young’s modulus (MPa) | Tensile strength (MPa) | Elongation (%) | N (mol/m3) | Mc (g/mol) |
|---|---|---|---|---|---|---|
| CUPE0.6 | 1.2253 ± 0.0207 | 2.99 ±0.53 | 16.02 ± 2.39 | 252.37± 35.12 | 402± 71 | 3125± 588 |
| CUPE0.9 | 1.2008± 0.0541 | 5.84±1.84 | 32.10 ± 2.69 | 278.24± 10.12 | 786± 248 | 1637± 404 |
| CUPE1.2 | 1.1717± 0.0211 | 29.82 ±2.67 | 33.35 ± 3.26 | 260.87 ± 22.22 | 4012± 359 | 294± 27 |
| CUPE1.2, 1 day, 80 °C | 1.1944± 0.0155 | 4.84 ±0.90 | 28.75 ± 1.28 | 265.43 ± 26.144 | 651± 121 | 1885± 316 |
| CUPE0.6, 1 day, 80 °C | 1.2114± 0.0168 | 2.53 ±0.30 | 15.62 ± 2.75 | 291.26 ± 17.90 | 341± 40 | 3589± 406 |
Polymerization conditions were 80 °C treatment for 2 days, unless otherwise specified.
3.3. Scaffold fabrication
Both particulate leaching and thermal induced phase separation techniques could be used to fabricate CUPE scaffolds. CUPE scaffolds fabricated by salt-leaching method are normally thicker than 500 μm and contain a lot of dead pores (data not shown) as many other salt-leached polymer scaffolds [16,32]. The phase separation method resulted in the formation of two dimensional, highly interconnective, thin scaffold sheets (Fig. 6A and B, 150 μm thick). Pore size in TIPS scaffolds was dependent on the quenching temperature and ranged between 50 μm and 200 μm(Fig. 6B). SEM and LIVE/DEAD assay indicated that an even cell distribution could be achieved on the thin CUPE scaffolds prepared using TIPS (Fig. 6C and D).
Fig. 6.

(A) Thin CUPE1.2 TIPS scaffold taken by a digital camera; (B) SEM image of CUPE1.2 TIPS scaffold; (C) SEM image of the NIH 3T3 fibroblast-seeded CUPE1.2 TIPS scaffold; and (D) fluorescence image (4×) of 3T3 fibroblasts on CUPE1.2 TIPS scaffold stained by LIVE/DEAD assay. Scaffolds obtained were thin (150 μm), porous and highly interconnective. Cells even distributed on the scaffold.
3.4. In vitro degradation studies
Degradation profiles of the different polymers are presented in Fig. 7. Polymers with higher isocyanate content exhibited faster degradation rates in both PBS and NaOH. CUPE1.2 demonstrated a mass loss of 16 ± 0.32% after two months in PBS, while CUPE0.6 lost only 8.13 ± 2.04% of its initial mass under similar conditions (Fig. 7a). Increasing the polymerization time made the CUPE polymers more resistant to degradation (Fig. 7b). Degradation rate was observed to be a function of the choice of diol used in the synthesis, the isocyanate content, and the post-polymerization conditions. Increasing the hydrophilicity of the diol used in the synthesis resulted in faster degradation rates as evidenced by the faster rate of degradation of the CUPE polymers containing poly (ethylene glycol) (PEG) in different ratios (Fig. 7c).
Fig. 7.

Degradation studies of CUPE in (a) PBS at 37 °C(N = 6) for 8 weeks and (b,c) 0.05 M NaOH solution at room temperature. The different polymers used in the PBS degradation study were polymerized at 80 °C for 2 days (a). CUPE0.9 polymer was used to study the effect of different post-polymerization conditions on the degradation rate of CUPEs (b). Effect of diol composition used for CUPE synthesis on degradation profiles was also studied. All the different CUPE polymers listed were polymerized for 2 days at 80 °C (c). **p < 0.01; N = 5.
3.5. Biocompatibility evaluation
Biocompatibility was initially evaluated by observing cell adhesion on CUPE films. NIH 3T3 fibroblasts and human aortic smooth muscle cells (HASMCs) were used as model cell lines. SEM images show that the cells attached and proliferating on the CUPE films (Fig. 8). Moreover, the in vitro appearance of the cells was consistent with that reported in literature.
Fig. 8.

SEM images of HASMCs (A) and fibroblasts (B) growing on the surface of CUPE1.2 films after three days in culture. Seeding density of 3 × 105 cells were used per film (7 mm diameter). Images were taken at 400 × magnification.
Cell adhesion and proliferation were quantitatively evaluated using the MTT assay. Results indicated that initially (at day 1 and day 3) a larger number of cells attached to the CUPE surface compared to PLLA control films (Fig. 9). Cells continued to proliferate and increased in number over the 1-week study period, wherein both CUPE and PLLA had comparable cell numbers.
Fig. 9.
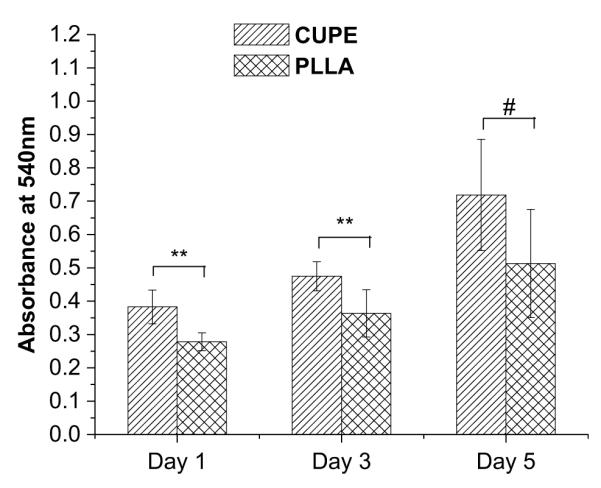
Comparison of growth and proliferation of HASMCs on PLLA (relative control) and CUPE1.2 films. MTT absorption was measured at 570 nm; N = 6. **p = 0.01 and #p > 0.05.
Foreign body response was studied by subcutaneous implantation of CUPE films and salt-leached scaffolds in Sprague Dawley rats with PLLA as a relative control. Both CUPE and PLLA implants were surrounded by well defined fibrous capsules at both 1-week and 4-week time points (Fig. 10). The fibrous capsules surrounding the CUPE implants (film: 245.3 ± 13.46 μm; scaffold: 436.6 ± 29.42 μm) (Fig. 10A,B) were substantially thinner than those surrounding the PLLA implants (film: 314.23 ± 17.48 μm, p < 0.001 compared to CUPE film; scaffold: 584.63 ± 49.83, p < 0.001 compared to CUPE scaffold) (Fig. 10C,D) at the 1-week time point. There was a significant reduction in capsule thickness at 4 weeks, for both the CUPE and PLLA implants. Fibrous capsules around CUPE films and scaffolds showed a reduction of 14% and 47.8%, respectively (Fig. 10E,F), compared to the 1-week implants. There was a reduction of 28% and 58% in the thickness of the fibrous capsules surrounding the PLLA films and scaffolds, respectively (Fig. 10G,H), at 4 weeks. Capsule thickness was comparable for both CUPE and PLLA implants at 4 weeks. These results suggest that CUPE implants may incite a weaker inflammatory response than PLLA implants. It should also be noted that no necrotic tissues were found on both CUPE and PLLA implants at both time points.
Fig. 10.

Histology of in vivo response to CUPE1.2 film (A,E) and salt-leached scaffold discs (B,F). PLLA films (C,G) and scaffolds (D,H) served as control. P and F are used to indicate the regions of polymer and fibrous capsule, respectively. All images were taken at 10× magnification. On the 1-week samples, although all samples were covered by a well defined fibrous capsule, CUPE implants were consistently surrounded by a thinner fibrous capsule as opposed to PLLA implants. In the case of the 4-week implants, overall, all the implants appear to yield similar extent of tissue response.
3.6. Platelet adhesion and activation
Platelets were found to adhere to both CUPE and PLLA (Fig. 11). However, there was a distinct difference in morphology of the adhered platelets. Platelets on CUPE were mostly round without any cytoplasmic extensions (Fig. 11A and B). On the other hand, majority of the platelets on PLLA discs were in the spread dendritic phase, a morphology, which is characterized by flattened platelets (Fig. 11C and D) with extensive pseudopodial processes. This extended morphology of platelets has been demonstrated as an indicator of platelet activation [33]. LDH assay was used to quantify adhered platelet numbers on PLLA and CUPE surfaces. Absorbance values of both CUPE and PLLA were normalized with respect to TCP. The normalized absorbance of PLLA samples (344.6 ± 32.4%) was significantly greater than that of the CUPE samples (212.3 ± 88.2%) (Fig. 12). The degree of platelet activation was assessed by using flow cytometry to measure the expression of P-selectin on the membranes of activated platelets. As shown in Fig. 13, the results showed that activation degree of the platelets exposed to CUPE is less (~13%) than that of the platelets exposed to PLLA (N = 8, p < 0.05).
Fig. 11.

Evaluation of Platelet adhesion and aggregation using SEM. Platelets on CUPE1.2 (A,B) and PLLA (C,D) discs were observed at same magnification. (A,C) 2000 ×, scale bar = 20 μm; (B,D) 5000×, scale bar = 10 μm. Platelets on CUPE1.2 were spherical and those on PLLA were spread.
Fig. 12.
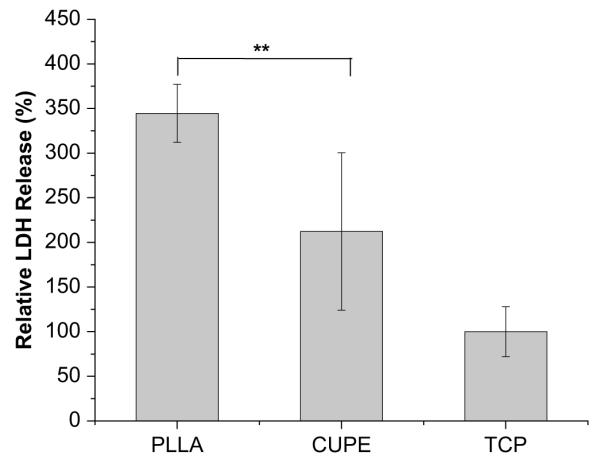
Quantification of platelet adhesion on the surface of the PLLA, CUPE1.2 and TCP by lactate dehydrogenase assays. TCP served as control and all readings were normalized to LDH activity of TCP. The absorption at 490 nm relates linearly with adhered platelet numbers. **p < 0.01, all groups have N = 8.
Fig. 13.
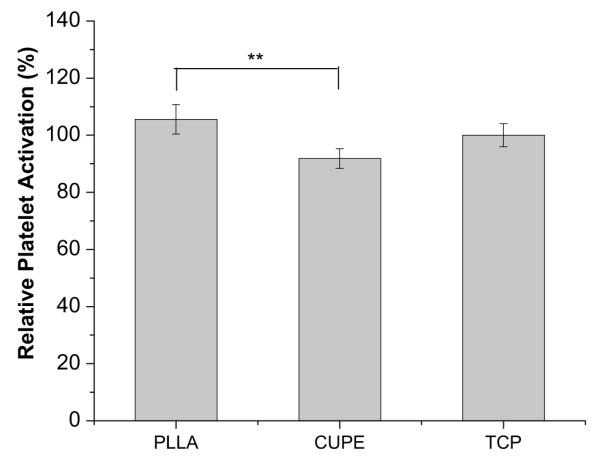
Determination of platelet activation by measuring the expression of membrane P-selectin on the platelet plasma membrane. Membrane P-selectin on platelets incubated with CUPE1.2 and PLLA samples (N = 8) was tagged with fluorophore conjugated CD62p (membrane P-selectin) monoclonal antibodies. The expression was analyzed by flow cytometry. Readings were normalized with respect to P-selectin expression on platelets exposed to TCP (control samples). *p < 0.05 and **p < 0.01. N = 8 in all groups.
4. Discussion
The poor mechanical properties of currently existing polyester elastomers like poly(diol citrates) and PGS place a limit on their utility for engineering tissues like ligaments and tendons which are high load bearing tissues. The weak nature of these materials also raises questions about how these scaffolds stand up to surgical handling. The polymer industry has created strong polymers like polyamides and polyurethanes in which strong hydrogen bonding between the amide bonds and urethane bonds, respectively, plays an important role in determining the mechanical strength of the material. In this work, we synthesized and evaluated a new generation of biodegradable polyester elastomers, crosslinked urethane-doped polyester (CUPE) elastomers by combining the advantages of crosslinked polyesters and polyurethanes. CUPE is a crosslinked polymer network, in which, ester bonds act as inter-chain crosslinks and urethane bonds are doped in the polymer chains.
In the present paper, pre-CUPE was synthesized by the chain extension of pre-poly(1,8-octanediol citrate) (pre-POC) using 1,6-hexamethyl diisocyanate (HDI) as a chain extender. HDI was chosen for the synthesis of CUPE because it has previously been used in the synthesis of various biodegradable polyurethanes (BPUs) [21,34–38] and has shown to result in higher molecular weight polyurethanes compared to other diisocyanates which are commonly used in BPU synthesis [21,39]. The molar concentration of HDI incorporated was varied to examine the effect of different degrees of chain extension on polymer properties. As revealed by material characterization techniques, the resulting CUPE polymers have a wide range of mechanical properties, degradation rates and surface properties.
Additional urethane peaks and amide peaks observed in the FT-IR spectrum of the CUPE pre-polymer were absent in the POC spectrum. This confirmed the incorporation of urethane linkages in the polymer chain. The presence of free carboxylic acid and hydroxyl groups also allows for further chain extension or the possibility of biofunctionalization, if needed.
Initial contact angles of the different CUPE pre-polymers were not affected by differences in the amount of HDI incorporated into the polymer chain (data not shown). This is likely due to the fact that the urethane bonded segment forms a small portion of the polyester chain and may not greatly influence the wettability of the material. Moreover, the urethane groups which are inserted into the polymer backbone are polar in nature and would offset the hydrophobicity of the methyl groups in HDI. From these observations, it was hypothesized that the contact angle of the material was instead determined by the diol that was used during synthesis. This hypothesis was tested by incorporating different molar ratios of polyethylene glycol (PEG) in the diol segment of the polymer chain. Changing the bulk composition of the polymer chain by incorporation of more hydrophilic PEG in the diol segment reduced the initial contact angle. CA-PEGU0/1 showed the smallest initial contact angle (42.78 ± 1.51°), because of the bulk PEG in the polymer chain. The initial contact angle increased when some portion of or all the PEGs was replaced with the relatively more hydrophobic 1.8-octanediol.
The low Tg of all the CUPE polymers and the absence of crystallinity confirmed by X-ray diffraction spectroscopy (data not shown) demonstrated their amorphous nature at 37 °C. The Tg of a polymer is a measure of the ease of chain mobility. In the presence of factors which restrict the movement of the polymer chains, the Tg increases. Typically, polymers with stronger intermolecular forces have more restricted chain mobility and hence higher glass transition temperatures. The Tg of all the CUPEs was observed to be higher than the Tg of POC [5]. This is attributed to the additional hydrogen bonding between urethane bonds in the CUPE network compared to the POC network, which contributes to more restricted chain mobility. The Tg of the different CUPE polymers increased with increasing isocyanate content during synthesis. Higher Tg of CUPE1.2 is attributed to the larger number of urethane bonds in the backbone of CUPE1.2 and therefore increased inter-chain hydrogen bonding. The introduction of ester crosslinks upon post-polymerization further restricted the chain mobility and increased the Tg of the CUPE in comparison to the pre-CUPE. Higher molecular weight and intermolecular hydrogen bonding also increased the thermal stability of the CUPEs with higher isocyanate content. Crosslinked polymers were more thermally stable compared to their corresponding pre-polymers due to the presence of thermally labile ester crosslinks. From Table 1 it can be seen that for similar conditions, the CUPE1.2 had higher density of inter-chain crosslinks, which could also contribute to the higher glass transition temperature and decomposition temperatures.
Incorporation of diisocyanate and subsequent participation of urethane bonds as inter-chain crosslinks could explain the significantly lower density of CUPE1.2 compared to CUPE0.6. Under reaction conditions excess diisocyanate in the reaction mixture would be forced to react with the hydroxyl and carboxyl pendant groups of the citric acid monomer forming urethane bonds and amide bonds, respectively. The incorporation of a long chain (in this case, 6 carbon chain) crosslinks between the polymer molecules would increase the inter-chain volume and subsequently result in reduced density.
Mechanical properties of CUPE could be modified by varying three different parameters: (a) choice of diol, (b) choice of isocyanate and its molar ratio used during synthesis and (c) post-polymerization conditions. This allows greater flexibility in controlling the strength and elasticity of the material as compared to other network polyesters, which rely on choice of monomer and post-polymerization conditions alone [4,5]. Higher tensile strength of CUPE could be obtained by increasing the amount of isocyanate used during synthesis (Table 1) or post-polymerization time and temperature (Fig. 5b,c). The tensile strength of CUPE films varied from 14.6 ± 1.004 MPa to 41.07 ± 6.85 MPa, and elongation at break up to 337 ± 6% were obtained under the synthesis conditions investigated. This is a dramatic improvement over the previously characterized poly(diol citrates) which had tensile strength values in the range of 2.93 ± 0.09 MPa to 11.15 ± 2.62 MPa [16]. The elongation of the polymers can be tailored to match that of the soft tissues such as arteries and veins which exhibit ultimate elongation of up to 260% [40]. The mechanical properties of a scaffold are directly related to that of the polymer used to fabricate it; CUPE could be used to make stronger scaffolds for a more diverse range of tissue engineering applications as opposed to poly(diol citrates). Interestingly, although CUPE1.2 had the highest degree of crosslinking, it also exhibited the fastest degradation in both phosphate buffered saline (PBS) (Fig. 7a) and 0.05 M NaOH (Fig. 7b). This could be attributed to the lower density and consequently larger inter-chain volume of the CUPE1.2 polymer, as seen from Table 1. The higher inter-chain volume would allow greater access to the hydrolytically labile ester bonds in the crosslinks and the polymer backbone. CUPEs containing PEG in the backbone degraded fastest due to their hydrophilic nature as compared to CUPE with 1,8-octanediol only. Thus, it is possible to formulate a large family of CUPE polymers by varying the choice and the molar composition of diols (including macrodiols such as PEG), isocyanate (HDI, BDI, lysine diisocyanate) during synthesis. This family would show a wide control on their mechanical properties and degradability. The details of CUPE family will be the topic of future publications.
The biocompatibility of CUPE was evaluated by both in vitro cell culture and in vivo animal implantation studies. The growth and proliferation of 3T3 fibroblasts and HASMCs in the in vitro study demonstrated good material—cell interaction. Quantitative assays further confirmed that cells grew and proliferated on CUPE films.
In vivo studies were conducted to assess the acute and chronic tissue responses to CUPE compared with a commonly used degradable polymer — PLLA. The degree of inflammation was assessed by measuring the thickness of the fibrotic capsule surrounding the implants at 1-week and 4-week time points. The presence of a thinner capsule 1 week after implantation suggests that CUPE may be less pro-inflammatory compared to PLLA.
Hemocompatibility of CUPE was evaluated by determining the extent of platelet adhesion and activation upon material contact. Platelets in the blood may be stimulated by contact with an implant surface. The subsequent formation of thrombi is one of the main causes of the failure of blood contacting devices like arterial stents and synthetic vascular grafts [41,42]. Thromboresistance is therefore one of the primary design requirements for an ideal synthetic vascular graft material [43–46]. Results from the platelet adhesion studies indicated that CUPE adhered fewer platelets compared to PLLA (Fig. 12). Moreover, it was also observed that platelets were bound passively on CUPE samples, as evidenced by their round morphology (Fig. 11A,B). Active binding with morphological changes associated with platelet activation was observed in platelets bound to PLLA (Fig. 11C,D). CUPE was also observed to activate lesser platelet numbers compared to PLLA, as indicated by flow cytometry studies (Fig. 13). The reduced tendency to adhere and activate platelets compared to PLLA implied that CUPE is more thromboresistant than PLLA and is, therefore, less likely to initiate thrombosis when used as a vascular graft substitute in vivo.
Compared to poly(diol citrates) and PGS, CUPE also demonstrated more versatile processability. Only limited methods can be employed for the fabrication of porous poly(diol citrates) or PGS scaffolds, with particulate/salt leaching being the only one utilized method so far. Due to their low molecular weights, pr-epolymers of poly(diol citrates) and PGS are not suitable for other scaffold processing technologies such as freeze-drying or electrospinning because of their intrinsic sticky nature. The CUPE pre-polymer, however, comprises of long polymer chains as a result of the chain extension carried out by the urethane bonds in the polymer backbone. The higher molecular weight of the CUPE pre-polymers allows the use of other scaffold fabrication technologies such as thermal induced phase separation (freeze-drying) and potentially electrospinning (fiber fabrication). In order to demonstrate this point, porous CUPE scaffolds were fabricated by both salt leaching and TIPS. The TIPS scaffolds also demonstrated good cell growth and proliferation with an even cell distribution. Thus, the evenly cell-distributed soft and elastic CUPE scaffolds can potentially be stacked into 3-dimensional (3-D) tissue constructs with even cell distribution throughout the construct. Further studies on the tissue construction based on 3-D CUPE scaffold sheets are underway and will be the subject of future publications.
5. Conclusion
We have developed a new class of soft but strong biodegradable elastomers, crosslinked urethane-doped polyesters (CUPEs). CUPEs demonstrated a wide range of mechanical properties and tunable degradation properties, and have greater processability than the currently available crosslinked polyester networks in designing scaffolds with desired mechanical properties. CUPEs could be synthesized by a simple and cost-effective method and have been shown to be anti-thrombotic and only slightly inflammatory. The introduction of CUPEs should contribute to biomaterials science and meet the increasing need for functional biomaterials in tissue engineering and other biomedical applications.
Acknowledgements
This work was supported in part by American Heart Association Beginning Grant-in-Aid award (J.Y.), UTA Research Enhancement fund (J.Y.), NIH R21 HL 082644 (K.N.) and NIH R01 GM074021 (L.T). Authors also thank Ashwin Nair for his helps on the histology analysis.
References
- [1].Lukashev ME, Werb Z. ECM signalling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998;8(11):437–41. doi: 10.1016/s0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- [2].Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006;20(7):811–27. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- [3].Stegemann JP, Hong H, Nerem RM. Mechanical, biochemical, and extracellular matrix effects on vascular smooth muscle cell phenotype. J Appl Physiol. 2005;98(6):2321–7. doi: 10.1152/japplphysiol.01114.2004. [DOI] [PubMed] [Google Scholar]
- [4].Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. 2002;20(6):602–6. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- [5].Yang J, Webb AR, Pickerill SJ, Hageman G, Ameer GA. Synthesis and evaluation of poly(diol citrate) biodegradable elastomers. Biomaterials. 2006;27(9):1889–98. doi: 10.1016/j.biomaterials.2005.05.106. [DOI] [PubMed] [Google Scholar]
- [6].Poirier Y, Nawrath C, Somerville C. Production of polyhydroxyalkanoates, a family of biodegradable plastics and elastomers, in bacteria and plants. Biotechnology (NY) 1995;13(2):142–50. doi: 10.1038/nbt0295-142. [DOI] [PubMed] [Google Scholar]
- [7].Bettinger CJ, Bruggeman JP, Borenstein JT, Langer RS. Amino alcohol-based degradable poly(ester amide) elastomers. Biomaterials. 2008;29(15):2315–25. doi: 10.1016/j.biomaterials.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pego AP, Poot AA, Grijpma DW, Feijen J. Copolymers of trimethylene carbonate and epsilon-caprolactone for porous nerve guides: synthesis and properties. J Biomater Sci Polym Ed. 2001;12(1):35–53. doi: 10.1163/156856201744434. [DOI] [PubMed] [Google Scholar]
- [9].Pego AP, Poot AA, Grijpma DW, Feijen J. Physical properties of high molecular weight 1,3-trimethylene carbonate and D,L-lactide copolymers. J Mater Sci Mater Med. 2003;14(9):767–73. doi: 10.1023/a:1025084304766. [DOI] [PubMed] [Google Scholar]
- [10].Younes HM, Bravo-Grimaldo E, Amsden BG. Synthesis, characterization and in vitro degradation of a biodegradable elastomer. Biomaterials. 2004;25(22):5261–9. doi: 10.1016/j.biomaterials.2003.12.024. [DOI] [PubMed] [Google Scholar]
- [11].Amsden B, Wang S, Wyss U. Synthesis and characterization of thermoset biodegradable elastomers based on star-poly(epsilon-caprolactone-co-D,L-lactide) Biomacromolecules. 2004;5(4):1399–404. doi: 10.1021/bm034538j. [DOI] [PubMed] [Google Scholar]
- [12].Motlagh D, Yang J, Lui KY, Webb AR, Ameer GA. Hernocompatibility evaluation of poly(glycerol-sebacate) in vitro for vascular tissue engineering. Biomaterials. 2006;27(24):4315–24. doi: 10.1016/j.biomaterials.2006.04.010. [DOI] [PubMed] [Google Scholar]
- [13].Schappacher M, Fabre T, Mingotaud AF, Soum A. Study of a (trimethylenecarbonate-co-epsilon-caprolactone) polymer part 1: preparation of a new nerve guide through controlled random copolymerization using rare earth catalysts. Biomaterials. 2001;22(21):2849–55. doi: 10.1016/s0142-9612(01)00029-1. [DOI] [PubMed] [Google Scholar]
- [14].Pego AP, Vleggeert-Lankamp CL, Deenen M, Lakke EA, Grijpma DW, Poot AA, et al. Adhesion and growth of human Schwann cells on trimethylene carbonate (co)polymers. J Biomed Mater Res A. 2003;67(3):876–85. doi: 10.1002/jbm.a.10074. [DOI] [PubMed] [Google Scholar]
- [15].Sundback CA, Shyu JY, Wang Y, Faquin WC, Langer RS, Vacanti JP, et al. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials. 2005;26(27):5454–64. doi: 10.1016/j.biomaterials.2005.02.004. [DOI] [PubMed] [Google Scholar]
- [16].Yang J, Motlagh D, Webb AR, Ameer GA. Novel biphasic elastomeric scaffold for small-diameter blood vessel tissue engineering. Tissue Eng. 2005;11(1112):1876–86. doi: 10.1089/ten.2005.11.1876. [DOI] [PubMed] [Google Scholar]
- [17].Noyes FR, Grood ES. Strength of anterior cruciate ligament in humans and Rhesus-monkeys. J Bone Joint Surg Am. 1976;58(8):1074–82. [PubMed] [Google Scholar]
- [18].Guan J, Sacks MS, Beckman EJ, Wagner WR. Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: synthesis, characterization and cytocompatibility. Biomaterials. 2004;25(1):85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- [19].Guan J, Wagner WR. Synthesis, characterization and cytocompatibility of polyurethaneurea elastomers with designed elastase sensitivity. Biomacromolecules. 2005;6(5):2833–42. doi: 10.1021/bm0503322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guan J, Sacks MS, Beckman EJ, Wagner WR. Synthesis, characterization, and cytocompatibility of elastomeric, biodegradable poly(ester-urethane)ureas based on poly(caprolactone) and putrescine. J Biomed Mater Res. 2002;61(3):493–503. doi: 10.1002/jbm.10204. [DOI] [PubMed] [Google Scholar]
- [21].Skarja GA, Woodhouse KA. Synthesis and characterization of degradable polyurethane elastomers containing and amino acid-based chain extender. J Biomater Sci Polym Ed. 1998;9(3):271–95. doi: 10.1163/156856298x00659. [DOI] [PubMed] [Google Scholar]
- [22].Alperin C, Zandstra PW, Woodhouse KA. Polyurethane films seeded with embryonic stem cell-derived cardiomyocytes for use in cardiac tissue engineering applications. Biomaterials. 2005;26(35):7377–86. doi: 10.1016/j.biomaterials.2005.05.064. [DOI] [PubMed] [Google Scholar]
- [23].Yang J, Webb AR, Ameer GA. Novel citric acid-based biodegradable elastomers for tissue engineering. Adv Mater. 2004;16(6):511–6. [Google Scholar]
- [24].Wei G, Ma PX. Structure and properties of nano-hydroxyapatite/polymer composite scaffolds for bone tissue engineering. Biomaterials. 2004;25(19):4749–57. doi: 10.1016/j.biomaterials.2003.12.005. [DOI] [PubMed] [Google Scholar]
- [25].Nam SY, Park TG. Porous biodegradable polymeric scaffolds prepared by thermally induced phase separation. J Biomed Mater Res. 1999;47(1):8–17. doi: 10.1002/(sici)1097-4636(199910)47:1<8::aid-jbm2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- [26].Zhang Z, Wang Z, Liu S, Kodama M. Pore size, tissue ingrowth, and endothelialization of small-diameter microporous polyurethane vascular prostheses. Biomaterials. 2004;25(1):177–87. doi: 10.1016/s0142-9612(03)00478-2. [DOI] [PubMed] [Google Scholar]
- [27].Guan J, Fujimoto KL, Sacks MS, Wagner WR. Preparation and characterization of highly porous, biodegradable polyurethane scaffolds for soft tissue applications. Biomaterials. 2005;26(18):3961–71. doi: 10.1016/j.biomaterials.2004.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Motlagh D, Allen J, Hoshi R, Yang J, Lui K, Ameer G. Hemocompatibility evaluation of poly(diol citrate) in vitro for vascular tissue engineering. J Biomed Mater Res A. 2007;82(4):907–16. doi: 10.1002/jbm.a.31211. [DOI] [PubMed] [Google Scholar]
- [29].Berman CL, Yeo EL, Wencel-Drake JD, Furie BC, Ginsberg MH, Furie B. A platelet alpha granule membrane protein that is associated with the plasma membrane after activation. Characterization and subcellular localization of platelet activation-dependent granule-external membrane protein. J Clin Invest. 1986;78(1):130–7. doi: 10.1172/JCI112542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25(26):5681–703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- [31].Jayakumar R, Lee YS, Nanjundan S. Synthesis and characterization of calcium containing polyurethane ureas. J Appl Polym Sci. 2003;90(13):3488–96. [Google Scholar]
- [32].Yang J, Shi GX, Bei JZ, Wang SG, Cao YL, Shang QX, et al. Fabrication and surface modification of macroporous poly(L-lactic acid) and poly(L-lactic-co-glycolic acid) (70/30) cell scaffolds for human skin fibroblast cell culture. J Biomed Mater Res. 2002;62(3):438–46. doi: 10.1002/jbm.10318. [DOI] [PubMed] [Google Scholar]
- [33].Allen RD, Zacharski LR, Widirstky ST, Rosenstein R, Zaitlin LM, Burgess DR. Transformation and motility of human platelets: details of the shape change and release reaction observed by optical and electron microscopy. J Cell Biol. 1979;83(1):126–42. doi: 10.1083/jcb.83.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kylma J, Seppala JV. Synthesis and characterization of a biodegradable thermoplastic poly(ester-urethane) elastomer. Macromolecules. 1997;30:2876–82. [Google Scholar]
- [35].Tatai L, Moore TG, Adhikari R, Malherbe F, Jayasekara R, Griffiths I, et al. Thermoplastic biodegradable polyurethanes: the effect of chain extender structure on properties and in-vitro degradation. Biomaterials. 2007;28(36):5407–17. doi: 10.1016/j.biomaterials.2007.08.035. [DOI] [PubMed] [Google Scholar]
- [36].Lee CR, Grad S, Gorna K, Gogolewski S, Goessl A, Alini M. Fibrin-polyurethane composites for articular cartilage tissue engineering: a preliminary analysis. Tissue Eng. 2005;11(910):1562–73. doi: 10.1089/ten.2005.11.1562. [DOI] [PubMed] [Google Scholar]
- [37].Grad S, Kupcsik L, Gorna K, Gogolewski S, Alini M. The use of biodegradable polyurethane scaffolds for cartilage tissue engineering: potential and limitations. Biomaterials. 2003;24(28):5163–71. doi: 10.1016/s0142-9612(03)00462-9. [DOI] [PubMed] [Google Scholar]
- [38].Chia SL, Gorna K, Gogolewski S, Alini M. Biodegradable elastomeric polyurethane membranes as chondrocyte carriers for cartilage repair. Tissue Eng. 2006;12(7):1945–53. doi: 10.1089/ten.2006.12.1945. [DOI] [PubMed] [Google Scholar]
- [39].Skarja GA, Woodhouse KA. In vitro degradation and erosion of degradable, segmented polyurethanes containing an amino acid-based chain extender. J Biomater Sci Polym Ed. 2001;12(8):851–73. doi: 10.1163/156856201753113060. [DOI] [PubMed] [Google Scholar]
- [40].Lee MC, Haut RC. Strain rate effects on tensile failure properties of the common carotid-artery and jugular veins of ferrets. J Biomech. 1992;25(8):925–7. doi: 10.1016/0021-9290(92)90232-p. [DOI] [PubMed] [Google Scholar]
- [41].Kannan RY, Salacinski HJ, Butler PE, Hamilton G, Seifalian AM. Current status of prosthetic bypass grafts: a review. J Biomed Mater Res B Appl Biomater. 2005;74(1):570–81. doi: 10.1002/jbm.b.30247. [DOI] [PubMed] [Google Scholar]
- [42].Kuchulakanti PK, Chu WW, Torguson R, Ohlmann P, Rha SW, Clavijo LC, et al. Correlates and long-term outcomes of angiographically proven stent thrombosis with sirolimus- and paclitaxel-eluting stents. Circulation. 2006;113(8):1108–13. doi: 10.1161/CIRCULATIONAHA.105.600155. [DOI] [PubMed] [Google Scholar]
- [43].Isenberg BC, Williams C, Tranquillo RT. Small-diameter artificial arteries engineered in vitro. Circ Res. 2006;98(1):25–35. doi: 10.1161/01.RES.0000196867.12470.84. [DOI] [PubMed] [Google Scholar]
- [44].Tranquillo RT. The tissue-engineered small-diameter artery. Ann N Y Acad Sci. 2002;961:251–4. doi: 10.1111/j.1749-6632.2002.tb03094.x. [DOI] [PubMed] [Google Scholar]
- [45].Ratcliffe A. Tissue engineering of vascular grafts. Matrix Biol. 2000;19:353–7. doi: 10.1016/s0945-053x(00)00080-9. [DOI] [PubMed] [Google Scholar]
- [46].Hoenig MR, Campbell GR, Rolfe BE, Campbell JH. Tissue-engineered blood vessels: alternative to autologous grafts? Arterioscler Thromb Vasc Biol. 2005;25(6):1128–34. doi: 10.1161/01.ATV.0000158996.03867.72. [DOI] [PubMed] [Google Scholar]