

Abstract
Deficiencies in mismatch repair (MMR) are associated with carcinogenesis. Rhodium metalloinsertors bind to DNA base mismatches with high specificity and inhibit cellular proliferation preferentially in MMR-deficient cells versus MMR-proficient cells. A family of chrysenequinone diimine complexes of rhodium with varying ancillary ligands that serve as DNA metalloinsertors has been synthesized, and both DNA mismatch binding affinities and antiproliferative activities against the human colorectal carcinoma cell lines HCT116N and HCT116O, an isogenic model system for MMR deficiency, have been determined. DNA photocleavage experiments reveal that all complexes bind to the mismatch sites with high specificities; DNA binding affinities to oligonucleotides containing single base CA and CC mismatches, obtained through photocleavage titration or competition, vary from 104 to 108 M−1 for the series of complexes. Significantly, binding affinities are found to be inversely related to ancillary ligand size and directly related to differential inhibition of the HCT116 cell lines. The observed trend in binding affinity is consistent with the metalloinsertion mode where the complex binds from the minor groove with ejection of mismatched base pairs. The correlation between binding affinity and targeting of the MMR-deficient cell line suggests that rhodium metalloinsertors exert their selective biological effects on MMR-deficient cells through mismatch binding in vivo.
Introduction
The mismatch repair (MMR) pathway corrects single base errors and insertion/deletion loops that arise during DNA synthesis, increasing the fidelity of DNA replication by a factor of 50–1000.1 If uncorrected, mismatches are converted to mutations in subsequent cycles of DNA replication, and cells with MMR deficiencies, not surprisingly, exhibit elevated mutation rates.2–4 Germline mutations in hMLH1 or hMSH2, essential genes for MMR in humans, dramatically increase the risk of developing hereditary nonpolyposis colon cancer (HNPCC), the most common type of inherited colon cancer.5,6 HNPCC is marked by early onset and the presence of cancers in several other tissue types.6 Roughly 15% of sporadic colorectal cancer cases have also been linked to MMR deficiency.7 Epigenetic silencing of the MMR genes has been identified as the cause of MMR deficiency in these cases.8 In addition to colorectal cancer, mismatch repair deficiencies have been found in approximately 16% of solid tumors of all tissue types.3,9
Importantly, MMR deficiency confers resistance or tolerance to many of the anti-cancer agents currently in clinical use.10,11 Alkylation by the commonly used chemotherapeutic agents N-methyl-N-nitrosourea (MNU) and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) at the O6 position of guanine nucleotides triggers an apoptotic response after recognition of O6-meG:C and O6-meG:T base pairs by the MMR pathway, while MMR-deficient cells tolerate this DNA methylation.10–12 Failure to recognize DNA adducts is also involved in the resistance of MMR-deficient cells to the platinum compounds cisplatin and carboplatin.10–13 The incorporation of anti-metabolites such as 5-fluorouracil and 6-thioguanine into DNA triggers cell cycle arrest and apoptosis through the MMR pathway, and consequently MMR-deficient cells are resistant to these agents as well.14,15 Other studies have shown low-level resistance to the type I topoisomerase poisons camptothecin and topotecan in hMLH1 deficient lines and to the type II topoisomerase poisons doxorubicin, epirubicin, and mitoxantrone in hMLH1 or hMSH2 deficient lines.16 It has also been hypothesized that treatment regimens with agents such as cisplatin might enrich tumors for MMR-deficient cells,17 and it has been shown that a substantial portion of secondary, or therapy-related, leukemias show signs of MMR deficiency.17,18 Collectively, these results show the broad involvement of MMR in mediating drug response, the effects of MMR deficiency on this response, and the need to develop therapeutic agents that specifically target MMR-deficient cells.
Our laboratory has previously developed bulky rhodium complexes that target DNA mismatches in vitro.19–23 These octahedral complexes contain an expansive tetracyclic aromatic ligand that can be accommodated preferentially by DNA at a thermodynamically destabilized mismatch site. The first-generation compound, Rh(bpy)2chrysi3+ (chrysi = 5,6-chrysenequinone diimine), binds 80% of DNA mismatches with typical binding constants of 106 M−1 and remarkable specificity for mismatched DNA; in a 2.6 kb DNA fragment DNA photocleavage reveals specific targeting of the mismatch.19,20,23 Subsequent work led to the incorporation of nitrogen atoms into the intercalating ligand and a 50-fold increase in binding affinity for the second-generation compound, Rh(bpy)2phzi3+ (phzi = benzo[a]-phenazine-5,6-quinonediimine).21 A high-resolution crystal structure of Rh(bpy)2chrysi3+ bound to single AC mismatches within a DNA oligonucleotide duplex reveals a distinctive binding mode at the mismatched site.24 We had previously determined that tris(chelate) complexes of Rh with a planar aromatic ligand bind to well-matched DNA by partial intercalation of the planar ligand from the major groove side into the base pair stack.25 However, binding to the mismatched site involves instead insertion of the expansive ligand into the DNA duplex from the minor groove side at the mismatched site with ejection of the mismatched bases out of the DNA stack; the inserted ligand stacks fully with adjacent base pairs. NMR studies of Rh(bpy)2chrysi3+ bound to an oligonucleotide containing a CC mismatch confirm this metalloinsertion mode for the complex at mismatched sites in solution.26
The in vivo effects of Rh(bpy)2chrysi3+ and Rh(bpy)2phzi3+ have been characterized in the isogenic cell lines HCT116N and HCT116O.27 The HCT116 cell line is a colorectal carcinoma line deficient in the hMLH1 gene. Two derivative cell lines, HCT116N and HCT116O, have been made through transfection of human chromosome 3 (ch3) and human chromosome 2 (ch2), respectively. The presence of a functional copy of ch3 restores MMR proficiency in the HCT116N line, while the HCT116O line transfected with ch2 remains MMR deficient.28 The mismatch recognition compounds developed within our laboratory were shown to selectively inhibit the proliferation of the repair-deficient HCT116O line.27
Recent work within our laboratory on luminescent ruthenium complexes has also shown that these tris(chelate) complexes are taken up inside cells through passive diffusion facilitated by the membrane potential.29 Variations in ancillary ligands have dramatic effects on cellular uptake, with increased lipophilicity facilitating uptake. Uptake can also be increased through functionalization with a nuclear localizing peptide.30
Here we examine the effects of ancillary ligand variation in the Rh(L)2chrysi3+ family on the ability of these complexes to target DNA mismatches in vitro and in vivo. Importantly, we establish that the differential inhibition of cellular proliferation in MMR-deficient cells is correlated with mismatch binding affinity.
Experimental Procedures
Materials
RhCl3 was purchased from Pressure Chemical, Inc. (Pittsburgh, PA). [Rh(NH3)5Cl]Cl2 was obtained from Strem Chemical, Inc. (Newburyport, MA). 2,2′-Dipyridylamine (HDPA), 4,7-diphenyl-1,10-phenanthroline (DIP), and Sephadex ion-exchange resin were obtained from Sigma-Aldrich (St. Louis, MO). Sep-Pak C18 solid-phase extraction cartridges were purchased from Waters Chemical Co. (Milford, MA). Phosphoramidites were purchased from Glen Research (Sterling, VA). Media and supplements were purchased from Invitrogen (Carlsbad, CA). BrdU, antibodies, buffers, and peroxidase substrate were purchased in kit format from Roche Molecular Biochemicals (Mannheim, Germany). All commercial materials were used as received.
Oligonucleotide Synthesis
Oligonucleotides were synthesized on an Applied Biosystems 3400 DNA synthesizer using standard phosphoramidite chemistry. DNA was synthesized with a 5′-dimethoxytrityl (DMT) protecting group. The oligonucleotides were cleaved from the beads by reaction with concentrated ammonium hydroxide at 60 °C overnight. The resulting free oligonucleotides were purified by HPLC using a C18 reverse-phase column (Varian, Inc.) on a Hewlett-Packard 1100 HPLC. The DMT group was removed by reaction with 80% acetic acid for 15 min at room temperature. The DMT-free oligonucleotides were precipitated with absolute ethanol and purified again by HPLC. Positive identification of the oligonucleotides and their purity were confirmed by MALDI-TOF mass spectrometry. Quantification was performed on a Beckman DU 7400 spectrophotometer using the extinction coefficients at 260 nm (ε260) estimated for single-stranded DNA.
Synthesis and Characterization of Metal Complexes
Chrysene-5,6-dione, [Rh(bpy)2chrysi]Cl3, and [Rh(phen)2chrysi]Cl3 were prepared according to previously reported procedures.19,20,31
[Rh(NH3)4chrysi]OTf3
[Rh(NH3)6]OTf3 was prepared as described by Sargeson.32,33 [Rh(NH3)6]OTf3 was reacted with a limiting amount of chrysene quinone in a 3:1 acetonitrile:water mixture with excess sodium hydroxide as a catalyst to form [Rh(NH3)4chrysi]OTf3. Acetonitrile was removed in vacuo, followed by filtration to remove unreacted chrysenequinone. The product was separated from unreacted [Rh(NH3)6]OTf3 by solid-phase extraction on a C18 cartridge and eluted with 1:1:0.001 acetonitrile:water:TFA. 1H NMR (DMSO-d6, 50 °C, 300 MHz): δ 13.30 (s), 12.32 (s), 8.876 (t, 1H, 7.7 Hz), 8.787 (d, 1H, 7.9 Hz), 8.57–8.51 (m, 2H), 8.358 (dd, 1H, 8.9 Hz, 4.6 Hz), 8.145 (d, 1H, 7.7 Hz), 7.85–7.70 (m, 4H), 4.73–4.54 (broad m, 6H), 3.862 (s, 3H) ppm. UV/vis (H2O, pH 5): 263 nm (60 900 M−1 cm−1), 283 nm (38 100 M−1 cm−1), 326 nm (12 600 M−1 cm−1), 413 nm (12 000 M−1 cm−1). MALDI-MS (cation): 425 m/z (M − 2H+) obsd, 427 m/z calcd.
rac-[Rh(HDPA)2(chrysi)]OTf3
[Rh(NH3)4(chrysi)]OTf3 (15 mg, 0.02 mmol) was reacted with HDPA (20 mg, 0.12 mmol, excess) in 20 mL of ethanol and 20 mL of water. The dark red solution was heated under reflux for 16 h. The reaction mixture turned reddish brown upon heating. Ethanol was removed under vacuum, and the resulting solution was filtered to remove any residue. The filtrate was concentrated on a Sep-Pak C18 cartridge, eluting with 1:1:0.01 acetonitrile:water:TFA, lyophilized, and purified on an alumina column, eluting with 2% methanol in dichloromethane. The fractions were collected and dried under vacuum to give an orange-brown solid (8 mg, 39%). 1H NMR (DMSO-d6, 300 MHz): δ 12.84 (s, 1H), 12.34 (s, 1H), 11.78 (s, 1H), 10.32 (d, 1H, 8.7 Hz), 8.63 (d, 1H, 6.9 Hz), 8.40 (d, 1H, 8.4 Hz), 8.31 (d, 1H, 9.3 Hz), 8.14 (m, 2H), 8.07 (d, 1H, 8.7 Hz), 8.04 (d, 1H, 5.4 Hz), 7.94 (m, 4H), 7.77 (m, 5H), 7.58 (m, 2H), 7.48 (d, 1H, 8.1 Hz), 7.41 (d, 1H, 8.4 Hz), 7.32 (s, 1H), 7.14 (m, 2H), 7.04 (t, 1H, 6.8 Hz), 6.98 (t, 1H, 6.9 Hz), 6.81 (t, 1H, 6.5 Hz) ppm. UV/vis (H2O, pH 5): 287 nm (42 200 M−1 cm−1), 321 nm (23 000 M−1 cm−1), 442 nm (8800 M−1 cm−1). ESI-MS (cation): 699.2 m/z (M − 2H+), 350.1 m/z (M − H2+) obsd, 699.2 m/z (M − 2H+) calcd.
rac-[Rh(DIP)2(NH3)2]OTf3
RhCl3 and 2 equiv of DIP were combined in 1:1 ethanol:water and refluxed overnight. The solvent was removed in vacuo, and the product was recrystallized by dissolving in acetonitrile at 60 °C and cooling to −20 °C. The precipitate was collected by filtration, washed in diethyl ether, and dissolved in neat triflic acid. The solution was again cooled and added dropwise to NH4OH at −20 °C. The pale white precipitate was collected by filtration and washed with a small amount of water to give [Rh(DIP)2(NH3)2]OTf3.
rac-[Rh(DIP)2chrysi]Cl3
rac-[Rh(DIP)2(NH3)2]OTf3 was combined with a 10% excess of 5,6-chrysenequinone and a catalytic amount of NaOH in acetonitrile and stirred at room temperature overnight. The condensation was terminated by addition of a stoichiometric amount of HCl. The solvent was removed in vacuo, and the product was purified by alumina column chromatography. Unbound chrysi ligand eluted first with ethyl acetate, and the purified product then eluted with acetonitrile. Finally, the compound was dissolved in 3:2 MeCN:H2O, and the triflate counterion was exchanged for chloride ion with Sephadex QAE-125 ion-exchange resin. UV/vis (H2O, pH 5): 290 nm (104 000 M−1 cm−1), 335 nm (43 900 M−1 cm−1), 373 nm (22 300 M−1 cm−1). ESI-MS (cation): 1020.9 m/z (M − 2H+), 511.0 m/z (M − H2+) obsd, 1023 m/z (M − H2+) calcd.
Photocleavage Titrations
The oligonucleotide was 32P-labeled at the 5′-end by incubating DNA with 32P-ATP and polynucleotide kinase (PNK) at 37 °C for 2 h, followed by purification using gel electrophoresis. A small amount of the labeled DNA (less than 1% of the total amount of DNA) was added to 2 μM DNA in 100 mM NaCl, 20 mM NaPi, pH 7.1 buffer. The DNA hairpin was annealed by heating at 90 °C for 10 min and cooling slowly to room temperature over a period of 2 h. Racemic rhodium complex solutions ranging from nanomolar to micromolar concentration were made in Milli-Q water. Annealed 2 μM DNA (10 μL) and 10 μL of Rh solution at each concentration were mixed in a microcentrifuge tube and incubated at 37 °C for 10 min. A light control (LC), in which the DNA was mixed with 10 μL of water and irradiated, and a dark control (DC), in which the DNA was mixed with the highest concentration of rhodium complex without irradiation, were also prepared. The samples were left in the heat block and irradiated on an Oriel (Darmstadt, Germany) 1000-W Hg/Xe solar simulator (340–440 nm) for 5 min. The irradiated samples were dried and electrophoresed in a 20% denaturing polyacrylamide gel. The gel was then exposed to a phosphor screen, and the relative amounts of DNA in each band were quantitated by phosphorimagery (ImageQuant).
Binding Constant Determination
The fraction of DNA cleaved in each lane on the gel was normalized and plotted against the log of the concentration of rhodium complex. At least three photocleavage titrations were carried out for each racemic metal complex. The pooled data were fit to a sigmoidal curve using OriginPro 6.1. The resulting midpoint value (i.e., the log of [rhodium complex] at the inflection point of the curve) was converted to units of concentration ([Rh50%]). The dissociation constant was calculated according to KD = [Rh50%] − 0.5[DNA], and the binding constant was defined as KB = 1/KD. The errors were derived from the errors associated with the midpoint values. For complexes that did not photocleave DNA, a binding competition titration was carried out with a constant amount (1 μM) of rac-Rh(bpy)2(chrysi)3+ added to each sample. The binding and dissociation constants of the non-photocleaving complex were calculated by solving simultaneous equlibiria involving DNA, Rh(bpy)2(chrysi)3+, and the complex in question in Mathematica 6.0.
Cell Culture
HCT116N and HCT116O cells were grown in RPMI medium 1640 supplemented with 10% FBS, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 units/mL penicillin, 100 μg/mL streptomycin, and 400 μg/mL Geneticin (G418). Cells were grown in tissue culture flasks and dishes (Corning Costar, Acton, MA) at 37 °C under 5% CO2 atmosphere.
Cellular Proliferation ELISA
HCT116N and HCT116O cells were plated in 96-well plates at 2000 cells/well and allowed 24 h to adhere. The cells were then incubated with rhodium complexes for the durations specified. For incubation less than 72 h, the Rh-containing medium was replaced with fresh medium, and the cells were grown for the remainder of the 72 h period. Cells were labeled with BrdU 24 h before analysis. The BrdU incorporation was quantified by antibody assay according to established procedures.34,35 Cellular proliferation was expressed as the ratio of the amount of BrdU incorporated by the treated cells to that of the untreated cells.
Results
Binding Affinities for Metal Complexes at Single Base Mismatches
The binding constants of the family of Rh(L)2chrysi3+ complexes at a CC and AC mismatch in a 29-mer DNA hairpin with the sequence 5′-GGCAGGXATGGCTTTTTGCCATCCCTGCC-3′ (X = C or A, underline denotes the mismatch) were measured. The hairpin sequence allows cleavage site determination on either strand around the DNA mismatch site. By irradiating samples of DNA titrated with varying concentrations of a rhodium complex, a photocleavage titration curve is obtained from which the binding constant of the rhodium complex is determined. A typical autoradiogram, taken after electrophoresis through a denaturing gel, of samples in a photocleavage titration with rac-Rh(bpy)2chrysi3+ at a CC mismatch is shown in Figure 2. The position of the photocleavage bands indicates that Rh(bpy)2chrysi3+ cleaves one base neighboring the mismatch site near the 3′-end. For this DNA sequence, we observed cleavage on only one strand as reported earlier.23 This cleavage pattern is found for both rac-Rh(phen)2chrysi3+ and rac-Rh(DIP)2chrysi3+ and holds for the AC mismatch for rac-Rh(bpy)2chrysi3+ and rac-Rh(phen)2chrysi3+. No other photocleavage bands are observed, demonstrating the high specificity of Rh(L)2chrysi3+ complexes binding to the mismatch. The photocleavage titration curve is generated from the autoradiogram by quantifying the amount of photocleavage relative to the total amount of DNA at each Rh concentration. Pooled data from at least three repeats were fitted to a sigmoidal curve (Figure 2) for determination of the midpoint ([Rh50%]) and the dissociation constant (KD). The KD value for rac-Rh(bpy)2chrysi3+ at a CC mismatch is found to be 30 nM. For rac-Rh(phen)2chrysi3+ and rac-Rh(DIP)2chrysi3+ at a CC mismatch, KD values of 320 nM and 11 μM are found, respectively (corresponding KB values are shown in Table 1). For the AC mismatch with both rac-Rh(bpy)2chrysi3+ and rac-Rh(phen)2chrysi3+, KD values are somewhat higher, as we expect given the greater thermodynamic stability of an AC mismatch versus a CC mismatch. rac-Rh(DIP)2chrysi3+ does not yield any photocleavage up to 100 μM; thus, its KD value is estimated to be greater than that.
Figure 2.
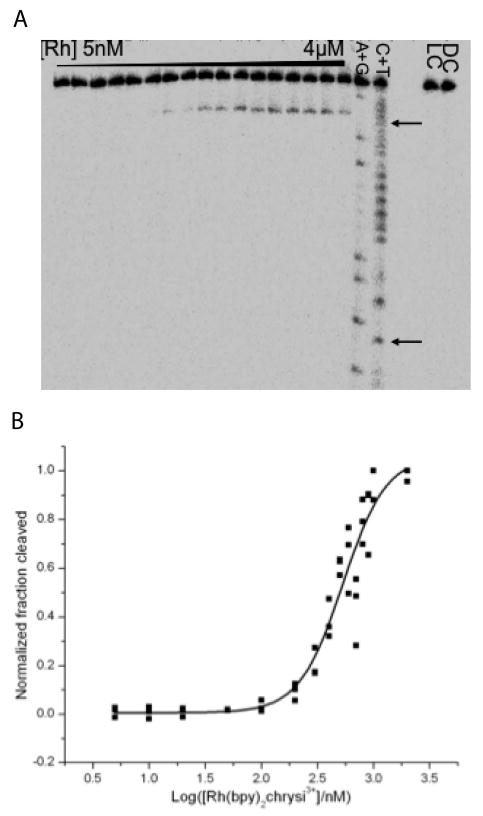
Binding affinities determined through DNA photocleavage. The DNA hairpin sequence is 5′-GGCAGGXATGGCTTTTTGCCATCCCTGCC-3′ (X = C or A, underline denotes the mismatch). Samples were irradiated and electrophoresed through a 20% denaturing PAGE gel. A light control (LC, without rhodium) and dark control (DC, without irradiation) were included. A representative autoradiogram of a photocleavage titration with rac-Rh(bpy)2chrysi3+ (A, arrows indicate positions of mismatched bases) and a representative sigmoidal curve fit of pooled data from photocleavage titrations for binding constant determination (B) are shown.
Table 1.
| Complex | Ancillary ligand | KB(CC)/M-1 | KB (AC)/M-1 |
|---|---|---|---|
| Rh(NH3)4chrysi3+ | NH3 | 1.0 × 108 | 3.4 × 106 |
| Rh(bpy)2chrysi3+ | 3.4 × 107 | 2.2 × 106 | |
| Rh(HDPA)2chrysi3+ | 2.0 × 107 | 2.6 × 106 | |
| Rh(phen)2chrysi3+ | 3.2 × 106 | 1.4 × 106 | |
| Rh(DIP)2chrysi3+ |  |
9.1 × 104 | < 104 |
Uncertainties are estimated to be 10%. Binding constants are determined from photocleavage or binding competition titrations using a DNA hairpin with the sequence 5′-GGCAGGXATGGCTTTTTGCCATCCCTGCC-3′ (X = C or A, underline denotes the mismatch). Samples were irradiated with a solar simulator (340–440 nm) at 37 °C in 50 mM NaCl, 10 mM NaPi, pH 7.1, as described in the Experimental Procedures.
For the polypyridyl complexes, values are given for racemic mixtures.
As with phenanthrenequinone diimine complexes of rhodium containing saturated amine ligands, Rh(NH3)4 chrysi3+ and rac-Rh(HDPA)2chrysi3+ promote relatively little DNA cleavage upon irradiation.36 As a result, their binding affinities were determined through binding competition titrations with 1 μM rac-Rh(bpy)2chrysi3+. On the basis of the binding constant of Rh(bpy)2chrysi3+, the binding constant of Rh(NH3)4chrysi3+ is calculated by solving simultaneous equilibria at the inflection point of the photocleavage titration curve. Through this competitive titration, the binding constant of Rh(NH3)4chrysi3+ at a CC mismatch is found to be 1.0 × 108 M−1. At an AC mismatch, KB of Rh(NH3)4chrysi3+ is 3.4 × 106 M−1. From similar binding competition titrations, the binding constant of rac-Rh(HDPA)2chrysi3+ is found to be 2.0 × 107 M−1 at a CC mismatch and 2.6 × 106 M−1 at an AC mismatch. It is apparent that the binding affinity correlates inversely with complex size; the smallest complex, Rh(NH3)4chrysi3+, shows the highest affinities for mismatched sites. The binding constants for the entire series of Rh(L)2chrysi3+ complexes are summarized in Table 1.
Inhibition of Cellular Proliferation by Enzyme-Linked Immunosorbent Assay (ELISA)
An ELISA for DNA synthesis was used to quantify the effects of the metalloinsertors on the proliferation of HCT116N cells (MMR-proficient) and HCT116O cells (MMR-deficient).27 Both cell lines were incubated with 0–25 μM of each member of the Rh(L)2chrysi3+ family except Rh(DIP)2chrysi3+, which was administered at 0–5 μM concentrations due to its greater uptake characteristics. Incubations were performed for 12, 24, 48, or 72 h. After the 12, 24, and 48 h incubations, the medium containing Rh was replaced with fresh medium, and the cells were grown for the remainder of the 72 h period. The extent of cellular proliferation is expressed as the ratio of BrdU incorporated by the rhodium-treated cells as compared to untreated controls. Figure 3 shows representative data for Rh(NH3)4chrysi3+ at various incubation times. No significant preferential inhibition of the HCT116O cell line is seen at incubation times less than 24 h, consistent with previous results for Rh(bpy)2chrysi3+, with the exception of Rh(DIP)2chrysi3+, which displays a small differential effect at 12 h.27 With longer incubation times, however, Rh(NH3)4chrysi3+ displays a strong differential effect with preferential inhibition of the MMR-deficient HCT116O cell line over the MMR-proficient HCT116N cell line. In particular, 48 h treatment with 10 μM Rh(NH3)4chrysi3+ inhibits the proliferation of the HCT116O line by 82 ± 2% while exerting little to no effect on the HCT116N line (7 ± 6% inhibition).
Figure 3.
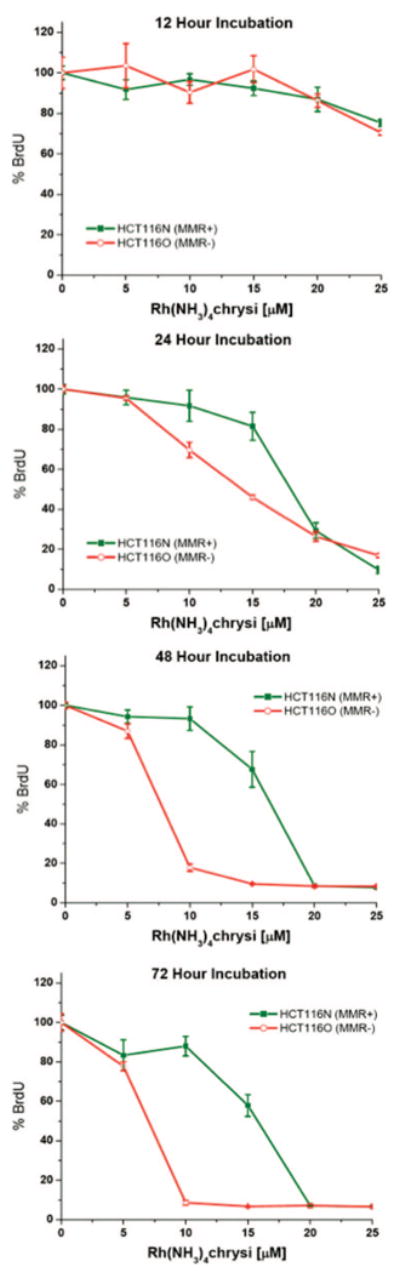
Inhibitory effects of Rh(NH3)4chrysi3+ as a function of incubation time on cellular proliferation. Shown are plots of BrdU incorporation (a measure of DNA synthesis and therefore cellular proliferation) normalized to the BrdU incorporation of untreated cells as a function of rhodium concentration. Standard error bars for five trials are shown. MMR-proficient HCT116N cells (green) and MMR-deficient HCT116O cells (red) were plated and allowed 24 h to adhere before incubation with 0–25 μM Rh(NH3)4chrysi3+ for 12, 24, 48, or 72 h. At the end of the 12, 24, and 48 h incubations, the medium containing Rh was replaced with fresh medium for the remainder of the 72 h, followed by ELISA analysis. BrdU was added to the medium 24 h prior to analysis.
Figure 4 shows the ELISA results for members of the metalloinsertor family as a function of incubation time. We have shown previously that the Λ-enantiomer of Rh(bpy)2chrysi3+ is biologically inactive27 and that structurally binding to a mismatch site is enantiospecific for the Δ-isomer.24 For this reason, treatment with the 10 μM achiral tetraammine complex was compared to treatment with 20 μM racemic mixtures of the Rh(L)2chrysi3+ complexes (L = HDPA, bpy, or phen). The differential effect of rhodium treatment between the cells lines was quantified by subtracting the normalized percentages of cellular proliferation for each cell line. Notably, the optimal incubation time for each compound is inversely related to the hydrophobicity of the ancillary ligands, with rac-Rh(phen)2chrysi3+ exhibiting an optimal incubation time of 24 h. This trend also continues with rac-Rh(DIP)2chrysi3+, which exhibits differential effects in as little as 12 h at concentrations as low as 2 μM (Figure 5). Based on the early effect at 12 h, the HDPA complex may have different uptake characteristics; the analogous Ru complex has not yet been examined using flow cytometry. With the exception of the HDPA complex, this variation in activity with incubation time for the family of complexes parallels closely results seen earlier for uptake in HeLa cells by Ru(bpy)2dppz2+, Ru(phen)2dppz2+, and Ru(DIP)2dppz2+, where the most rapid uptake is apparent with the lipophilic DIP complex.29
Figure 4.
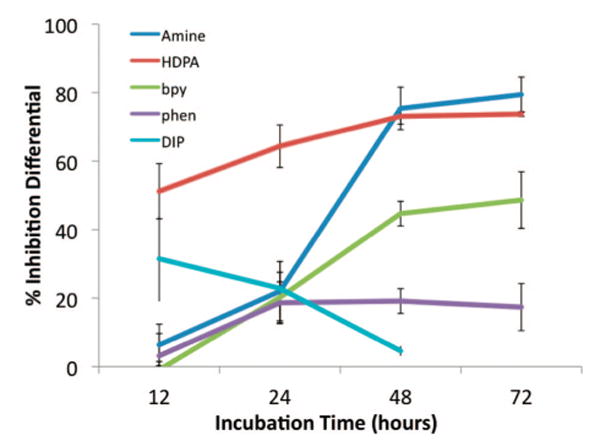
Inhibitory effects of rhodium metalloinsertors as a function of incubation time. Shown are plots of BrdU incorporation normalized to the BrdU incorporation of untreated cells as a function of rhodium concentration. The inhibition differential is the difference of the normalized percentages of cellular proliferation for each cell line, with standard error bars (SN–O = √(SN2 + SO2)). ELISA analyses were performed as in Figure 3. Cells were incubated with no rhodium, 2 μM rac-Rh(DIP)2chrysi3+, 10 μM Rh(NH3)4chrysi3+, or 20 μM rac-Rh(L)2chrysi3+ (L = HDPA, bpy, or phen).
Figure 5.
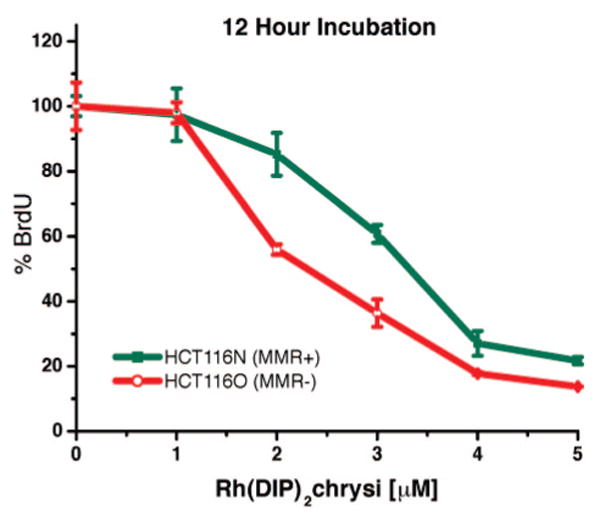
Inhibitory effects of rac-Rh(DIP)2chrysi3+. Shown are plots of BrdU incorporation normalized to the BrdU incorporation of untreated cells as a function of rhodium concentration. Standard error bars for five trials are shown. MMR-proficient HCT116N cells (green) and MMR-deficient HCT116O cells (red) were plated and allowed 24 h to adhere before incubation with 0–5 μM rac-Rh(DIP)2chrysi3+ for 12 h. At the end of the incubation, the medium containing Rh was replaced with fresh medium, and cells were grown for an additional 60 h before ELISA analysis. BrdU was added to the medium 24 h prior to analysis.
Figure 6 summarizes the differential effects on cell proliferation and the incubation time for the family of complexes. Clear correlations with the binding constants for these complexes are evident (Table 1). Significantly, the differential effect in inhibiting cell proliferation in MMR-deficient cells is directly correlated to the binding affinity of the compound for DNA mismatches. Rh(NH3)4chrysi3+ (KB = 1 × 108 M−1 at a CC mismatch), for example, shows the largest differential effect in inhibiting proliferation of MMR-deficient versus -proficient HCT116 cells after 72 h (79 ± 5%), while Rh(phen)2chrysi3+ (KB = 3.2 × 106 M−1 at a CC mismatch) shows a small differential effect (17 ± 7%). The DIP complex is rapidly taken up by the cells but also shows only a small differential inhibitory effect correlating with its poor specific binding at the mismatch site.
Figure 6.
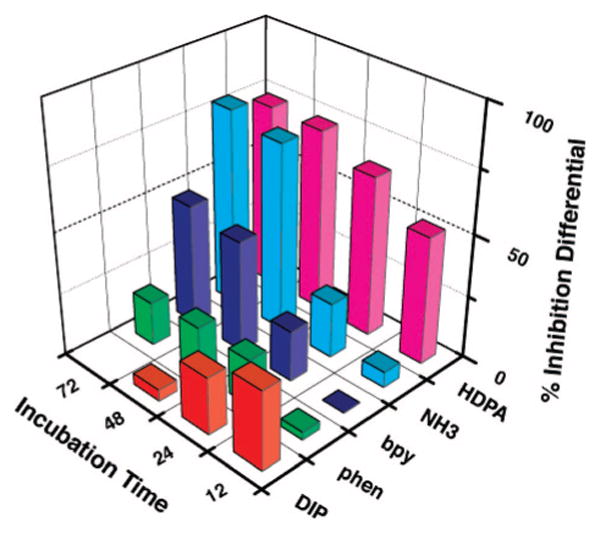
Inhibitory effects of rhodium metalloinsertors as a function of metal complex identity. Shown are bar graphs of BrdU incorporation normalized to the BrdU incorporation of untreated cells as a function of rhodium concentration. The inhibition differential is the difference of the normalized percentages of cellular proliferation for the two cell lines, HCT116O versus HCT116N. ELISA analyses were performed as in Figure 3. Cells were incubated with no rhodium, 2 μM rac-Rh(DIP)2chrysi3+, 10 μM Rh(NH3)4chrysi3+, or 20 μM rac-Rh(L)2chrysi3+ (L = HDPA, bpy, or phen). A correlation between mismatch binding affinity and differential inhibition of MMR-deficient cells is evident.
Discussion
A clear trend emerges when comparing the binding constants of the series of rhodium complexes to mismatched sites: the DNA mismatch binding affinity increases as the size of the ancillary ligand decreases. This trend is consistent with what we have learned from the structural studies, specifically that mismatch binding by insertion via the minor groove is subject to stringent space constraints. With major groove intercalation, the base rise is increased and the major groove offers space to accommodate the ancillary ligands. In contrast, with insertion, there is no increase in base pair rise; the mismatched bases are instead ejected and replaced by the deeply inserted chrysi ligand. Moreover, the minor groove, small even for hydrophobic groove binding molecules, offers little space for the ancillary ligands. While little enantioselectivity is apparent for intercalation of bpy complexes into B-form DNA, Δ-Rh(bpy)2chrysi3+ binds enantiospecifically to single base mismatches.24,37 Thus steric interactions of the ancillary ligands are seen as an extremely important factor governing the binding affinity of a metal complex at the mismatch site.
We have previously demonstrated that mismatch binding affinity is correlated with thermodynamic destabilization over all mismatch identities and sequence contexts.20,23 Here, we see for all the complexes that binding to the CC mismatch is tighter than binding to the AC mismatch. This is consistent with our previous observations, since AC is the thermodynamically more stable mismatch and, in this case, is estimated to stabilize the hairpin duplex by ∼0.5 kcal/mol relative to one containing a CC mismatch.38 We assume, then, that the general trend holds for all members of this metalloinsertor family. This stabilization is translated into a higher dissociation constant (smaller binding affinity) for the entire series of rhodium complexes. This decrease in binding affinity depends upon the greater energy required to eject the mismatched bases from the base pair stack, as evident crystallographically and by NMR.24,26 Nonetheless, for the family of chrysi complexes, the inverse relationship between the size of the ancillary ligand and the binding affinity still holds, with the smallest complex Rh(NH3)4chrysi3+ showing the highest affinity and that of the largest complex, Rh(DIP)2chrysi3+, more than 2 orders of magnitude lower.
Figure 7 compares the crystal structure of Δ-Rh(bpy)2chrysi3+ bound to the mismatch site24 with models of Δ-Rh(DIP)2chrysi3+ and Rh(NH3)4chrysi3+ similarly bound via the minor groove through metalloinsertion. Preserving the DNA conformation from the crystal structure, we see that Δ-Rh(DIP)2chrysi3+ runs into substantial steric hindrance, as its axial phenyl rings extend up and down into the groove, directly clashing with the bases. However, its equatorial phenyl rings do not pose any steric problems, as they point away from the DNA. These observations are supported by the small binding constant measured for Rh(DIP)2chrysi3+. Rh(phen)2chrysi3+, intermediate in size, shows binding affinities for the mismatches that are an order of magnitude lower than those of the bpy derivative but more than an order of magnitude higher than those of the DIP complex. Rh(HDPA)2chrysi3+ is slightly larger in size than the bpy derivative, but the HDPA ligands are more flexible, and there is an opportunity for hydrogen bonding; as a result, Rh(bpy)2chrysi3+ and Rh(HDPA)2chrysi3+ have comparable affinities for the mismatch. Analogously, the large binding constant of Rh(NH3)4chrysi3+ can be mostly attributed to its small size. Here it is reasonable to suggest that the axial ammines may also hydrogen bond with the neighboring base pairs to form additional stabilizing interactions. Nonetheless, as evident in Figure 7, the small cone size of the tetraammine structure clearly facilitates deep insertion within the minor groove site. In fact, the clear inverse correlation of binding affinity with ancillary ligand size, and the finding that Rh(DIP)2chrysi3+, despite its cumbersome size, is able to bind at all to a mismatch site with some specificity, corroborate our understanding of the driving force and the dynamics of mismatch recognition: the π-stacking between the inserted chrysi ligand and the adjacent bases provides the major stabilizing force for binding, and both the metal complex and DNA distort their conformations to accommodate each other in the bound state.
Figure 7.
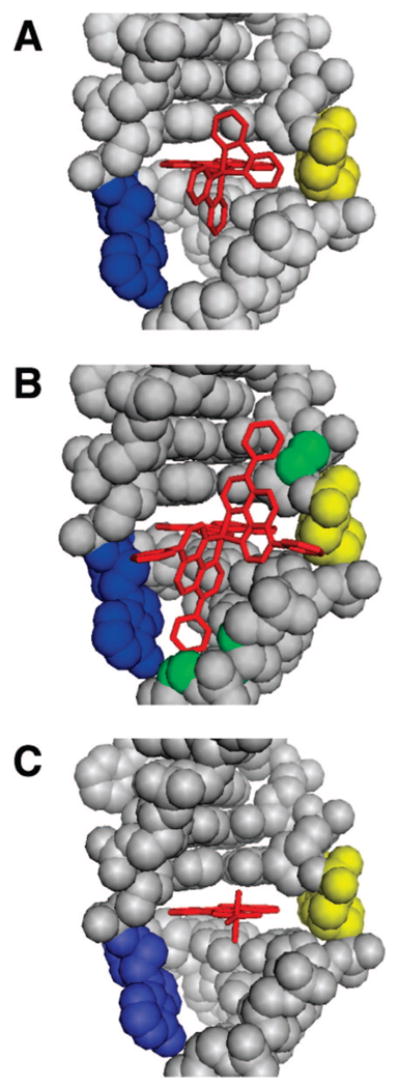
Crystal and model structures of rhodium metalloinsertors bound to the mismatch site. Rhodium insertors (red) are shown bound to the DNA (gray) from the minor groove at the mismatch site with the bases (adenine in blue, cytosine in yellow) ejected and the chrysi ligand stacked fully with the adjacent base pairs. The crystal structure of Δ-Rh(bpy)2chrysi3+ bound to the CA mismatch is shown in panel (A), along with structural models of Δ-Rh(DIP)2chrysi3+ (B) and Δ-Rh(NH3)4chrysi3+ (C) binding based on the crystal structure. Superposition of the DIP complex upon the rhodium center of the bpy complex leads to steric clashes with the sugar–phosphate backbone (possible atoms involved in green), whereas the tetraammine complex is easily accommodated.
Importantly, the DNA mismatch binding affinities of the Rh(L)2chrysi3+ family correlate well with the differential biological effects seen between the repair-proficient HCT116N and repair-deficient HCT116O cell lines. This correlation supports the hypothesis that DNA mismatches are the target of rhodium metalloinsertors in vivo. Because of this correlation, we may attribute the preferential inhibitory effect on MMR-deficient cells to binding of the complexes to DNA mismatches. Since the MMR-deficient cells contain more mismatches, the tighter binding complexes would be expected to display a greater inhibitory effect. It should be noted that finding any inhibitory effect with these complexes was at first surprising, since they bind DNA noncovalently and might be expected to be readily displaced. Although the mechanism of inhibition is not yet fully understood, it is likely that protein recognition of the metal-mismatch complex, perhaps by RNA polymerase or topoisomerase, may generate a covalent protein–DNA lesion and contribute to the cellular response.
The differential inhibitory effect seen with Rh(HDPA)2chrysi3+ cannot be understood simply on the basis of binding affinities. Despite having essentially the same mismatch binding affinity as Rh(bpy)2chrysi3+, the HDPA complex preferentially inhibits the MMR-deficient cell line almost as well as Rh(NH3)4chrysi3+ with long incubation times; with short times of incubation, the differential inhibitory effect by Rh(HDPA)2chrysi3+ is greatest. Both the HDPA ligand and the amine group have the potential to form hydrogen bonds. This hydrogen bonding capability and flexibility of the ligands might serve to make them more effective inhibitors of any protein–DNA interactions. Indeed, ruthenium complexes bearing HDPA ligands have been shown to exhibit DNA binding and cytotoxicity.39
Certainly, as with any pharmaceutical design, cellular uptake must also be considered. In the case of the HDPA complex, based upon the variations in inhibitory effect with incubation time, the amine ligands may facilitate nuclear uptake. Dppz analogues with the HDPA ligands have not yet been examined with respect to their uptake characteristics. For the bpy complexes, the 48 h incubation time required for Rh(bpy)2chrysi3+ to exert its anti-proliferative effect matches the 48 h requirement observed for Ru(bpy)2dppz2+ uptake in HeLa cells.27,29 The more lipophilic Rh(DIP)2chrysi3+ here is found to exert anti-proliferative effects at much shorter incubation times and lower concentrations, which also matches the accelerated uptake observed for Ru(DIP)2dppz2+. Cellular uptake is surely a rate-limiting factor in biological activity of the rhodium metalloinsertors, yet cellular uptake is not the only challenge: proper subcellular localization must also be achieved in order for any drug to act upon its target. It has been well established that lipophilic cations preferentially target the mitochondria, whereas hydrophilic cations do not.40,41 It may be that Rh(HDPA)2chrysi3+ and Rh(NH3)4chrysi3+ lack the lipophilicity required for mitochondrial accumulation, allowing a greater proportion of these compounds to reach the nucleus once inside the cell. This difference in intracellular partitioning could then account for the differential effects of Rh(HDPA)2chrysi3+.
In the development of octahedral rhodium complexes as anti-cancer agents, the choice of ancillary ligand can be seen as a design tradeoff, with the binding affinity for a DNA mismatch greatly outweighing uptake properties as the critical factor in the successful targeting of repair- deficient cells. Beyond their effects on DNA binding and overall cellular uptake, it is highly likely that the ancillary ligands affect the cellular response in other ways, including the potential for hydrogen bonding and differences in uptake and intracellular distribution. Here we are confronted with a tradeoff that may seem inevitable: more hydrophobic ligands facilitate cellular uptake but impede mismatch binding. Perhaps this tradeoff can be avoided by making conjugates arranged with functional moieties tethered with consideration of the structure of the DNA-bound complex associated snugly in the minor groove. Most importantly, these data support the contention that the cell-specific inhibitory effect we observe depends upon binding to the DNA mismatch inside the cell. This cell-specific strategy thus represents a promising direction in the development of small metal complexes that react preferentially in MMR-deficient cells, those susceptible to cancerous transformation.
Supplementary Material
Figure 1.
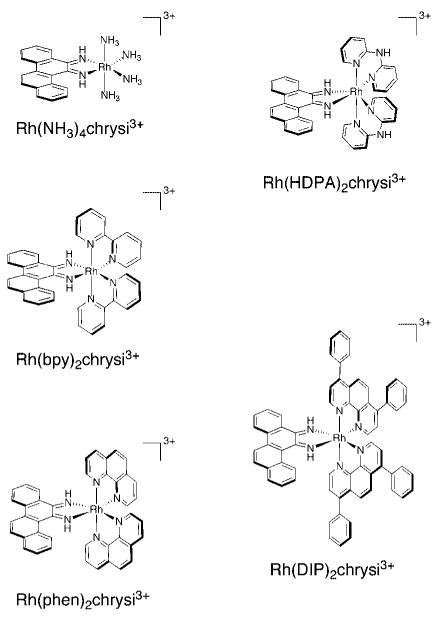
Rh(L)2chrysi3+ family of metalloinsertors.
Acknowledgments
Financial support for this work from the NIH (GM33309 to J.K.B.) is gratefully acknowledged. The authors also thank Dr. Jonathan Hart for his synthesis of [Rh(NH3)4chrysi]OTf3.
Footnotes
Supporting Information Available: Figures S1 and S2, showing inhibitory effects of rac-Rh(HDPA)2chrysi3+ and rac-Rh(phen)2chrysi3+ as a function of incubation time on cellular proliferation. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Iyer RR, Pluciennik A, Burdett V, Modrich PL. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 2.Loeb LA. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- 3.Bhattacharya NP, Skandalis A, Ganesh A, Groden J, Meuth M. Proc Natl Acad Sci U S A. 1994;91:6319–6323. doi: 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strauss BS. Mutat Res. 1999;437:195–203. doi: 10.1016/s1383-5742(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 5.Papadopoulos N, Lindblom A. Hum Mutat. 1997;10:89–99. doi: 10.1002/(SICI)1098-1004(1997)10:2<89::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 6.Peltomaki P. Hum Mol Genet. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 7.Lawes DA, SenGupta S, Boulos PB. Eur J Surg Oncol. 2003;29:201–212. doi: 10.1053/ejso.2002.1399. [DOI] [PubMed] [Google Scholar]
- 8.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JJ, Markowitz S, Willson JKV, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Proc Natl Acad Sci U S A. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arzimanoglou II, Gilbert F, Barber HRK. Cancer. 1998;82:1808–1820. doi: 10.1002/(sici)1097-0142(19980515)82:10<1808::aid-cncr2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 10.Pors K, Patterson LH. Curr Top Med Chem. 2005;5:1133–1149. doi: 10.2174/156802605774370883. [DOI] [PubMed] [Google Scholar]
- 11.Valentini AM, Armentano M, Pirrelli M, Caruso ML. Cancer Treatment Rev. 2006;32:607–618. doi: 10.1016/j.ctrv.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Carethers JM, Hawn MT, Chauhan DP, Luce MC, Marra G, Koi M, Boland CR. J Clin Invest. 1996;98:199–206. doi: 10.1172/JCI118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehme A, Christen RD, Howell SB. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 14.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aebi S, Fink D, Gordon R, Kim HK, Zheng H, Fink JL, Howell SB. Clin Cancer Res. 1997;3:1763–1767. [PubMed] [Google Scholar]
- 16.Fedier A, Schwarz VA, Walt H, Carpini RD, Haller U, Fink D. Int J Cancer. 2001;93:571–576. doi: 10.1002/ijc.1356. [DOI] [PubMed] [Google Scholar]
- 17.Fink D, Aebi S, Howell SB. Clin Cancer Res. 1998;4:1–6. [PubMed] [Google Scholar]
- 18.Karran P, Offman J, Bignami M. Biochimie. 2003;85:1149–1160. doi: 10.1016/j.biochi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Jackson BA, Barton JK. J Am Chem Soc. 1997;119:12986–12987. [Google Scholar]
- 20.Jackson BA, Barton JK. Biochemistry. 2000;39:6176–6182. doi: 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- 21.Junicke H, Hart JR, Kisko J, Glebov O, Kirsch I, Barton JK. Proc Natl Acad Sci U S A. 2003;100:3737–3742. doi: 10.1073/pnas.0537194100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeglis BM, Pierre VP, Barton JK. Chem Commun. 2007;44:4565–4579. doi: 10.1039/b710949k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson BA, Alekseyev VY, Barton JK. Biochemistry. 1999;38:4655–4662. doi: 10.1021/bi990255t. [DOI] [PubMed] [Google Scholar]
- 24.Pierre VC, Kaiser JT, Barton JK. Proc Natl Acad Sci U S A. 2007;104:429–434. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kielkopf CL, Erkkila KE, Hudson BP, Barton JK, Rees DC. Nat Struct Biol. 2000;7:117–121. doi: 10.1038/72385. [DOI] [PubMed] [Google Scholar]
- 26.Cordier C, Pierre VC, Barton JK. J Am Chem Soc. 2007;129:12287–12295. doi: 10.1021/ja0739436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart JR, Glebov O, Ernst RJ, Kirsch IR, Barton JK. Proc Natl Acad Sci U S A. 2006;103:15359–15363. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 29.(a) Puckett CA, Barton JK. J Am Chem Soc. 2007;129:46–47. doi: 10.1021/ja0677564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Puckett CA, Barton JK. Biochemistry. 2008;47:11711–11716. doi: 10.1021/bi800856t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunner J, Barton JK. Biochemistry. 2006;45:12295–12302. doi: 10.1021/bi061198o. [DOI] [PubMed] [Google Scholar]
- 31.Zeglis BM, Barton JK. Nat Protoc. 2007;2:357–371. doi: 10.1038/nprot.2007.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon NE, Lawrance GA, Lay PA, Sargeson AM. Inorg Chem. 1983;22:846–847. [Google Scholar]
- 33.Dixon NE, Lawrance GA, Lay PA, Sargeson AM. Inorg Chem. 1984;23:2940–2947. [Google Scholar]
- 34.Reitmar AH, Risley R, Bristow RG, Wilson T, Ganesh A, Jang A, Peacock J, Benchimol S, Hill RP, Mak TW, Fishel R, Meuth M. Cancer Res. 1997;57:3765–3771. [PubMed] [Google Scholar]
- 35.Gratzner HG. Science. 1982;218:474–475. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 36.Krotz AH, Kuo LY, Shields TP, Barton JK. J Am Chem Soc. 1993;115:3877–3882. [Google Scholar]
- 37.Barton JK. Science. 1986;233:727–734. doi: 10.1126/science.3016894. [DOI] [PubMed] [Google Scholar]
- 38.SantaLucia J, Jr, Hicks D. Annu Rev Biophys Biomol Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 39.Rajendiran V, Murali M, Suresh E, Palaniandavar M, Periasamy VS, Akbarsha MA. Dalton Trans. 2008:2157–2170. doi: 10.1039/b715077f. [DOI] [PubMed] [Google Scholar]
- 40.Napolitano SM, Aprille JR. Adv Drug Delivery Rev. 2001;49:63–70. doi: 10.1016/s0169-409x(01)00125-9. [DOI] [PubMed] [Google Scholar]
- 41.Murphy MP, Smith RAJ. Adv Drug Delivery Rev. 2000;41:235–250. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.