

Abstract
The DNA-binding properties of Ru(bpy)2(eilatin)2+ have been investigated to determine if the sterically expansive eilatin ligand confers specificity for destabilized single-base mismatches in DNA. Competitive DNA photocleavage experiments employing a sequence-neutral metallointercalator, Rh(bpy)2(phi)3+ (phi = 9,10-phenanthrenequinonediimine), and a mismatch-specific metalloinsertor, Rh(bpy)2(chrysi)3+ (chrysi = chrysene-5,6-quinonediimine), reveal that the eilatin complex binds to a CC mismatched site with an apparent binding constant of 2.2(2) × 106 M−1. Nonetheless, the selectivity in binding mismatched DNA is not high: competitive titrations with Rh(bpy)2(phi)3+ show that the complex binds also to well-matched B-form sites. Thus, Ru(bpy)2(eilatin)2+, despite containing the extremely expansive eilatin ligand, displays lower selectivity for the mismatch than does Rh(bpy)2(chrysi)3+, a metalloinsertor containing the smaller, though still bulky, chrysene-5,6-quinonediimine ligand. In summary, the size and shape of the eilatin ligand allow stacking with both well-matched and mismatched DNA.
Introduction
Octahedral metal complexes that bind DNA have received considerable attention based upon their potential utility as nucleic acid probes and chemotherapeutics.1 Our laboratory has long focused on developing the DNA recognition capabilities of metal complexes, and recently we have designed bulky octahedral complexes that specifically target single-base mismatches.2
Single-base DNA mismatches occur in the cell as a result of polymerase errors, genotoxic chemicals, or UV-induced damage.3 Left unrepaired, mismatches can, upon replication, lead to potentially harmful single-base mutations. In response to this continual threat to the integrity of its DNA, the cell has evolved a complex enzymatic mismatch repair machinery that recognizes and repairs these sites of damage as they are formed.4 If this machinery is disabled, however, mismatches and consequent mutations will accumulate in the genome, often with dire consequences. Indeed, mutations in mismatch repair genes have been implicated in 80% of hereditary nonpolyposis colon cancers.5
Unlike sequence-specific metal complexes in which ancillary ligands bear the recognition elements,1 mismatch-specific metal complexes, most notably Rh(bpy)2(chrysi)3+ (chrysi = chrysene-5,6-quinonediimine) and Rh(bpy)2(phzi)3+ (phzi = benzo[a]phenazine-5,6-quinonediimine), depend upon the intercalating ligand for their recognition properties.6 Aromatic steric bulk is the key. While the intercalating ligands of complexes that bind well-matched DNA, such as Rh(bpy)2(phi)3+ (phi = 9,10-phenanthrenequinonediimine), slide easily between the base pairs, the more sterically expansive ligands of mismatch-specific complexes are simply too wide to fit into well-matched B-form DNA (Figure 1). The complexes bearing these bulky ligands are, however, able to bind mismatched sites, which are thermodynamically destabilized relative to well-matched DNA because of the impaired hydrogen bonding and stacking of the mismatched bases. Rh(bpy)2(chrysi)3+, for example, has been shown to bind 80% of all mismatches in all possible sequence contexts. Moreover, this mismatch binding is highly specific: the Rh complex was shown to target a single-base mismatch within a 2700 bp DNA fragment.7 More destabilized mismatches are bound more tightly than less destabilized sites, with highly stable, G-containing sites escaping recognition altogether.
Figure 1.
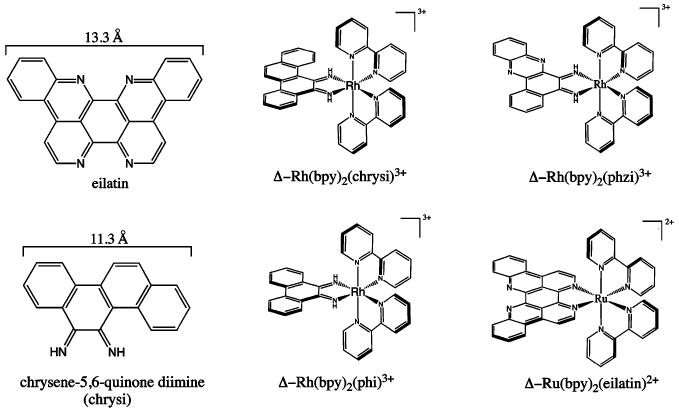
Chemical structures of eilatin, 9,10-chrysene-5,6-quinone diimine, Δ-Rh(bpy)2(chrysi)3+, Δ-Rh(bpy)2(phzi)3+, Δ-Rh(bpy)2(phi)3+, and Δ-Ru(bpy)2(eilatin)2+. Width approximations made using ChemDraw 3D with energy-minimized structures.
Importantly, recent structural studies have revealed the basis for this targeting and for the correlation between the binding affinity and thermodynamic instability of a mismatch.8 Interestingly, these complexes do not bind mismatches via traditional metallointercalation; Rh(bpy)2(phi)3+, for example, binds from the major groove, inserts a ligand into the base stack, and, in so doing, increases the base pair rise of the helix.9 Instead, these bulkier metal complexes bind the mismatched site through insertion of the expansive ligand into the duplex from the minor groove so as to eject the mismatched bases into the major groove. For metalloinsertion, the inserted ligand replaces the extruded bases in the DNA π stack.8 Because this metalloinsertion occurs via the narrow minor groove and yields no increase in the base-pair rise, the binding site for the metal complex is sterically restrictive. Consequently, the binding is enantiospecific for Δ-Rh(bpy)2(chrysi)3+: the right-handed helix can only accommodate the right-handed enantiomer.
These bulky metalloinsertors have been employed successfully in a range of applications, yet we continue to seek new mismatch-specific complexes in an effort to target the more stable base mismatches.10,11 We considered that augmenting the size of the bulky aromatic ligand might provide this increase in mismatches targeted because a greater surface area for π stacking might yield the boost in binding affinity required for the targeting of more thermodynamically stable mismatched sites. Here we describe binding of Ru(bpy)2(eilatin)2+, a complex bearing a singularly expansive ligand (Figure 1), to matched and mismatched duplex DNA.
Eilatin is a highly symmetric, heptacylic marine alkaloid first isolated in 1988 from the Red Sea tunicate Eudistoma sp.12 While the molecule itself has proven to be of significant interest to both synthetic and biological chemists, it is, however, eilatin coordinated to an octahedral metal complex that offers the possibility of high-affinity metalloinsertion. Ru(bpy)2(eilatin)2+ has been prepared and characterized spectroscopically by Kol and co-workers.13 Moreover, studies with nucleic acids by Tor and co-workers have revealed binding to folded RNAs and nonspecific association with calf thymus DNA.14 Our laboratory has previously examined the binding of luminescent Ru complexes to DNA and RNA, most notably the light-switch compound Ru(phen)2(dppz)2+ (dppz = dipyridophenazine).15 Yet here, our interest is primarily predicated on the shape characteristics of the ligand and its potential applications as a specific probe for mismatched DNA. Our studies show, however, that steric bulk alone is insufficient to achieve site-specificity.
Experimental Section
Metal Complexes
Eilatin was synthesized from kynuramine (Aldrich) via the facile biomimetic pathway of Kashman et al. and purified via preparatory thin-layer chromatography (silica, 96:4 CH2Cl2/MeOH).16 Ru(bpy)2(eilatin)2+ was synthesized as reported and purified via an ion exchange column (Aldrich Sephadex CM25) and reverse-phase high-performance liquid chromatography [HP1100 HPLC system with Varian DynaMax C18 semipreparative column, gradient of 15:85 TO 60:40 H2O (0.1% TFA)/MeCN (0.1% TFA) over 60 min].13 No extinction coefficients in buffer have been reported. Inductively coupled plasma mass spectrometry was employed to determine aqueous extinction coefficients of ε = 64 000 M−1 cm−1 (244 nm), 68 000 M−1 cm−1 (287 nm), and 38 000 M−1 cm−1 (426 nm) for [Ru(bpy)2(eilatin)]Cl2. Because few enantioselective effects were evident in binding Ru(bpy)2(eilatin)2+ to well-matched oligonucleotides, racemates were employed in all experiments used here.14a,b Moreover, dimerization or aggregation of Ru(bpy)2(eilatin)2+ in solution over the concentrations utilized for experiments does not appear to be an issue because, in this concentration range, no deviations from Beer's law are found. Some aggregation associated with DNA binding may arise however.13 [Rh(bpy)2(chrysi)]Cl3 and [Rh(bpy)2(phi)]Cl3 were synthesized and purified via published protocols17 and also employed as racemic mixtures. All reagents and solvents were used as received.
Oligonucleotide Synthesis and Photocleavage
Oligonucleotides were synthesized from phosphoramidites on an ABI 3400 DNA synthesizer. Following synthesis, the oligonucleotides were purified both with and without dimethoxytrityl protecting groups via reverse-phase HPLC [HP1100 HPLC system with Varian DynaMax C18 semipreparative column, gradient of 5:95 to 45:55 MeCN/50 mM NH4OAc(aq) over 30 min for DMT-ON purification and 2:98 to 17:83 55 MeCN/50 mM NH4OAc(aq) over 30 min for DMT-OFF purification]. All PAGE experiments described employed denaturing 20% polyacrylamide gels (National Diagnostics) and were performed according to published procedures.17 DNA strands were radioactively labeled with [γ-32P]-ATP (MP Biomedicals) using literature protocols and purified by 20% denaturing PAGE.17 All irradiations were performed using an Oriel Instruments solar simulator (300–440 nm). Gels were developed using Molecular Dynamics phosphorimaging screens and a Molecular Dynamics Storm 820 phosphorimager and subsequently visualized and quantified with Molecular Dynamics ImageQuant software.
Results and Discussion
Characterization of Site Selectivity of Ru(bpy)2(eilatin)2+
Owing to the short excited-state lifetime of Ru(bpy)2(eilatin)2+, direct methods such as DNA photocleavage or singlet oxygen sensitization could not be used to characterize the sites targeted by the Ru complex within the DNA duplex.18 Instead, competition experiments were employed. We first utilized Rh(bpy)2(phi)3+, which binds duplex DNA with little site-selectivity,19 in order to probe where the Ru complex binds through competitive inhibition. A synthetic 36-mer oligonucleotide was synthesized with complements featuring a guanine (EL-M) or a cytosine (EL-MM) across from a central cytosine (bold) to form matched and mismatched strands: 5′-CGCTACGTCTATATGCATGATCCTAAGTGACAGTAC-3′. After synthesis and purification, the reverse strand (not shown) was radioactively labeled with 32P-ATP at its 5′ terminus via standard protocols.17 Then, samples (1 μM) of radiolabeled EL-M and EL-MM DNA in buffer (50 mM NaCl, 10 mM NaPi, pH 7.1) were incubated with 8 μM Rh(bpy)2(phi)3+ and irradiated for 20 min using a solar simulator in the presence of variable amounts of Ru(bpy)2(eilatin)2+. A concentration of 8 μM Rh(bpy)2(phi)3+ provides 1 Rh molecule/4 base pairs, enough to saturate the entire oligonucleotide with Rh complexes.
Autoradiography of the resultant gel reveals that, with photoactivation, Rh(bpy)2(phi)3+ promotes cleavage on the EL-M DNA at six discrete sites (with base numbers from 3′-end): C19, G22, C27, C29, T32, and C33 (Figure 2). Interestingly, EL-MM DNA is cleaved at the same locations by Rh(bpy)2(phi)3+ but also displays two more cleavage bands: T13 and C16. The C16 position is the mismatched site. The somewhat curious cleavage at T13 may result from local conformational changes created by the nearby mismatch in the EL-MM sequence, leading to hyperreactivity.20
Figure 2.
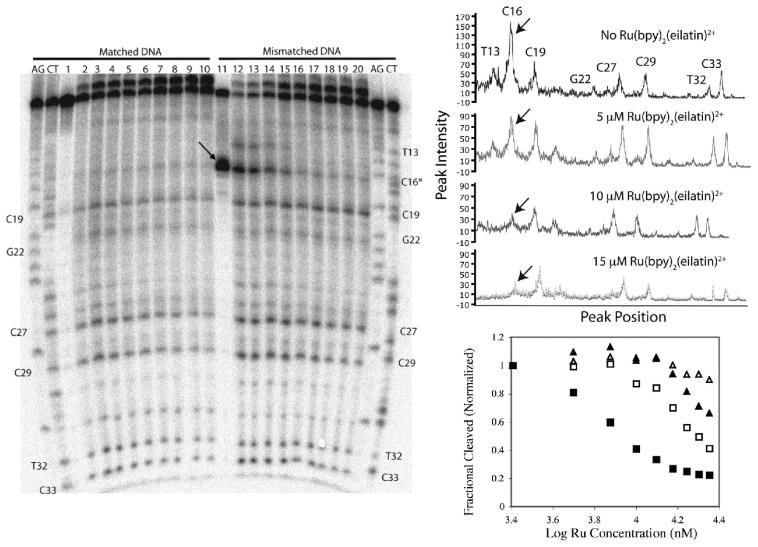
Competitive binding of Ru(bpy)2(eilatin)2+ to matched and mismatched DNA monitored using Rh(bpy)2(phi)3+ photocleavage. Left: Denaturing polyacrylamide gel showing the competition of Rh(bpy)2(phi)3+ and Ru(bpy)2(eilatin)2+ for matched (left) and mismatched (right) DNA of the sequence 3′-GCGATGCAGATATACCTACTAGGATTCACTGTCATG-32P-5′. All samples were prepared with 1 μM DNA, 50 mM NaCl, 10 mM NaPi, and pH 7.1 and, unless otherwise stated, irradiated for 20 min on a solar simulator. Left and right AG and CT lanes are Maxam–Gilbert sequencing reactions for matched and mismatched DNA, respectively. Lanes 1–10 employ matched DNA and lanes 11–20 mismatched DNA. Sample conditions: lanes 1 and 11, 1 μM Rh(bpy)2(chrysi); lanes 2–10 and 12–20, 8 μM Rh(bpy)2(phi)3+. Lanes 3–10 and 13–20 also contain increasing amounts of Ru(bpy)2(eilatin)2+, beginning with 2.5 μM Ru(bpy)2(eilatin)2+ in lanes 3 and 13 and increasing in increments of 2.5–22.5 μM in lanes 10 and 20. The arrow marks the mismatch site. Right top: Line plots of lanes 14, 16, 18, and 20 in gel. The arrow marks the mismatched site. Right bottom: Quantitation of the Rh(bpy)2(phi)3+ cleavage band intensity as a function of the Ru(bpy)2(eilatin)2+ concentration. Symbols: filled square, C16; empty square, C29; filled triangle, C27; empty triangle, C19.
Figure 2 also shows the effect of increasing Ru(bpy)2(eilatin)2+ concentrations on Rh(bpy)2(phi)3+ photocleavage by gel autoradiography and a corresponding line plot. With increasing concentrations of Ru(bpy)2(eilatin)2+, on both the matched and mismatched duplexes, all of the Rh(bpy)2(phi)3+ cleavage bands lessen in intensity, indicating that Ru(bpy)2(eilatin)2+ is competing with, and eventually inhibiting, Rh binding at all sites. At these Ru concentrations, this nonspecific inhibition of Rh photocleavage cannot be accounted for primarily through light absorption by the Ru complex but instead must reflect competitive binding of the Ru complex to well-matched DNA sites. Increasing concentrations of Ru(bpy)32+, a metal complex that binds DNA very weakly and has extinction coefficients similar to those of Ru(bpy)2(eilatin)2+ over the spectral range of interest, have no effect on the photocleavage intensities of Rh(bpy)2(phi)3+ and Rh(bpy)2(chrysi)3+ in the salient concentration range.15 Moreover, this Ru(bpy)32+ control also excludes the likelihood that Ru(bpy)2(eilatin)2+ inhibits Rh photocleavage by quenching of the Rh excited state. For the well-matched duplex, photocleavage with 8 μM Rh(bpy)2(phi)3+ is fully inhibited at ∼15 μM Ru(bpy)2(eilatin)2+. Nonspecific duplex binding occurring in the micromolar range is therefore comparable for the two complexes.
Interestingly, however, on the mismatched duplex, site preferences for both Rh(bpy)2(phi)3+ and Ru(bpy)2(eilatin)2+ are evident. In the absence of Ru, Rh photocleavage on the mismatched duplex is most intense at the mismatched site, C16. However, with increasing Ru, it is photocleavage at this mismatched site that is preferentially inhibited, and cleavage at the mismatch site is competed out at noticeably lower concentrations of Ru(bpy)2(eilatin)2+ (∼5 μM). This differential inhibition is most evident in the line plot and gel quantitation of the titration (Figure 2). The higher photocleavage for Rh(bpy)2(phi)3+ in the absence of Ru actually reflects a slightly higher affinity for the mismatched versus matched site, a common characteristic of classical intercalators.7b,21 Preferential inhibition of Rh photocleavage by Ru(bpy)2(eilatin)2+ may similarly reflect this preferential stacking with a mismatched site. Indeed, the gel quantitation shows that binding to the mismatch is less than an order of magnitude tighter than to matched sites. Curiously, the T13 cleavage site is also competed out well before the other matched locations by the Ru complex. Because hyperreactivity of Rh(bpy)2(phi)3+ at T13 depends on the nearby C16 mismatch, it appears it is similarly affected in competition with Ru(bpy)2(eilatin)2+.
Determination of the Binding Affinity of Ru(bpy)2(eilatin)2+ to the Mismatch
While competition with a nonspecific intercalator provides qualitative information about site preference, quantitative data regarding site-specific affinity can be determined by competition with the mismatch-specific metalloinsertor, Rh(bpy)2(chrysi)3+. For this second competition experiment, a similar but shorter 5′-32P-labeled oligonucleotide was synthesized to minimize binding to matched DNA. Complements containing a guanine and cytosine across from the central cytosine (bold) were also synthesized to afford matched (ES-M) and mismatched (ES-MM) duplexes: 5′-32P-TTAGGATCATCCATATA-3′. A titration employing 1 μM mismatched DNA in buffer (50 mM NaCl, 10 mM NaPi, pH 7.1) and variable Rh(bpy)2(chrysi)3+ was first used to obtain a mismatch-specific binding constant for the Rh complex of 1.7(2) × 106 M−1.
Given a known binding constant for Rh(bpy)2(chrysi)3+, the competition experiment yields the quantitative binding affinity for the mismatched site. The competition experiment was performed using 3 μM ES-MM DNA and 3 μM Rh(bpy)2(chrysi)3+ in 50 mM NaCl, 10 mM NaPi, and pH 7.1 along with increasing concentrations of Ru(bpy)2(eilatin)2+ (0–20 μM). The samples were then irradiated for 15 min on the solar simulator and subsequently eluted through a denaturing polyacrylamide gel. The resultant gel clearly shows initially strong Rh(bpy)2(chrysi)3+ photocleavage at the mismatch site that is inhibited by increasing concentrations of Ru(bpy)2(eilatin)2+ (Figure 3). From these titration data, we can extract a CC mismatch-specific binding constant for Ru(bpy)2(eilatin)2+ of 2.2(2) × 106 M−1.22 It is interesting that the Ru affinity for this mismatched site is comparable to that of Rh(bpy)2(chrysi)3+. Note that this value reflects binding to a 15-mer that contains additional matched sites to which the Ru complex may also bind (albeit likely at higher Ru concentrations). As a result, the binding affinity for the mismatched site must be considered in the context of competition also with matched sites.
Figure 3.
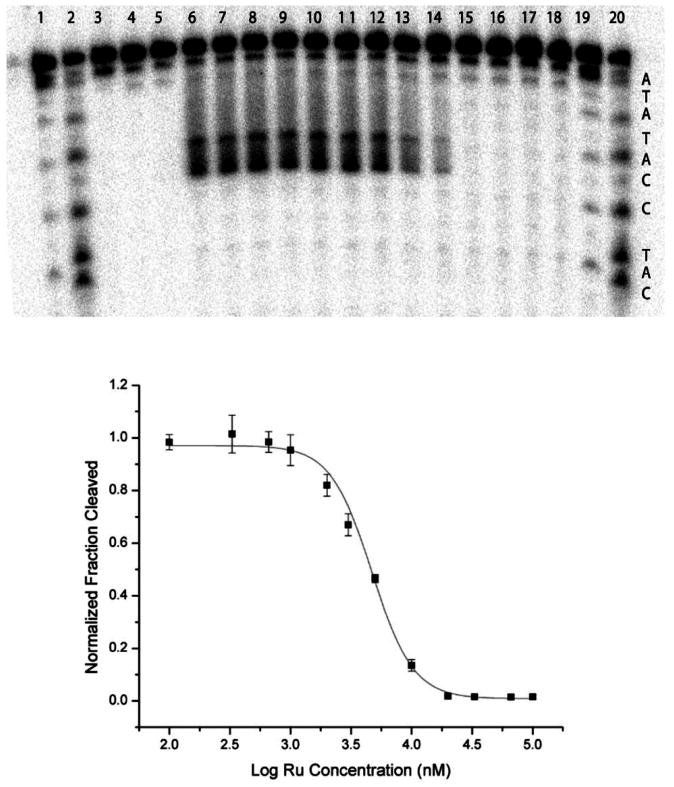
Competitive binding of Ru(bpy)2(eilatin)2+ to mismatched DNA monitored using Rh(bpy)2(chrysi)3+ photocleavage. Top: Denaturing polyacrylamide gel of a competition experiment between Rh(bpy)2(chrysi)3+ and Ru(bpy)2(eilatin)2+ for a CC mismatch in the oligonucleotide 5′-32P-TTAGGATCATCCATATA-3′. AG and CT lanes are Maxam–Gilbert sequencing reactions. All samples contained a 3 μM mismatched duplex in a buffer of 50 mM NaCl, 10 mM NaPi, and pH 7.1 and were irradiated for 10 min using a solar simulator unless otherwise stated. Sample conditions: lane 1, DNA only irradiated without Rh; lane 2, 3 μM Rh(bpy)2(chrysi)3+ without irradiation; lane 3, 3 μM Ru(bpy)2(eilatin)2+ irradiated without Rh; lanes 4–16, 3 μM Rh(bpy)2(chrysi)3+ and increasing concentrations of Ru(bpy)2(eilatin)2+, 0, 0.1, 0.33, 0.66, 1, 2, 3, 5, 10, 20, 33, 66, and 100 μM, respectively. Wide photocleavage bands do not reflect nonspecific photocleavage at more than one site but rather the multiple products produced by hydrogen abstraction upon photoactivated cleavage at the mismatched site. Bottom: Plot of a fraction cleaved against the Ru(bpy)2(eilatin)2+ concentration for four trials of the competition experiment.
Implications for the Design of Bulky Metalloinsertors
Taken together, the two competition experiments described clearly indicate that while Ru(bpy)2(eilatin)2+ does show some preference in binding the CC mismatch, the bulky complex also displays significant binding to well-matched B-form DNA sites. The site specificity of the Ru complex for a mismatch is therefore significantly less than that of Rh(bpy)2(chrysi)3+.7 A comparison of the measured mismatched-site dissociation constant [KD = 460(9) nM] to those obtained for matched sites supports this assertion; with matched DNA, binding is in the low micromolar range.14 Thus, the selectivity of the complex for mismatched sites is modest (ratio of binding mismatched versus matched ≤ 10). It is noteworthy that earlier it was suggested that Ru(bpy)2(eilatin)2+ may bind preferentially to large structural motifs in folded RNAs.14 Binding of the hydrophobic and large cationic Ru complex may arise with a range of nucleic acid structures.
The ability of Ru(bpy)2(eilatin)2+ to bind both matched and mismatched DNA prompts the consideration of how the Ru complex may interact structurally with matched and mismatched sites. Figure 4 shows schematic illustrations of Ru(bpy)2(eilatin)2+ bound to mismatched DNA in comparison to Rh(bpy)2(chrysi)3+ and to matched DNA in comparison to Rh(bpy)2(phi)3+. Binding of the Rh complexes to their target sites is based upon crystal structures8,9 and shows access from the minor groove side for metalloinsertion into a mismatched site and from the major groove side for access by metallointercalation. For the Rh complexes, it is apparent that these binding modes permit complete stacking of the inserting ligand between the base pairs. Moreover, the ancillary ligands of the octahedral complexes provide a barrier to both deeper insertion and significant rotation in the pocket. The complexes are bound so that the dyad axis of the base pairs bisects the immine–Rh–immine angle. In this mode, binding of the complexes is optimized for stacking, at both the mismatched and matched sites. As illustrated in Figure 4, the Ru complex is also well situated within a mismatched site for substantial stacking overlap, despite the large size of the eilatin ligand. Overlap with the base pairs is quite comparable for the chrysi and eilatin ligands, consistent with their similar binding affinity for the CC mismatch. Significantly for Ru(bpy)2(eilatin)2+, however, the complex can still stack well within a matched site although rotated relative to the bound Rh complex. The eilatin ligand is sufficiently expansive that substantial stacking is available between the base pairs without a straight-on orientation of the complex. It is noteworthy that we have seen previously for Ru(bpy)2dppz2+ fluorescence and NMR results that are consistent with a mixture of straight-on and side-on orientations in matched duplex DNA.23 Here, at the matched site, the eilatin complex can easily rotate within the intercalation site and maintain significant overlap with the bases above and below. Indeed, the stacking area appears comparable to that of the phi complex, just as their binding affinities for matched sites are similar. The great expanse of the eilatin permits this significant stacking without the axial ligands serving as a barrier to rotation. Thus, while binding to a mismatched site by Ru(bpy)2(eilatin)2+ is possible, binding to the matched site is not precluded.
Figure 4.
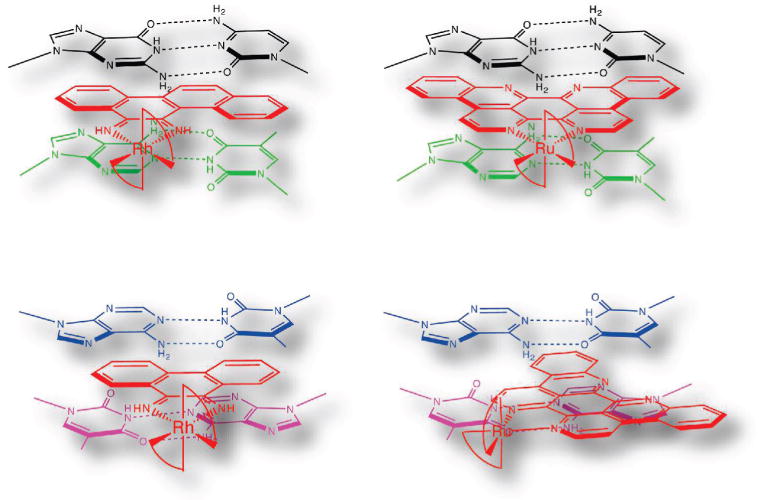
Schematic illustrations of Ru(bpy)2(eilatin)2+ (right) bound to mismatched (top) and matched (bottom) DNA sites based on the crystal structures of chrysi (top left) and phi (bottom left) complexes of Rh bound to mismatched and matched DNA, respectively. For binding to the mismatched site, the metal complexes are oriented from the minor groove side, whereas for binding to the matched site, the association is from the major groove side.
These studies show that simply increasing the expanse of a metalloinsertor is not sufficient to gain an increase in specific binding to a mismatched site in duplex DNA. While binding to the mismatched site is well accommodated by a bulkier ligand, the increased expanse also provides a stacking area for the complex at a matched site if the ligand is particularly expansive. In a comparison of the family of bulky metal complexes, similar affinities in binding-mismatched DNA are observed for those bearing the phzi and chrysi ligands versus that containing the even more expansive eilatin ligand. The stacking interaction is defined by the surface area of the base pairs above and below the metalloinsertor, not just the size of the metalloinsertor. With the eilatin complex, a substantial affinity and stacking area are also available at the matched site. Thus, for this expansive complex, instead, we see that specificity for a single base-pair mismatch is lost. That the eilatin ligand extends considerably from the metal center in two directions is likely responsible for this loss in specificity for mismatched sites and gain in affinity for matched DNA; these structural characteristics of Ru(bpy)2(eilatin)2+ allow the complex to bind matched DNA in a manner that Rh(bpy)2(chrysi)3+ and Rh(bpy)2(phzi)3+ cannot. As a consequence, then, these experiments teach us something simple about the design of mismatch-recognition ligands: bulky is good, but bulkier is not necessarily better.
Supplementary Material
Acknowledgments
We thank the NIH (Grant GM33309) for their financial support. We also thank Drs. Y. Kashman and Y. Tor for helpful correspondence.
Footnotes
Supporting Information Available: Rh(bpy)2(chrysi)3+ binding constant determination gel and plot, Ru(bpy)32+/Rh(bpy)2(chrysi)3+ competition experiment gel, and Rh(bpy)2(chrysi)3+/Ru(bpy)2(eilatin)2+ competition experiment plot. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Erkilla KE, Odom DT, Barton JK. Chem Rev. 1999;99:2777. doi: 10.1021/cr9804341. [DOI] [PubMed] [Google Scholar]
- 2.Zeglis BM, Pierre VP, Barton JK. Chem Commun. 2007:4565. doi: 10.1039/b710949k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Modrich P. Annu Rev Genet. 1991;25:229. doi: 10.1146/annurev.ge.25.120191.001305. [DOI] [PubMed] [Google Scholar]; (b) Kolodner R. Genes Dev. 1996;10:1433. doi: 10.1101/gad.10.12.1433. [DOI] [PubMed] [Google Scholar]
- 4.(a) Harfe BD, Jinks-Robertson S. Annu Rev Genet. 2000;34:359. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]; (b) Modrich P. J Biol Chem. 2006;281:30305. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Kolodner RD. Trends Biochem Sci. 1995;20:397. doi: 10.1016/s0968-0004(00)89087-8. [DOI] [PubMed] [Google Scholar]; (b) Arizamanoglou II, Gilbert F, Barber HR. Cancer. 1998;82:1808. doi: 10.1002/(sici)1097-0142(19980515)82:10<1808::aid-cncr2>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]; (c) Loeb LA, Loeb KR, Anderson JP. Proc Natl Acad Sci U S A. 2003;100:776. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Jackson BA, Barton JK. J Am Chem Soc. 1997;119:12986. [Google Scholar]; (b) Junicke H, Hart JR, Kisko J, Glebov O, Kirsch IR, Barton JK. Proc Natl Acad Sci U S A. 2003;100(7):3737. doi: 10.1073/pnas.0537194100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Jackson BA, Alekseyev VY, Barton JK. Biochemistry. 1999;38:4656. doi: 10.1021/bi990255t. [DOI] [PubMed] [Google Scholar]; (b) Jackson BA, Barton JK. Biochemistry. 2000;39:6176. doi: 10.1021/bi9927033. [DOI] [PubMed] [Google Scholar]
- 8.(a) Pierre VP, Kaiser JT, Barton JK. Proc Natl Acad Sci U S A. 2007;104:429. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cordier C, Pierre VP, Barton JK. J Am Chem Soc. 2007;129:12287. doi: 10.1021/ja0739436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kielkopf CL, Erkilla KE, Hudson BP, Barton JK, Rees DC. Nat Struct Biol. 2000;7:117–121. doi: 10.1038/72385. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hart JR, Johnson MD, Barton JK. Proc Natl Acad Sci U S A. 2004;101:14140. doi: 10.1073/pnas.0406169101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hart JR, Glebov O, Ernst RJ, Kirsch IL, Barton JK. Proc Natl Acad Sci U S A. 2006;103:15359. doi: 10.1073/pnas.0607576103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brunner J, Barton JK. J Am Chem Soc. 2006;128:6772. doi: 10.1021/ja0612753. [DOI] [PubMed] [Google Scholar]; (d) Zeglis BM, Barton JK. J Am Chem Soc. 2006;128:5654. doi: 10.1021/ja061409c. [DOI] [PubMed] [Google Scholar]; (e) Schatzschneider U, Barton JK. J Am Chem Soc. 2004;126:8631. doi: 10.1021/ja048543m. [DOI] [PubMed] [Google Scholar]
- 11.(a) Ruba E, Hart JR, Barton JK. Inorg Chem. 2004;43:4570. doi: 10.1021/ic0499291. [DOI] [PubMed] [Google Scholar]; (b) Petitjean A, Barton JK. J Am Chem Soc. 2004;126:14728. doi: 10.1021/ja047235l. [DOI] [PubMed] [Google Scholar]
- 12.Rudi A, Benayahu Y, Goldberg I, Kashman Y. Tetrahedron Lett. 1988;29:6655. [Google Scholar]
- 13.(a) Rudi A, Kashman Y, Gut D, Lellouche F, Kol M. Chem Commun. 1997:17. [Google Scholar]; (b) Gut D, Goldberg I, Kol M. Inorg Chem. 2003;42:3483. doi: 10.1021/ic020703c. [DOI] [PubMed] [Google Scholar]; (c) Bergman SD, Gut D, Kol M, Sabatini C, Barbieri A, Barigelletti F. Inorg Chem. 2005;44:7943. doi: 10.1021/ic051022v. [DOI] [PubMed] [Google Scholar]; (d) Gut D, Goldberg I, Kol M. Inorg Chem. 2003;42:3483. doi: 10.1021/ic020703c. [DOI] [PubMed] [Google Scholar]; (e) Gut D, Rudi A, Kopilov J, Goldberg I, Kol M. J Am Chem Soc. 2002;124:5449. doi: 10.1021/ja012636b. [DOI] [PubMed] [Google Scholar]
- 14.(a) Luedtke NW, Hwang JS, Glazer EC, Gut D, Kol M, Tor Y. ChemBioChem. 2002;3:766. doi: 10.1002/1439-7633(20020802)3:8<766::AID-CBIC766>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (b) Luedtke NW, Hwang JS, Nava E, Gut D, Kol M, Tor Y. Nucl Acids Res. 2003;31:5732. doi: 10.1093/nar/gkg758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Friedman AE, Chambron JC, Sauvage JP, Turro NJ, Barton JK. J Am Chem Soc. 1990;112:4960. [Google Scholar]; (b) Jenkins Y, Friedman AE, Turro NJ, Barton JK. Biochemistry. 1992;31:10809. doi: 10.1021/bi00159a023. [DOI] [PubMed] [Google Scholar]
- 16.Gellerman G, Babad M, Kashman Y. Tetrahedron Lett. 1993;34:1827. [Google Scholar]
- 17.Zeglis BM, Barton JK. Nat Prot. 2007;2:357. doi: 10.1038/nprot.2007.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chow CS, Barton JK. Methods Enzymol. 1992;212:219. doi: 10.1016/0076-6879(92)12014-h. [DOI] [PubMed] [Google Scholar]
- 19.Pyle AM, Long EC, Barton JK. J Am Chem Soc. 1989;111:4520. [Google Scholar]
- 20.Lim MH, Lau IH, Barton JK. Inorg Chem. 2007;46:9528. doi: 10.1021/ic701598k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson BA. PhD Dissertation. California Instititue of Technology; Pasadena, CA: 2000. [Google Scholar]
- 22.(a) Garbett NC, Chaires JB. Methods Cell Biol. 2008;84:3. doi: 10.1016/S0091-679X(07)84001-5. [DOI] [PubMed] [Google Scholar]; (b) Hart JR. PhD Dissertation. California Institute of Technology; Pasadena, CA: 2006. [Google Scholar]
- 23.Dupureur CM, Barton JK. J Am Chem Soc. 1994;116:10286. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.