

Abstract
A previous study in nonhuman primates demonstrated that genetic immunization against the rhesus cytomegalovirus phosphoprotein 65-2 (pp65-2) and glycoprotein B (gB) antigens both stimulated antigen-specific antibodies and CD8 T cell responses, and significantly reduced plasma viral loads following intravenous challenge with RhCMV. It was also noted in this study that weak CD4 T cell and neutralizing antibody responses were generated by DNA alone. To broaden the type of immune responses, a DNA prime/protein boost strategy was used in seronegative macaques, consisting of four DNA immunizations against pp65-2, gB, and immediate-early 1 (IE1), followed by two boosts with formalin-inactivated RhCMV virions. This heterologous prime/boost strategy elicited robust antigen-specific CD4 and CD8 T cell responses in addition to biologically relevant neutralizing antibody titers. Animals were challenged with RhCMV delivered into four sites via a subcutaneous route. Skin biopsies of one of the inoculation sites 7 days post challenge revealed marked differences in the level of RhCMV replication between the vaccinated and control monkeys. Whereas the inoculation site in the controls was noted for a prominent inflammatory response and numerous cytomegalic, antigen-positive (IE1) cells, the inoculation site in the vaccinees was characterized by an absence of inflammation and antigen-positive cells. All five vaccinees developed robust recall responses to viral antigens, and four of them exhibited long-term viral immune responses consistent with effective control of viral expression and replication. These results demonstrate that a heterologous DNA prime/protein boost strategy greatly expands the breadth of antiviral immune responses and greatly reduces the level of viral replication at the primary site of challenge infection.
Keywords: Cytomegalovirus, Vaccine, Macaque
1. Introduction
Development of a human cytomegalovirus (HCMV) vaccine that confers protection in susceptible target populations remains a clinically relevant, yet unmet, objective despite more than 30 years of clinical trials and animal modeling [1,2]. The absence of an FDA-approved vaccine is a combination of multiple factors, including the complexity of HCMV natural history, the challenges and expense of conducting sufficiently powered clinical trials, and incompletely defined correlates of protective immunity. The Institute of Medicine placed a HCMV vaccine into its highest priority for development because the benefits of a vaccine, primarily preventing the devastating impact of congenital HCMV infection, would far exceed the development costs [3]. Multiple strategies have been tried and are still ongoing, including the live attenuated Towne strain, recombinant subunit glycoprotein B (gB), heterologous viral expression vectors, naked DNA, and different combinations of these methods. While none of the strategies has yet led to FDA approval, results from human clinical trials and animal modeling in the murine, guinea pig [4–8], and rhesus macaque CMV systems have demonstrated immunogenicity, partial protection, and translational modalities for improved human studies.
Unlike studies with guinea pig CMV [4], previous CMV vaccine studies in both the murine and rhesus systems have demonstrated that while DNA is an effective prime for the immune system, it is a poor inducer of biologically relevant neutralizing titers and antigen-specific CD4 responses. Morello et al. [9] demonstrated that a protein boost with formalin-inactivated murine CMV (FI-MCMV) virions augmented protective immune responses primed by prior DNA vaccinations. DNA vaccination appears to be weakly immunogenic in humans since detectable immune responses in a Phase I trial stimulated detectable immune responses in 50% or less of the seronegative vaccine recipients [1,10]. However, a limited number of human studies suggest that DNA immunization may sufficiently prime the immune system for heterologous booster immunizations [11–13]. As single HCMV vaccine regimens have not yet elicited a level of protection commensurate with what is considered necessary to protect those at risk for primary infection, a combination of immunization modalities may be required. Limited studies in humans have proven equivocal about whether heterologous prime/boost strategies against HCMV broaden and strengthen anti-HCMV immune responses [14,15]. This study was initiated to determine whether a combination prime/boost strategy is applicable to CMV in a nonhuman primate setting. Building upon our prior study involving DNA immunization in rhesus macaques [16] and that of Morello et al. with MCMV [9], seronegative rhesus macaques were first immunized with plasmid expression vectors for RhCMV pp65-2, gB, and IE1 and then boosted with FI-RhCMV virions formulated in the water-in-oil adjuvant Montanide ISA 720 [17,18].
2. Materials and methods
2.1. DNA plasmids
The expression plasmids for the RhCMV pp65-2 (pND/65-2) and the transmembrane-deleted version of gB (pND/gBΔTM) have been described previously [16,19]. A cDNA for the RhCMV IE1 gene (exons 2–4) was reverse transcribed from RNA that was purified from rhesus dermal fibroblasts infected with RhCMV strain 68-1, and then cloned into the pND expression vector, generating pND/IE1 [20]. All expression plasmids were purified from bacteria using Endofree plasmid extraction kits (Qiagen). DNA was resuspended in PBS buffer (Invitrogen) at a concentration of 1 mg/ml and stored at −20 °C.
2.2. Animal protocols
All animal protocols were approved in advance by the Institutional Animal Care and Use Committee of the University of California, Davis (UC Davis), which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All animals were serologically negative for RhCMV prior to use in this study.
2.3. DNA and protein immunizations of rhesus macaques
Ten healthy, colony-bred, juvenile rhesus macaques (Macaca mulatta), approximately 1.5 years of age, from the California National Primate Research Center were grouped into two groups (five monkeys per group). Each monkey in the vaccine group, serologically confirmed to be RhCMV seronegative by ELISA, was immunized intramuscularly (IM) with 150 μg and intradermally (ID) with 50 μg of each plasmid according to published protocols [20]. The three plasmids (pND/gBΔTM, pND/65-2, and pND/IE1) were combined together for both the IM and ID injections. The IM immunization was given in the triceps muscles, and ID vaccination was given in multiple sites on the abdomen on weeks 0, 4, 8, and 12 (Fig. 1). Each monkey in the control group was mock-immunized by the same routes with the “empty” pND vector.
Fig. 1.
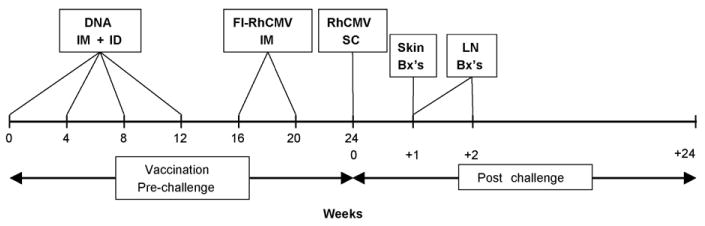
Schedules for DNA vaccination and RhCMV inoculation. Animals were vaccinated intramuscularly (IM) and intradermally (ID) four times at weeks 0, 4, 8, and 12 with separate expression plasmids for gBΔ/pp65-2/ IE1 followed by two IM vaccinations at weeks 16 and 20 with formalin-inactivated RhCMV (FI-RhCMV) formulated in the Montanide ISA 720 adjuvant. Control animals were mock-vaccinated at the same time points with an “empty” pND vector (DNA) followed by two vaccinations with saline formulated in Montanide ISA 720. All animals were challenged with RhCMV strain 68-1 by subcutaneously (SC) inoculating the animals at four sites with a total of 2 × 106 PFU of virus. Skin biopsies (Bx's) were obtained from one site at week +1 (relative to the time of challenge) and a second site at +10 days (not shown). Axillary lymph node (LN) biopsies were obtained at weeks +1 and +2. Blood draws and oral swabs were obtained throughout the 22 weeks of observation post challenge (not shown).
At week 16, 4 weeks after the last DNA immunization, each DNA-vaccinated animal was boosted with a formalin-inactivated preparation of RhCMV (FI-RhCMV), prepared as follows. A total of 18 T-175 flasks of primary dermal rhesus fibroblasts (∼90% confluency) were infected with RhCMV 68-1 (American Type Culture Collection) at an MOI of 0.5. When the infected cells exhibited 100% CPE, the supernatant was removed from the cells, clarified by a low speed centrifugation, and the cell-free supernatant was saved for subsequent processing. The cells were gently rinsed once with HBSS and removed from the surface by agitation. The cells were pelleted by a low speed centrifugation and resuspended in a 2 ml of growth medium. The resuspended cell mixture underwent three freeze/thaw cycles at −80°/+37°. Following a low-speed centrifugation to remove cell debris, the cell supernatant and disrupted cell supernatant were combined, and virions were collected by centrifugation (1 h at 9000 rpm in a Beckman JLA-16.250 rotor). The pelleted virions were resuspended in 9 ml total of HBSS and dispersed in a cup horn sonicator using four 30 s pulses on ice. The virus preparation was then inactivated with formalin according to the protocol that has been described for MCMV [21]. Briefly, the virus mixture was incubated with an equal volume of a 1:1000 dilution of formalin (37% Biotech grade, Fisher) for a final 1:2000 dilution of formalin. The virus/formalin mixture was incubated at 37 °C for 72 h with vigorous vortexing multiple times each day. The formalin was inactivated by the addition of an equimolar concentration of sodium bisulfite (i.e., 6.2 × 10−3 final molarity). The protein concentration was determined by the Bradford protein assay (Pierce), and aliquots were stored at −80°C. Complete inactivation of the FI-RhCMV preparation and suitability for injection into the DNA-primed monkeys were confirmed by an absence of both infectivity and toxicity on susceptible cells in culture. In addition, a seronegative rhesus macaque was immunized twice with 336 μg of FI-RhCMV. Absence of infectivity was demonstrated by the absence of the sustained anti-RhCMV immune response (see Fig. 2) that is a hallmark of both natural infection and experimental inoculation with infectious RhCMV [22,23].
Fig. 2.
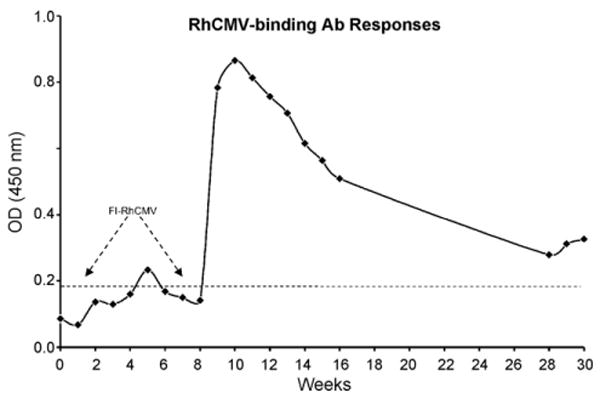
A RhCMV seronegative macaque was immunized twice with FI-RhCMV (336 μg of total protein per immunization) at weeks 0 and 8. RhCMV-binding antibody responses were quantified by ELISA and are presented as Absorbency (at 450 nm). The threshold for a sample to be considered positive is indicated by the dotted line. The slight rise in absorbency at weeks 28–30 reflect the normal fluctuations in antibody responses that would be expected with weekly sampling.
Each monkey was immunized on week 16 with 100 μg of FI-RhCMV in the presence of the water-in-oil Montanide ISA 720 adjuvant (Seppic Inc., Fairfield, NJ) [17,18]. FI-RhCMV was diluted with protein (in PBS) and repeatedly mixed through two syringes connected by a 20-gauge needle with ISA 720 at a ratio of 3:7 (protein:adjuvant) to achieve full emulsification. A sufficient volume of protein was used so that each animal was injected subcutaneously (SC) with 1 ml of emulsified protein containing 100 μg of FI-RhCMV. The FI-RhCMV/Montanide ISA 720 mixture was well-tolerated. There was no detectable local reactivity at the injection site (e.g., redness, swelling, apparent sensitivity to touch, obvious restricted arm movement), and the animals maintained normal activities (e.g., movement, feeding, grooming), suggesting the absence of significant systemic reaction to the protein/adjuvant mixture. Body temperatures were not recorded. The animals were boosted a second time with protein in adjuvant on week 20. Control animals were mock-immunized each time with PBS/ISA 720. Longitudinal blood draws were processed for analysis of post-vaccine immune responses.
Note that a group consisting of immunizations with just FI-RhCMV was not included since previous studies have demonstrated that FI-MCMV does not protect from infection, although immunization with the inactivated preparation significantly reduced mortality [21].
2.4. RhCMV challenge
All animals (vaccines and controls) were challenged on week 24 by a SC inoculation with 2 × 106 plaque forming units of RhCMV strain 68-1 (ATCC) that had been passage less than three times on primary rhesus dermal fibroblasts. This isolate was confirmed to contain an open UL36 reading frame (not shown) since a previous report noted that one genetic variant of the 68-1 strain had generated a truncated version of UL36 during passage in culture [24]. RhCMV68-1 also lacks three alpha chemokine-like open reading frames found in wild-type RhCMV naturally circulating in breeding colonies of rhesus macaques [25]. The virus was delivered in 4× 0.1 ml volumes in separate sites on the back of the animal, and the inoculation sites were marked with indelible ink. A skin biopsy of one of the inoculation sites was obtained from each monkey either on day +6 or +7, and a second biopsy of a second inoculation site was obtained on day +10. The biopsies (∼1 cm in diameter) included the epidermis, dermis, subcutis, and frequently, but not always, some of the underlying muscle. The biopsies were fixed in 2% paraformaldehyde and embedded in paraffin for histological and immunohistochemical analyses. Longitudinal post-challenge blood draws were processed for peripheral blood mononuclear cells (PBMC) and plasma, and oral and genital swabs were obtained for DNA analysis, according to published protocols [16].
2.5. DNA extraction and real-time PCR
DNA was extracted from oral and genital swab samples and plasma using QIAmp blood kit according to published protocol [26]. Each sample was processed according to the manufacture's instructions. The final elution volume was 200 μl. Extracted DNA was stored at −80 °C until real-time PCR analysis was performed. RhCMV DNA copies in plasma, oral and genital swabs were detected by a previously described real-time PCR assay for RhCMV 68-1 [27].
2.6. ELISA assays
Binding antibodies to total RhCMV antigens, gBΔTM, and pp65-2 were assayed by ELISA using extracts of RhCMV-infected rhesus dermal fibroblasts or 293T [28] cells transiently transfected separately with pND/gBΔTM, pND/pp65-2, or pND/IE1, following previously published protocols [19,29]. Briefly, 96-well microplates (Immulon 4 HBX, Dynex Technologies, Inc.) were coated overnight at 4°C with specific antigens (0.5 μg/well for pp65-2 and IE1 antigens, 0.25 μg/well for RhCMV and gBΔ antigens) and corresponding cell control extracts in 100 μl of coating buffer (0.05 M carbonate/bicarbonate buffer pH 9.6, Sigma, for pp65-2 and IE1; Hanks buffered salt solution (HBSS)/0.375% bicarbonate buffer (GIBCO) for RhCMV and gBΔ). The following day, each plate was washed six times with phosphate-buffered saline (PBS)/0.05% Tween 20 (PBS-T, wash buffer) and blocked with 300 μl/well of PBS containing 1% bovine serum albumin (BSA) for 2 h at room temperature. After washing six times, plasma/serum samples, diluted in 1% BSA/PBS-T (dilution buffer), were added to duplicate wells (100 μl/well) and incubated for 2 h at 25 °C. The samples were diluted as follows: 1:100 (Figs. 2 and 3A); 1:800 (Figs. 3B and C and 6). The plates were washed six times with PBS-T. Peroxidase-conjugated goat anti-monkey IgG (KPL, Inc.), diluted (1:100) to empirically determined optimal concentrations in 1% BSA/PBS-T, was added for 1 h. The plates were subsequently washed again with PBS-T and then incubated at 25 °C with 100 μl/well of tetramethylbenzidine liquid substrate (Sigma–Aldrich) for 30 min for color development. The reaction was terminated by addition of 0.5 M H2SO4 (50 μl/well), and the absorbance was recorded spectrophotometrically at a wavelength of 450 nm. For each sample, the absorbance at 450 nm (OD450) for control antigen-coated well was subtracted from that obtained for the antigen-coated well. A sample was considered positive if the net absorbance was 0.1 or greater.
Fig. 3.
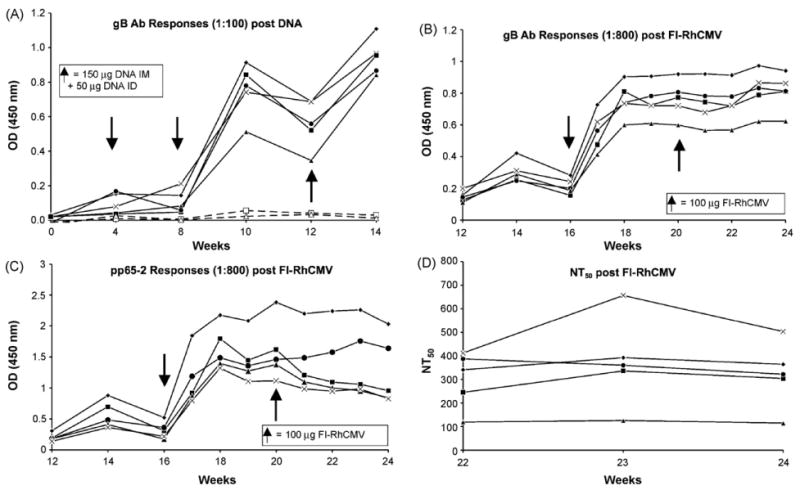
Antibody (Ab) responses to RhCMV gBΔTM post DNA ((A) 1:100 dilution of plasma) and FI-RhCMV ((B) 1:800 dilution), and to pp65-2 post FI-RhCMV ((C) 1:800 dilution). Arrows indicate times of immunization (in addition to week 0 in Fig. 1A), and dashed lines in (A) represent responses for two of the control animals immunized with the “empty” pND vector. The 50% neutralizing (NT50) Ab titers are shown (D) beginning at Week 22 (2 weeks after the second immunization with FI-RhCMV).
Fig. 6.
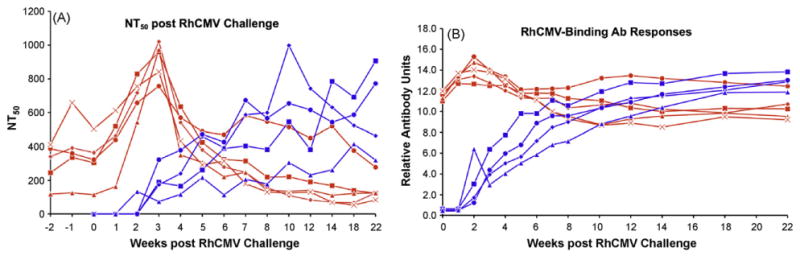
Post RhCMV challenge NT50 and RhCMV-binding antibody responses are presented for the vaccinated (red) and control (blue) animals. The Relative Antibody Units for the RhCMV-binding Ab responses were based on serial dilutions of a plasma sample from a naturally RhCMV-infected macaque. An arbitrary value of 1 was assigned to that optical density (450 nm) for the serial dilutions that was above background and that was within the linear range of the assay. The Relative Antibody Units for the plasma samples from the control and vaccinated monkeys were derived from the ratio of the optical densities for those samples relative to the minimally positive optical density for the serial dilutions. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
2.7. Neutralization assays
The neutralizing antibody titer of monkey plasma (EDTA anticoagulant) was measured in the absence of complement using an engineered variant of RhCMV strain 68-1 that expresses the enhanced green fluorescent protein (EGFP) [30]. Briefly, 2.5 × 104 PFU of RhCMV/EGFP were incubated with serial half-log dilutions (1:31–1:31,000) of heat-inactivated (56 °C, 30 min) plasma in a final volume of 500 μl (using DMEM/10% fetal bovine serum as needed to achieve the final volume). A pooled mixture of plasma from eight seronegative macaques served as a negative control and was incubated with virus identically as for the immune plasma samples. Virus and plasma were incubated for two hours at 37 °C, and the mixtures were added in triplicate (100 μl/well) to monolayers of Telo-RF cells [31], which had been seeded the day before at a density of 2 × 104 cells/well in Optilux black-walled, clear bottom 96-well plates (BD Falcon). Three wells of cells were incubated in growth medium only. After a 4-hr incubation, the virus/plasma mixture was removed, the cells were washed twice with pre-warmed HBSS, and 100 μl of complete medium was added to each well. The mean fluorescent intensity of each well was measured 48 h later on a SpectraMax M5 plate reader (Molecular Devices Corporation; Sunnyvale, CA) using the SoftMax Pro software (version 4.8). Nine individual readings were obtained per well (using a bottom read) and summed across the well. The excitation, emission, and cutoff wavelengths were 472, 507, and 495 nm, respectively. Data generated by the Softmax Pro software were imported into Excel, and the mean background fluorescence (measured from the wells treated with media only) was subtracted from the mean fluorescence for each sample analyzed (i.e., both immune and seronegative control samples). This generated the relative fluorescent unit (RFU) for each sample. The percent neutralization at each dilution of plasma was calculated as follows: (1 − (RFUimmune/RFUseroneg)) × 100. The 50% neutralization titer (NT50) was calculated by plotting the percent neutralization (Y-axis) versus the logarithm of the dilution (X-axis). A trendline equation was established for the linear portion of the data, from which the dilution of plasma resulting in 50% neutralization titer (NT50) was calculated (expressed as the reciprocal of the dilution).
2.8. Histology and immunohistochemistry
Four micron sections of the skin biopsies were either stained with hematoxylin and eosin for histology or processed for immunohistochemical detection of RhCMV IE1 protein according to published protocols [22].
2.9. Intracellular cytokine staining
Freshly isolated PBMC were assayed for their ability to secrete IFN-γ, TNF-α or IL-2 during in vitro restimulation with either sonicated RhCMV antigen preparation or overlapping peptide pools (15mers overlapping by 11 amino acids) representing the entire amino acid sequence of either pp65-2 or IE1 using a previously published protocol [32,33]. Briefly, PBMC were stimulated for 6 h with the peptide pool or medium in the presence of cross-linked antibodies to CD28 and CD49d (clones 28.2 and 9F10, respectively; BD Biosciences) at the final concentration of 1.0 μg/ml. Brefeldin-A at 10 μg/ml was added to the culture for the final 5 h of stimulation. After stimulation, the cells were surface-stained with conjugated antibodies to CD3, CD4 and CD8 for 20 min at room temperature. Subsequently, the cells were fixed and permeabilized with successive incubation with FACS Permeabilizing Solution (BD Biosciences). Permeabilized cells were then incubated with conjugated anti-IFN-γ, anti-TNF-α or anti-IL-2 for 20 min at room temperature, washed and fixed in 1% paraformaldehyde. Three hundred thousand lymphocyte events were collected on a FACS Aria (BD Biosciences), and the data were analyzed with FlowJo software (TreeStar, Ashland, OR).
3. Results
3.1. Immunogenicity, but lack of infectivity of FI-RhCMV
As a first step in determining whether a protein boost could augment protective immune responses primed by DNA, it was necessary to demonstrate (i) the immunogenicity of the FI-RhCMV preparation and (ii) a lack of infectious virus following formalin treatment of virus. Incubation of rhesus dermal fibroblasts with 13, 27, and 53 μg of FI-RhCMV resulted in a complete absence of viral cytopathic effect after 3 weeks in culture. Some transient cytotoxic effects were noted at the highest amount of protein assayed. Immunization of a seronegative macaque with 336 μg of FI-RhCMV adjuvanted in Montanide ISA 721 resulted in a minimal increase above background in RhCMV-binding antibody responses over the course of 8 weeks (Fig. 2). Previous work has demonstrated that had as few as 100 PFU of infectious virus been present in the FI-RhCMV preparation, there would have been in a demonstrable increase in antibody responses during this period of time (data not shown). A second immunization with FI-RhCMV (336 μg) at 8 weeks led to a rapid and vigorous increase in antibody responses one week later with peak antibody responses observed 10 weeks after the priming immunization (Fig. 2), confirming the immunogenicity of the FI-RhCMV preparation. Importantly, the antibody responses declined to a level just above background at weeks 28–30. The sharp drop in antibody responses after two immunizations was inconsistent with the presence of infectious virus in the FI-RhCMV preparation. RhCMV antibodies become detectable 2–4 weeks after live RhCMV infection and normally reach a plateau titer at 12–24 weeks, remaining relatively constant thereafter.
3.2. DNA prime/protein boost immunization
Having demonstrated the immunogenicity and lack of infectivity of the FI-RhCMV preparation, five RhCMV seronegative macaques were genetically immunized with plasmid expression vectors for RhCMV pp65-2, gBΔTM, and IE1, according to the timeline in Fig. 1. RhCMV gB encodes the majority of neutralizing epitopes, and pp65-2 induces cellular responses in the vast majority of RhCMV infected macaques [19,29]. Similar to host immune responses to HCMV infection, the RhCMV IE1 protein is a prominent target of CD8+ T cell responses in infected macaques [34]. Detectable humoral responses were detected following the third genetic immunization at week 8 (Fig. 3A). The antibody responses for gB peaked 2 weeks later and declined by the time of the fourth immunization at 12 weeks, a pattern which has been noted previously [16,20]. Immunization with the FI-RhCMV preparation at 16 weeks dramatically increased both gB and pp65-2 (Fig. 3A and B) antibody responses, although no further increase was observed following the second protein boost at 20 weeks. Importantly, biologically relevant values of 50% neutralizing antibody titers (NT50) were stimulated in four of the five vaccinees (range = 110–680) (Fig. 3D). The normative range of NT50 in long-term RhCMV seropositive macaques is 231–3348 (N = 24; mean = 973; median = 833) (unpublished data). All five vaccinees had detectable neutralizing antibodies at the time of RhCMV challenge. There was no apparent increase in IE1 antibody responses in any vaccinee following either FI-RhCMV booster immunization (Fig. 7). The virion purification protocol should have minimized the potential for IE1 contamination, and the absence of any booster effect on IE1 western blot responses is consistent with this interpretation. However, the presence of any IE1 protein in the FI-RhCMV preparation cannot be formally excluded, particularly since the IE1 protein is abundantly expressed during infection in culture.
Fig. 7.
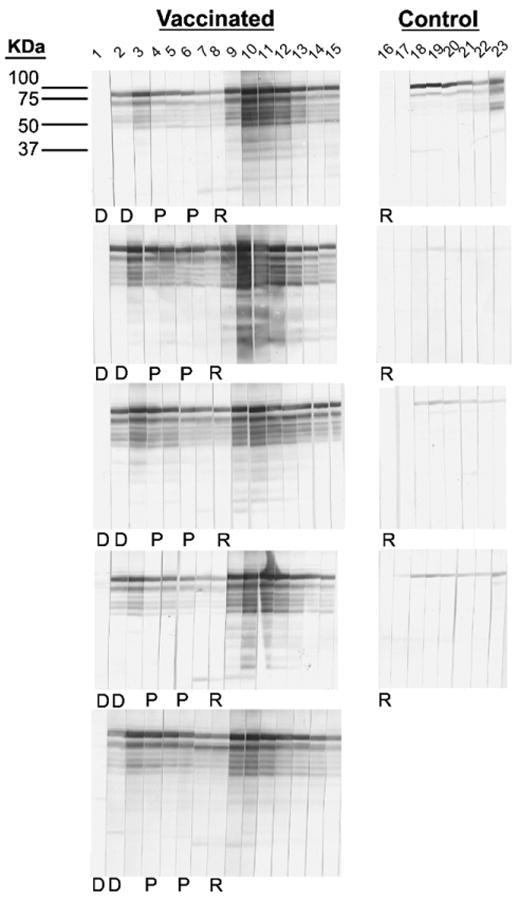
Western blot analysis of IE1-specific antibody responses post vaccination and RhCMV challenge. A longitudinal analysis of anti-IE1 antibody responses post vaccination and challenge in the vaccinees (left half) and post RhCMV challenge in the controls (right half) is shown. Lane 1: week 0; lane 2: week 12 post priming immunization; lane 3: week 14; lane 4: week 16; lane 5: week 18; lane 6: week 20; lane 7: week 22; lanes 8 and 16: week 24 (relative to the time of priming immunization)/week +0 (relative to the time of RhCMV challenge); lanes 9 and 17: week +1 (relative to the time of RhCMV challenge); lanes 10 and 18: week +2; lanes 12 and 19: week +3; lanes 13 and 20: week +4; lanes 14 and 21: week +6; lanes 14 and 22: week +8; lanes 15 and 23: week +10. D: DNA immunizations at weeks 0 and 12. P: FI-RhCMV immunizations at weeks 16 and 20. R: RhCMV challenge at week 24 (relative to the time of the priming immunizations).
Consistent with our previous studies, DNA immunization stimulated a predominant CD8+ T cell response 2 weeks after the last genetic immunization at week 12 (Fig. 4B and D compared to 4A and C). The frequency of pp65-2-specific CD8+T cells secreting IFN-γ or TNF-α ranged from 0.16 to 0.79%, or 0.02 to 0.64%, respectively. Very few antigen-specific CD4+ T cells were detected using either an overlapping pp65-2 peptide pool or disrupted RhCMV virions as antigen. IE1-specific responses were not assayed at this point due to unavailability of an overlapping IE1 peptide pool. Following the two protein immunizations, detectable IFNγ-and/or TNF-α secreting CD4+ T cells were detected after stimulation with the pp65-2 peptide pool or inactivated virions (Fig. 4A and C). IE1-specific T CD4+ cells were detected when assayed for the first time following the protein immunizations. Since prior studies have demonstrated that DNA immunization generally generates only CD8-specific T cell responses [16,35], the detection of CD4+-positive IE1-specific T cells was not anticipated when first analyzed after FI-RhCMV immunization. Although the virion preparation should have minimized the potential for non-virion proteins, such as IE1 to be present, low-level contamination of the FI-RhCMV immunogen with IE1 cannot be excluded. Western blot analysis of IE1 antibody responses demonstrated that this protein was a potent immunogen, stimulating vigorous antibody responses following genetic immunization (Fig. 7). However, there was no evidence of increased IE1 antibody responses following immunization with FI-RhCMV, consistent with the interpretation that there was no contaminating IE1 protein in the FI-RhCMV preparation. It is not known why IE1 stimulated both CD4+ and CD8+ T cell responses, whereas only CD8+ T cells were observed following genetic immunization with pp65-2 (Fig. 4). In contrast to increasing CD4+T cell responses after FI-RhCMV immunization, the dominant pp65-2 CD8+T cell responses after DNA immunization declined in some animals, while CD8+T cell responses to IE1 and the RhCMV lysate increased. No consistent temporal pattern was observed within an individual animal after the first and second protein immunizations. Importantly though, all animals developed detectable RhCMV-specific T cell responses to one or more antigens. At the time of challenge at 24 weeks, two of the five vaccinees still had detectable T cells responses, although the frequencies of antigen-specific cells in peripheral blood generally declined over time.
Fig. 4.
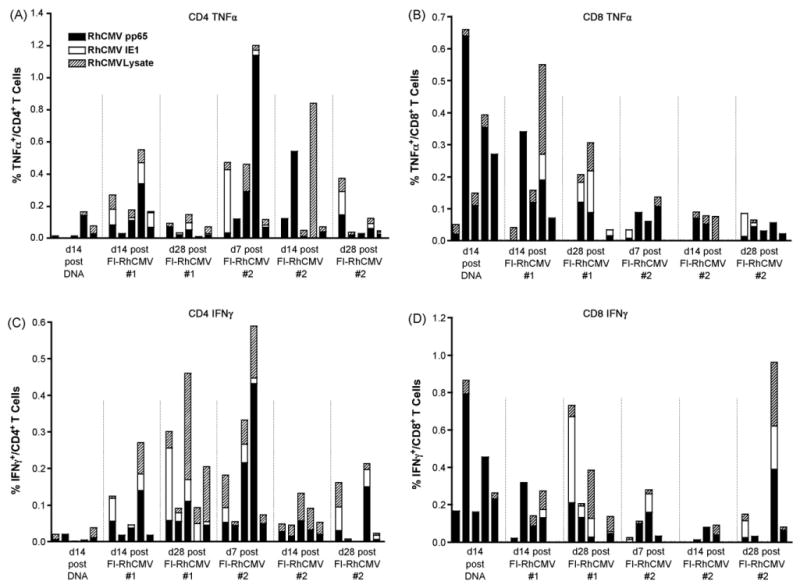
CD4+ (A and C) and CD8+ (B and D) T cell vaccine responses for TNF-α (A and B) and IFNγ (C and D) using overlapping peptide pools for RhCMV pp65-2 (RhCMV pp65) and IE1, and a lysate prepared from RhCMV-infected cells. The IE1 peptide pool was not available at the first assay timepoint 14 days after the last DNA immunization (d14 post DNA). The other timepoints are presented relative to the time of the first or second immunization with FI-RhCMV (#1 and #2, respectively). d28 post FI-RhCMV #1 and #2 represent the times for the second immunization with FI-RhCMV and RhCMV challenge (respectively).
3.3. Viral parameters of RhCMV challenge
All five of the vaccinated animals and five age-matched, pND-vaccinated controls were inoculated subcutaneously (SC) with RhCMV 24 weeks after the initial DNA immunization. Previous studies have used the intravenous (IV) route of inoculation [16]. The virus/host interactions that occur in the SC microenvironment may recapitulate those that would occur at a mucosal surface following natural exposure to virus more than would occur by direct delivery of virus to blood. Although macaques can be successfully infected orally [27], which is a likely natural route of transmission in the wild [23], there is no control over the titer of virus that actually crosses the oral mucosa to initiate infection. To assess the early host responses to viral challenge, 2 × 106 PFU of RhCMV were delivered SC into four sites. A biopsy of one inoculation site was obtained one week later, and a second biopsy was obtained again on day 10 post challenge.
Skin biopsies of an inoculation site 1 week post challenge revealed marked differences in the level of RhCMV replication between the vaccinated and control monkeys (Fig. 5A and B). Whereas the inoculation site in the controls was noted for a prominent, mononuclear inflammatory response and numerous cytomegalic, antigen-positive (IE1) cells, the inoculation site in all of the vaccinees was characterized by an absence of inflammation and antigen-positive cells. Inflammatory cells and cytomegalic/IE1-positive cells were mostly contained within the subcutis, although some spread to the underlying muscle was occasionally observed. Identical results were obtained in multiple sections from each biopsy, indicating that the absence of inflammation and IE1-positive cells in the vaccinees was not the result of a failure to sample the relevant portion of the skin biopsy. The results strongly support the contention that pre-challenge vaccination resulted in a marked suppression of early local replication at the site of inoculation. The biopsy of one site obtained at 10 days post challenge was histologically normal in all of the controls and vaccinees, characterized by a complete absence of inflammation and IE1 staining. The apparent absence of replicating virus at 10 days demonstrated that local replication was effectively contained by this point of time in the absence of prior immunity.
Fig. 5.
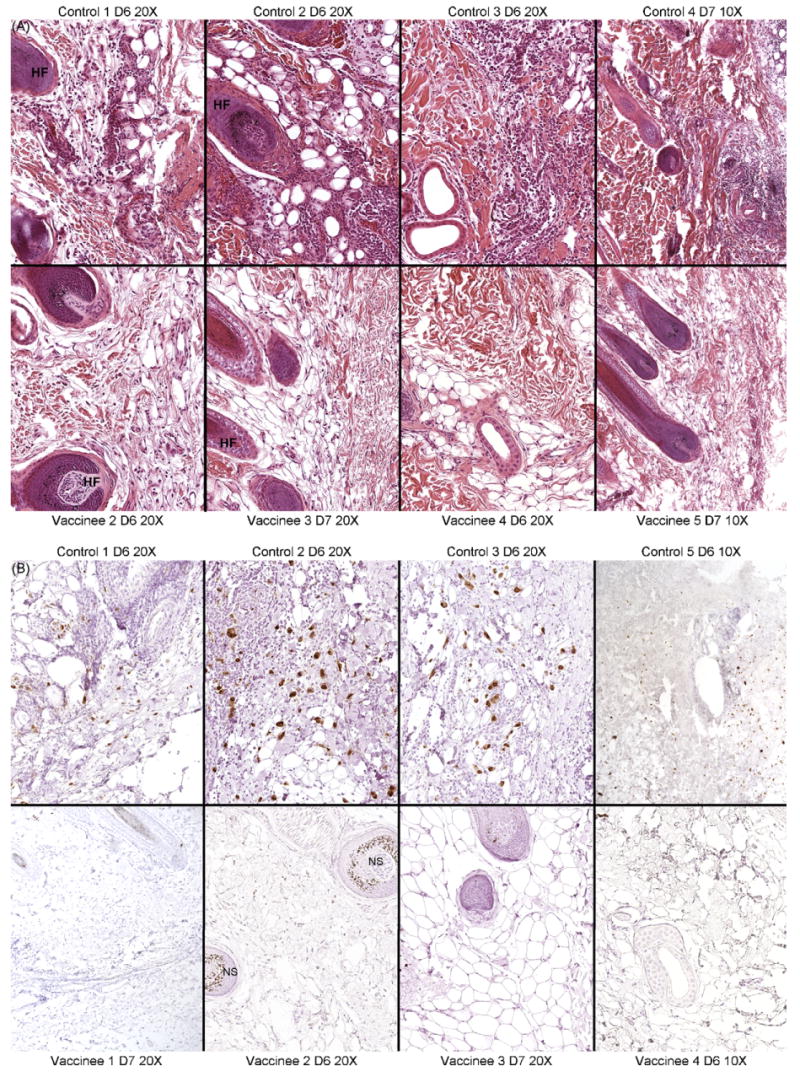
(A) Histological sections from one of the inoculations sites stained with hematoxylin and eosin (H and E). The representative sections from four animals from the control (top row) and vaccinated (bottom) groups obtained on either day 6 (D6) or 7 (D7) post inoculation are shown. Magnifications (10× or 20×) are indicated. HF: hair follicle. (B) Immunohistochemical staining for RhCMV IE1 in four of the five control and vaccinated animals, including the fifth animal of each group whose H and E section is not presented in (A). Antigen-positive cells are noted for brown-stained nuclei. Non-specific staining (NS) in some of the hair follicles is indicated.
Active local replication in three of the five control monkeys was accompanied by a low but detectable presence of viral DNA in plasma on day +7 (range = 760–1850 genome equivalents per ml of plasma); plasma-associated viral DNA was not detected in any of the vaccinated animals at any time post challenge. Viral DNA was also detected on day +14 for one of the same controls (765 copies/ml). The low copy numbers and less than 100% detection of RhCMV DNA in the plasma of the controls were not entirely unexpected. Previous studies investigating the patterns of RhCMV infection following SC inoculation failed to detect viral DNA in plasma following inoculation with either of two titers of RhCMV (102 or 104 PFU) (data not shown). Oral and genital swabs were collected from all animals following RhCMV challenge, and all were uniformly negative for detectable RhCMV DNA.
3.4. Immune parameters of RhCMV challenge
All five vaccinees developed vigorous antibody responses within one week of challenge with peak levels for neutralizing and total RhCMV antigen-binding antibodies observed at 3 weeks post challenge (Fig. 6). Antibody responses declined thereafter. In contrast, antiviral antibodies were first detected in the controls at 2 weeks post challenge with variable levels of increases observed through 22 weeks post challenge. Antibody results for just four of the controls are presented since one animal had to be euthanized 3 weeks after challenge for reasons unrelated to RhCMV. The contrast in the antibody responses between the two treatment groups was particularly salient for the neutralizing antibody titers. The NT50 titers for four of the five vaccinees had declined to titers below all four of the vaccinees by 10 weeks post challenge, a pattern that was maintained for the remainder of the study period through 22 weeks. The median NT50 titers for the vaccinated group at week 22 (125, range 84–278) was significantly lower than that of the controls (618, range 320–907) (P = 0.008). Similarly, the median value for the relative antibody units (Fig. 6B) of the vacinees was 2-fold lower than that of the controls at week 22. The antibody responses to RhCMV IE1 also demonstrated the utility of heterologous prime/boost strategies in augmenting immunogenicity. In this case, the DNA priming immunization was boosted by the expression of IE1 during RhCMV challenge. The IE1 protein is generally a poor immunogen for antibodies following either experimental inoculation (Fig. 7) or natural infection (data not shown), in contrast to its immunogenicity following DNA immunization. This is illustrated for the four control animals in which anti-IE1 antibodies detected by western blot ranged from absent (1 monkey) to weak (2 monkeys) to moderate (1 monkey) at any time post challenge (Fig. 7). In contrast, all five vaccines developed robust recall responses to IE1 within 2 weeks of challenge that exceeded the magnitude of the responses seen with DNA immunization alone.
Distinctions in the cellular responses were also observed between the control and vaccine groups. In general, cellular responses to RhCMV antigens were detected in the controls in both draining lymph nodes (axillary) and PBMC at 1 week post challenge, followed by declines in the magnitude of the responses by 2 weeks (Figs. 8–10). Further, the RhCMV-specific T cell responses were predominantly elicited by CD4+ T cells (Fig. 8), consistent with a primary CD4+ T cell response preceding the CD8+ effector T cell response. Strong and sustained RhCMV-specific CD8+ T cell responses, however, did not develop in any of the control animals, at least not to the extent that they became detectable in peripheral blood (Fig. 9). This apparent lack of effector cell induction in control animals was consistent with the detection of RhCMV IE1 positive cells and a higher degree of inflammation in tissue biopsies of controls compared to vaccinated animals at day 7 after RhCMV challenge. In contrast, in vaccines higher CD4+ and CD8+ responses were detected at 2 weeks in both draining LN and PBMC, compared to the responses observed 1 week post challenge (Figs. 8–10). The majority of RhCMV-specific CD4 and CD8 T cells secreted TNF-α in response to in vitro antigen stimulation (Figs. 8 and 9, top panels). Several animals had multifunctional RhCMV-specific T cells secreting both TNF-α and IFN-γ, but only few of these cells were also able to produce IL-2 (data not shown). The direct cytotoxic function was not evaluated in the current study. The data are indicative of early control of virus replication and delayed virus dissemination by vaccine-induced cellular and humoral responses. Importantly, the RhCMV-specific T cell responses in the vaccinees were equally well represented by CD4 and CD8+ T cells indicative of a true effector response sufficient to clear virally infected cells. The latter conclusion is supported by the fact that RhCMV-binding and neutralizing antibody titers were considerably higher and more rapidly induced in the vaccinated animals compared to unvaccinated controls, but declined after the acute phase to levels below those observed in the controls. The antibody responses after the acute phase of infection are consistent with a more effective control of persistent RhCMV infection in the vaccinated animals. Although we were not able to correlate viral shedding levels and immune responses in these animals, the tissue pathology clearly demonstrated that vaccine-induced immune responses changed the early kinetics of virus replication and allowed a rapid response to the RhCMV challenge.
Fig. 8.
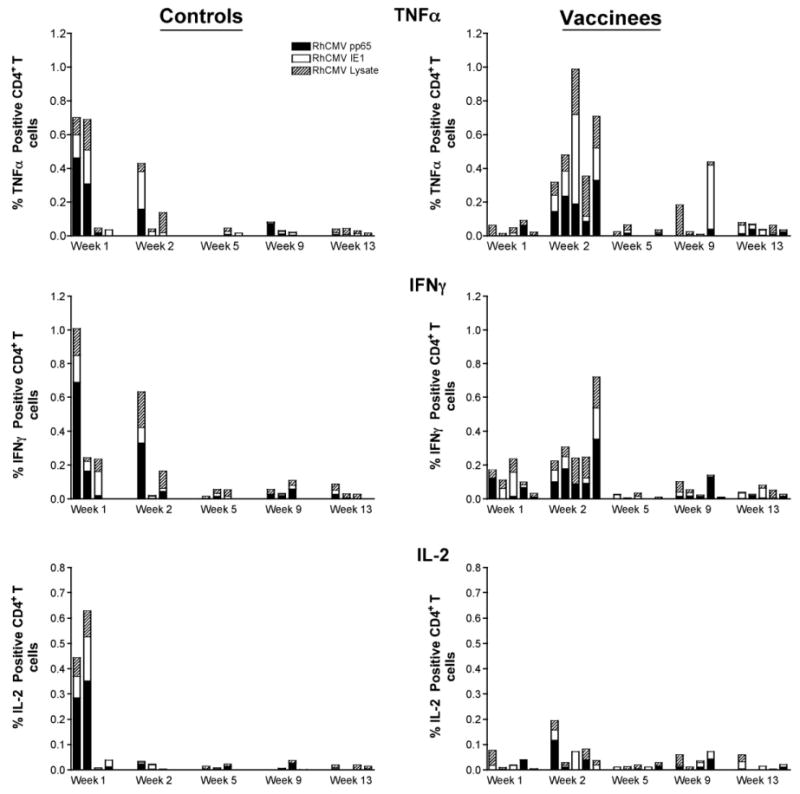
Peripheral RhCMV-specific CD4+ responses (TNF-α, IFNγ, and IL-2) in the control and vaccine groups post RhCMV challenge.
Fig. 10.
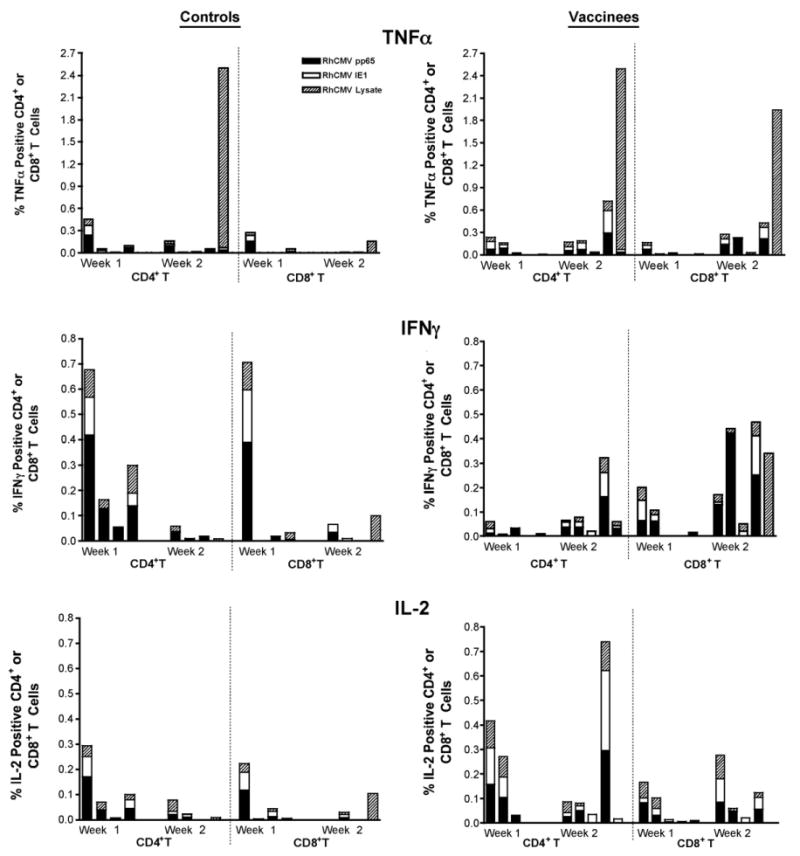
RhCMV-specific CD8+ responses (TNF-α, IFNγ, and IL-2) in the axillary lymph node of the control and vaccine groups post RhCMV challenge.
Fig. 9.
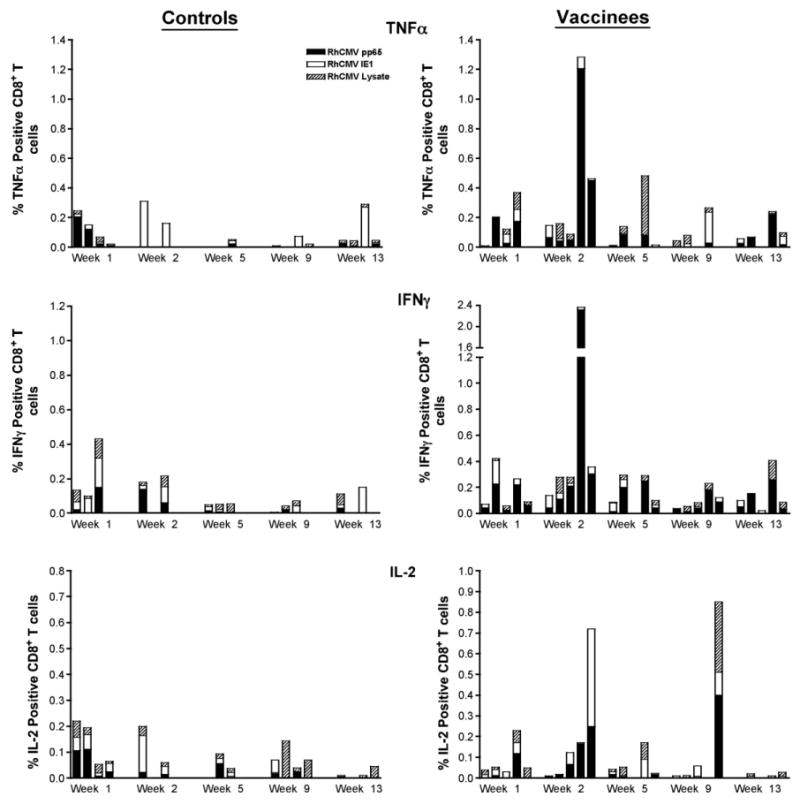
Peripheral RhCMV-specific CD8+ responses (TNF-α, IFNγ, and IL-2) in the control and vaccine groups post RhCMV challenge.
4. Discussion
The protective efficacy of a HCMV vaccine is likely to be determined by the presence of effective immune responses at the local site of challenge. The less potential there is for dissemination beyond the site of challenge, the less potential there will be for transmission. The profound differences in the extent of inflammation and viral gene expression at the subcutaneous site of challenge between the vaccinated and unvaccinated animals in the current study are consistent with the interpretation that a combination of DNA priming and inactivated virion boosting elicited immune responses that dramatically suppressed local RhCMV replication. The absence of detectable RhCMV DNA in plasma of all five vaccinated animals, compared to detection in three of the five controls, also supports the contention that dissemination of virus from the site of challenge to the periphery was limited.
Unfortunately, the results of this study did not enable a direct correlation between any of the vaccine-induce immune mechanisms and virological outcome. Shedding of RhCMV in saliva was not detected in any animal, control or vaccinated. Active shedding of virus in saliva and urine for years is a hallmark of animals infected with wild-type RhCMV [23,26,36], but our subsequent studies indicate that this is not a phenotype associated with the 68-1 strain of RhCMV that was used in this study (data not shown). The absence of shedding of 68-1 in unvaccinated control animals in this study stands in contrast with those of Price et al. [37] who observed persistent viruria beginning 6–9 weeks after SC inoculation. Previous work from our lab has demonstrated only sporadic, low level shedding of the 68-1 strain following intravenous challenge of either naïve or vaccinated macaques [16]. The 68-1 strain is pathogenic in fetal macaques [23,30,38,39] and in macaques coinfected with SIV [27]. Moreover, the virus disseminates to multiple cell types and can establish a persistent infection, as demonstrated by the presence of antigen-positive cells 6 months after inoculation [22] and the infrequent detection of virus in urine or oral swabs two years after challenge (data not shown). Future vaccine studies in the rhesus model should be directed at challenging animals with wild-type virus, either through horizontal transmission from naturally infected and RhCMV-secreting animals, or by direct inoculation with an isolate that exhibits a shedding phenotype. Studies performed as a result of the unexpected lack of RhCMV shedding in the current study suggest that wild type strains of RhCMV, which naturally circulate in breeding populations of macaques and which contain the full coding content of the ULb' region [25], are persistently shed in the saliva and urine of animals inoculated subcutaneously (unpublished observations).
The vaccine efficacy with regard to control of RhCMV replication could only be validated by the detection of IE1 positive cells in tissue biopsies and plasma viral DNA detection (see above). Indirect evidence for control of RhCMV replication was provided by the different kinetics and quality of the immune responses elicited in vaccine and control animals after RhCMV challenge. At the time of challenge, neutralizing antibody titers were present in all five vaccinees, whereas detectable cellular responses were present in only two. Importantly though, increased neutralizing antibody titers and memory T cell responses were observed within 1 and 2 weeks of challenge, respectively, suggesting that cellular and humoral responses contributed to the protective efficacy of the vaccine although the relative contribution to protective vaccine efficacy of each of these two antiviral effector mechanisms cannot be determined using the current study design. Binding and neutralizing antibody titers in vaccinees rose rapidly and declined between 3 and 4 weeks post challenge when antibody titers were still rising in control animals. While RhCMV-specific T cell responses developed within the first week post challenge in control animals, the quality of the response differed from the vaccinated animals. Control animals mounted predominantly IFNγ positive CD4+ T cell responses, whereas the T cell responses in vaccinated animals were characterized mainly by effector CD8+ T cells producing TNF-α and IFNγ. This type of a response pattern was found both in peripheral blood and the lymph nodes examined.
There are two possible caveats to this conclusion. First, only peripheral blood T cell responses were measured longitudinally. Second, while we sampled axillary lymph nodes at 1 and 2 weeks post challenge, it is possible that additional cervical and/or inguinal LN should have been sampled because the challenge inoculum was administered at four different sites and thus, multiple draining LN potentially served as local sites of primary virus replication and initiation of memory responses. However, the lack of IE1 positive cells at the site of RhCMV inoculation in vaccinated compared to control animals is consistent with the more pronounced effector response observed in vaccinees. Further, the significant reduction in the NT50 values and the decline in T cell responses after the acute post challenge phase is consistent with long-term control of viral gene expression and replication in the vaccinated monkeys.
The DNA prime/protein boost strategy employed for this study focused on those RhCMV antigens corresponding to the HCMV proteins eliciting the predominant neutralizing and cytotoxic immune responses in humans [19,29,34,40–42]. However, two immunizations with FI-RhCMV elicited de novo antibody responses to virion-associated proteins beyond boosting pp65-2 and gB responses. It is likely that these responses contributed at some level to the protective effect we observed. RhCMV gB encodes epitopes stimulating the majority of neutralizing antibodies, but not all, suggesting that other virion envelope glycoproteins in the FI-RhCMV preparation may have augmented the neutralizing response observed post challenge. In addition, Morello et al. [9,43–45] have demonstrated in the MCMV model that inclusion of antigens that otherwise are minor immunogens during natural infection and FI-MCMV induces long-term protection. FI-MCMV used alone protects from morbidity and mortality, although infectious virus can be recovered from the salivary glands of challenged mice [21]. Inactivated HCMV is immunogenic in guinea pigs [21] but is not under evaluation in human clinical trials [3]. It is not known whether the use of formalin-inactivated RhCMV is directly translatable to the human setting. However, the results of our study extend the previous work with MCMV [9,45] by demonstrating that the heterologous combination of DNA priming and inactivated virions stimulates demonstrable levels of protection in both non-human primates and mice. Prime boost strategies in humans, using a canarypox vector to express gB as a prime followed either by a recombinant gB subunit vaccine or the live attenuated Towne vaccine to boost, indicated that priming with the canarypox vector stimulated faster responses with the boost. Yet, comparable end-point titers can be achieved using multiple immunizations with the subunit gB vaccine alone [14,15].
A recent study from our lab extended the prime/boost concept to a single DNA priming immunization against RhCMV gB, pp65-2 and IE1, followed by two booster immunizations with recombinant modified vaccinia Ankara (MVA) expressing the same antigens [35]. While a single DNA immunization did not elicit detectable antiviral immune responses, a single MVA booster immunization stimulated both humoral and cellular immune responses above those observed in macaques once with only MVA (i.e., no DNA prime). However, there were no differences in immune responses at the time of challenge between animals immunized once with DNA followed by two treatments with MVA and those immunized twice with MVA. Peak plasma viral loads were reduced in the vaccinated animals from 26 to 99%, compared to unvaccinated controls following intravenous challenge, suggesting that either vaccine modality (DNA prime/MVA boost and MVA only) provided some measure of protection against RhCMV replication following challenge. However, there was no apparent protection against simultaneous SC challenge since the vaccinees exhibited a comparable level of replication at the site of inoculation to the unvaccinated controls. It is difficult to directly compare the results of this current study with those of our previous study. Not only were there differences in the numbers of immunizations (3 in this study versus 6 in our previous study), the routes of challenge were also different (SC versus IV/SC). Despite these distinctions, both prime/boost strategies elicited pre-challenge immune responses that exceeded those observed with DNA alone [16]. Like the single DNA immunization modality, both prime/boost regimens provided demonstrable levels of protection against RhCMV replication post challenge. The results to date highlight the importance of standardizing the routes of challenge, source of challenge virus, and assessment of the long-term control of RhCMV replication.
We have observed that, following immunization with DNA alone [16] or with either of two prime boost strategies (DNA/MVA [35] or DNA/FI-RhCMV), antiviral immune responses generally wane over time. This observation stands in contrast to the immune responses in RhCMV-infected macaques. Although antibody titers to some RhCMV proteins can change over time, total RhCMV-binding and neutralizing titers remain essentially unchanged over the span of many years (Oxford et al., manuscript in preparation). Further, cellular responses to RhCMV increase as the animals age from juveniles (3–5 years) to older adults (>14 years) (data not shown). This likely reflects the persistence of RhCMV in naturally infected animals in comparison to immunized animals that develop immune responses towards non-replicating antigens. One clinical implication of these nonhuman primate studies is that either frequent booster immunizations would be necessary or new immunization strategies optimized to elicit sustained levels of protective immunity would need to designed. It is worth noting that DNA immunization does stimulate the generation of long-lived memory T and B cells since booster immunizations one year after the previous booster provoke anamnestic responses. A recent paper by Berger et al. [46] performed adoptive transfer of either effector memory (TEM) or central memory (TCM) CD8+ T cells in pig-tailed macaques (Macaca nemestrina). Only TCM retained the capacity to home to the lymph nodes and persist long-term in the recipients, suggesting that markers for this pool of T cells (CD62L and CCR7) should be incorporated into future studies to assess the potential for long-term vaccine responses.
In summary, the heterologous combination of a DNA prime and FI-RhCMV boost increased both the breadth and magnitude of the pre-challenge immune responses compared with those observed in a previous study using only multiple DNA immunizations. This included the induction of RhCMV-specific CD4+ T cell responses and biologically relevant neutralizing antibody titers. Partial efficacy was evident by reduced frequencies of RhCMV positive cells at the site of inoculation at 1 week, lack of plasma viremia and the transient nature of RhCMV-specific memory responses in vaccinated animals compared to control animals. It remains to be determined whether the use of a formalin-inactivated virus booster immunization has clinical applicability for HCMV. Although recognized as a potential vaccine modality [21], formalin-inactivated HCMV is not in clinical trials [3]. This may stem in part from the incomplete protection afforded by FI-MCMV in murine studies [21]. It may also stem from the inherent difficulties in assuring complete inactivation of virus preparations [47–49]. Accordingly, the results of this study should be viewed as a paradigm for the utility of DNA priming and multi-protein booster immunizations (via recombinant proteins) in humans.
Acknowledgments
This work was supported by NIH grants to PAB (AI063356 and AI49342) and the California National Primate Research Center (RR000169).
References
- 1.Khanna R, Diamond DJ. Human cytomegalovirus vaccine: time to look for alternative options. Trends Mol Med. 2006 January;12(1):26–33. doi: 10.1016/j.molmed.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Plotkin SA. Is there a formula for an effective CMV vaccine? J Clin Virol. 2002 August;25(Suppl 2):S13–21. doi: 10.1016/s1386-6532(02)00093-8. [DOI] [PubMed] [Google Scholar]
- 3.Stratton KR, Durch JS, Lawrence RS. Vaccines for the 21st century: a tool for decision making. Washington, D.C.: National Academy Press; 2000. [PubMed] [Google Scholar]
- 4.Schleiss MR, Bourne N, Bernstein DI. Preconception vaccination with a glycoprotein B (gB) DNA vaccine protects against cytomegalovirus (CMV) transmission in the guinea pig model of congenital CMV infection. J Infect Dis. 2003 December;188(12):1868–74. doi: 10.1086/379839. [DOI] [PubMed] [Google Scholar]
- 5.Schleiss MR, Bourne N, Jensen NJ, Bravo F, Bernstein DI. Immunogenicity evaluation of DNA vaccines that target guinea pig cytomegalovirus proteins glycoprotein B and UL83. Viral Immunol. 2000;13(2):155–67. doi: 10.1089/vim.2000.13.155. [DOI] [PubMed] [Google Scholar]
- 6.Schleiss MR, Bourne N, Stroup G, Bravo FJ, Jensen NJ, Bernstein DI. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein B vaccine. J Infect Dis. 2004 April;189(8):1374–81. doi: 10.1086/382751. [DOI] [PubMed] [Google Scholar]
- 7.Schleiss MR, Jensen NJ. Cloning and expression of the guinea pig cytomegalovirus glycoprotein B (gB) in a recombinant baculovirus: utility for vaccine studies for the prevention of experimental infection. J Virol Methods. 2003 March;108(1):59–65. doi: 10.1016/s0166-0934(02)00258-6. [DOI] [PubMed] [Google Scholar]
- 8.Schleiss MR, Lacayo JC, Belkaid Y, McGregor A, Stroup G, Rayner J, et al. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis. 2007 March;195(6):789–98. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- 9.Morello CS, Ye M, Hung S, Kelley LA, Spector DH. Systemic priming-boosting immunization with a trivalent plasmid DNA and inactivated murine cytomegalovirus (MCMV) vaccine provides long-term protection against viral replication following systemic or mucosal MCMV challenge. J Virol. 2005 January;79(1):159–75. doi: 10.1128/JVI.79.1.159-175.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wloch MK, Smith LR, Boutsaboualoy S, Reyes L, Han C, Kehler J, et al. Safety and Immunogenicity of a bivalent cytomegalovirus DNA vaccine in healthy adult subjects. J Infect Dis. 2008 June;197(12):1634–42. doi: 10.1086/588385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat Med. 2003 June;9(6):729–35. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 12.Moorthy VS, Pinder M, Reece WH, Watkins K, Atabani S, Hannan C, et al. Safety and immunogenicity of DNA/modified vaccinia virus ankara malaria vaccination in African adults. J Infect Dis. 2003 October;188(8):1239–44. doi: 10.1086/378515. [DOI] [PubMed] [Google Scholar]
- 13.Vuola JM, Keating S, Webster DP, Berthoud T, Dunachie S, Gilbert SC, et al. Differential immunogenicity of various heterologous prime-boost vaccine regimens using DNA and viral vectors in healthy volunteers. J Immunol. 2005 January;174(1):449–55. doi: 10.4049/jimmunol.174.1.449. [DOI] [PubMed] [Google Scholar]
- 14.Adler SP, Plotkin SA, Gonczol E, Cadoz M, Meric C, Wang JB, et al. A canarypox vector expressing cytomegalovirus (CMV) glycoprotein B primes for antibody responses to a live attenuated CMV vaccine (Towne) J Infect Dis. 1999;180(3):843–6. doi: 10.1086/314951. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein DI, Schleiss MR, Berencsi K, Gonczol E, Dickey M, Khoury P, et al. Effect of previous or simultaneous immunization with canarypox expressing cytomegalovirus (CMV) glycoprotein B (gB) on response to subunit gB vaccine plus MF59 in healthy CMV-seronegative adults. J Infect Dis. 2002 March;185(5):686–90. doi: 10.1086/339003. [DOI] [PubMed] [Google Scholar]
- 16.Yue Y, Kaur A, Eberhardt MK, Kassis N, Zhou SS, Tarantal AF, et al. Immunogenicity and protective efficacy of DNA vaccines expressing rhesus cytomegalovirus glycoprotein B, phosphoprotein 65-2, and viral interleukin-10 in rhesus macaques. J Virol. 2007 February;81(3):1095–109. doi: 10.1128/JVI.01708-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aucouturier J, Dupuis L, Deville S, Ascarateil S, Ganne V. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev Vaccines. 2002 June;1(1):111–8. doi: 10.1586/14760584.1.1.111. [DOI] [PubMed] [Google Scholar]
- 18.Aucouturier J, Ganne V, Laval A. Efficacy and safety of new adjuvants. Ann N Y Acad Sci. 2000;916:600–4. doi: 10.1111/j.1749-6632.2000.tb05343.x. [DOI] [PubMed] [Google Scholar]
- 19.Yue Y, Zhou SS, Barry PA. Antibody responses to rhesus cytomegalovirus glycoprotein B in naturally infected rhesus macaques. J Gen Virol. 2003 December;84(Pt 12):3371–9. doi: 10.1099/vir.0.19508-0. [DOI] [PubMed] [Google Scholar]
- 20.Loomis-Huff JE, Eberle R, Lockridge KM, Rhodes G, Barry PA. Immunogenicity of a DNA vaccine against herpes B virus in mice and rhesus macaques. Vaccine. 2001;19:4865–73. doi: 10.1016/s0264-410x(01)00232-8. [DOI] [PubMed] [Google Scholar]
- 21.Tolpin MD, Starr SE, Arbeter AM, Plotkin SA. Inactivated mouse cytomegalovirus vaccine: preparation, immunogenicity, and protective effect. J Infect Dis. 1980 October;142(4):569–74. doi: 10.1093/infdis/142.4.569. [DOI] [PubMed] [Google Scholar]
- 22.Lockridge KM, Sequar G, Zhou SS, Yue Y, Mandell CM, Barry PA. Pathogenesis of experimental rhesus cytomegalovirus infection. J Virol. 1999;73:9576–83. doi: 10.1128/jvi.73.11.9576-9583.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barry PA, Chang WLW. Primate betaherpesviruses. In: Arvin A, Campadielli G, Moore P, Mocarski E, Roizman B, Whitley R, et al., editors. Human Herpesviruses: Biology, Therapy and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. pp. 1051–75. [PubMed] [Google Scholar]
- 24.McCormick AL, Skaletskaya A, Barry PA, Mocarski ES, Goldmacher VS. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virol. 2003;316:221–33. doi: 10.1016/j.virol.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Oxford KL, Eberhardt MK, Yang KW, Strelow L, Kelly S, Zhou SS, et al. Protein coding content of the U(L)b' region of wild-type rhesus cytomegalovirus. Virology. 2008 March;373(1):181–8. doi: 10.1016/j.virol.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huff JL, Eberle R, Capitanio J, Zhou SS, Barry PA. Differential detection of B virus and rhesus cytomegalovirus in rhesus macaques. J Gen Virol. 2003;84:83–92. doi: 10.1099/vir.0.18808-0. [DOI] [PubMed] [Google Scholar]
- 27.Sequar G, Britt WJ, Lakeman FD, Lockridge KM, Tarara RP, Canfield DR, et al. Experimental coinfection of rhesus macaques with rhesus cytomegalovirus and simian immunodeficiency virus: pathogenesis. J Virol. 2002;76(15):7661–71. doi: 10.1128/JVI.76.15.7661-7671.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein–Barr virus shuttle system. Mol Cell Biol. 1987 January;7(1):379–87. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yue Y, Kaur A, Zhou SS, Barry PA. Characterization. immunological analysis of the rhesus cytomegalovirus homologue (Rh112) of the human cytomegalovirus UL83 lower matrix phosphoprotein (pp65) J Gen Virol. 2006 April;87(Pt 4):777–87. doi: 10.1099/vir.0.81516-0. [DOI] [PubMed] [Google Scholar]
- 30.Chang WL, Tarantal AF, Zhou SS, Borowsky AD, Barry PA. A recombinant rhesus cytomegalovirus expressing enhanced green fluorescent protein retains the wild-type phenotype and pathogenicity in fetal macaques. J Virol. 2002 September;76(18):9493–504. doi: 10.1128/JVI.76.18.9493-9504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang WL, Kirchoff V, Pari GS, Barry PA. Replication of rhesus cytomegalovirus in life-expanded rhesus fibroblasts expressing human telomerase. J Virol Methods. 2002 October;104(2):135–46. doi: 10.1016/s0166-0934(02)00060-5. [DOI] [PubMed] [Google Scholar]
- 32.Abel K, Pahar B, Van Rompay KK, Fritts L, Sin C, Schmidt K, et al. Rapid virus dissemination in infant macaques after oral simian immunodeficiency virus exposure in the presence of local innate immune responses. J Virol. 2006 July;80(13):6357–67. doi: 10.1128/JVI.02240-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartigan-O'Connor DJ, Abel K, McCune JM. Suppression of SIV-specific CD4+ T cells by infant but not adult macaque regulatory T cells: implications for SIV disease progression. J Exp Med. 2007 October 11;204:2679–92. doi: 10.1084/jem.20071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan KS, Kaur A. Flow cytometric detection of degranulation reveals phenotypic heterogeneity of degranulating CMV-specific CD8+ T lymphocytes in rhesus macaques. J Immunol Methods. 2007 August;325(1–2):20–34. doi: 10.1016/j.jim.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue Y, Wang Z, Abel K, Li J, Strelow L, Mandarino A, et al. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med Microbiol Immunol. 2008;197:117–23. doi: 10.1007/s00430-008-0074-5. [DOI] [PubMed] [Google Scholar]
- 36.Asher DM, Gibbs CJ, Lang DJ, Gadjusek DC, Chanock RM. Persistent shedding of cytomegalovirus in the urine of healthy rhesus monkeys. Proc Soc Exp Biol Med. 1974;145:794–801. doi: 10.3181/00379727-145-37897. [DOI] [PubMed] [Google Scholar]
- 37.Price DA, Bitmansour AD, Edgar JB, Walker JM, Axthelm MK, Douek DC, et al. Induction and evolution of cytomegalovirus-specific CD4+ T cell clonotypes in rhesus macaques. J Immunol. 2008 January;180(1):269–80. doi: 10.4049/jimmunol.180.1.269. [DOI] [PubMed] [Google Scholar]
- 38.London WT, Martinez AJ, Houff SA, Wallen WC, Curfman BL, Traub RG, et al. Experimental congenital disease with simian cytomegalovirus in rhesus monkeys. Teratol. 1986;33:323–31. doi: 10.1002/tera.1420330311. [DOI] [PubMed] [Google Scholar]
- 39.Tarantal AF, Salamat S, Britt WJ, Luciw PA, Hendrickx AG, Barry PA. Neuropathogenesis induced by rhesus cytomegalovirus in fetal rhesus monkeys (Macaca mulatta) J Infect Dis. 1998;177:446–50. doi: 10.1086/514206. [DOI] [PubMed] [Google Scholar]
- 40.Kaur A, Kassis N, Hale CL, Simon M, Elliott M, Gomez-Yafal A, et al. Direct relationship between suppression of virus-specific immunity and emergence of cytomegalovirus disease in simian AIDS. J Virol. 2003 May;77(10):5749–58. doi: 10.1128/JVI.77.10.5749-5758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alford CA, Britt WJ. Cytomegalovirus. In: Roizman B, Whitley RJ, Lopez C, editors. The human herpesviruses. New York: Raven Press, Ltd.; 1993. pp. 227–55. [Google Scholar]
- 42.Lacey SF, La Rosa C, Zhou W, Sharma MC, Martinez J, Krishnan A, et al. Functional comparison of T cells recognizing cytomegalovirus pp65 and intermediate-early antigen polypeptides in hematopoietic stem-cell transplant and solid organ transplant recipients. J Infect Dis. 2006 November;194(10):1410–21. doi: 10.1086/508495. [DOI] [PubMed] [Google Scholar]
- 43.Morello CS, Cranmer LD, Spector DH. Suppression of murine cytomegalovirus (MCMV) replication with a DNA vaccine encoding MCMV M84 (a homolog of human cytomegalovirus pp65) J Virol. 2000;74(8):3696–708. doi: 10.1128/jvi.74.8.3696-3708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morello CS, Kelley LA, Munks MW, Hill AB, Spector DH. DNA immunization using highly conserved murine cytomegalovirus genes encoding homologs of human cytomegalovirus UL54 (DNA polymerase) and UL105 (helicase) elicits strong CD8 T-cell responses and is protective against systemic challenge. J Virol. 2007 July;81(14):7766–75. doi: 10.1128/JVI.00633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morello CS, Ye M, Spector DH. Development of a vaccine against murine cytomegalovirus (MCMV), consisting of plasmid DNA and formalin-inactivated MCMV, that provides long-term, complete protection against viral replication. J Virol. 2002 May;76(10):4822–35. doi: 10.1128/JVI.76.10.4822-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008 January;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nathanson N, Langmuir AD. The Cutter incident. Poliomyelitis following formaldehyde-inactivated poliovirus vaccination in the United States during the spring of 1955. II. Relationship of poliomyelitis to Cutter vaccine. Am J Hyg. 1963 July;78:29–60. doi: 10.1093/oxfordjournals.aje.a120328. [DOI] [PubMed] [Google Scholar]
- 48.Nathanson N, Langmuir AD. The Cutter incident. Poliomyelitis following formaldehyde-inactivated poliovirus vaccination in the United States during the spring of 1955. I. Background. Am J Hyg. 1963 July;78:16–28. doi: 10.1093/oxfordjournals.aje.a120327. [DOI] [PubMed] [Google Scholar]
- 49.Offit PA. The Cutter incident, 50 years later. N Engl J Med. 2005 April;352(14):1411–2. doi: 10.1056/NEJMp048180. [DOI] [PubMed] [Google Scholar]