

Abstract

Early studies led to the identification of 3β-(4-methoxyphenyl)tropane-2β-carboxylic acid methyl ester (5) with high affinity at the DAT (IC50 = 6.5 nM) and 5-HTT (Ki = 4.3 nM), while having much less affinity at the NET (Ki = 1110 nM). In the present study, we replaced the 4′-methoxy group of the 3β-phenyl ring with a bioisosteric 4′-methylthio group to give 7a. We also synthesized a number of 3β-(4-alkylthiophenyl)tropanes 7b-e, 3β-(4-methylsulfinylphenyl) and 3β-(4-methylsulfonylphenyl)tropane analogues 7f-h as well as the 3β-(4-alkylthiophenyl)nortropane derivatives 8-11 to further characterize the structure-activity relationship of this type of compound for binding at monoamine transporters. With exception of the 4′-methylsulfonyl analogue 7h, all the tested compounds possessed high binding affinities at the 5-HTT. The Ki values ranged from 0.19 nM to 49 nM. The 3β-(4-methylthiophenyl)tropane 7a and its N-(3-fluoropropyl) analogue 9a and N-allyl analogue 10a are the most selective compounds for the 5-HTT over the NET (NET/5-HTT = 314-364) in the series. However, none of the compounds showed selectivity similar to 5 for both the DAT and 5-HTT relative to the NET. This study provided useful SAR information for rational design of potent and selective monoamine transporter inhibitors.
Keywords: Monoamine transporters, 3-Phenyltropanes, 3-Phenylnortropanes, Cocaine, Addiction
1. Introduction
Monoamine neurotransmitter transporters are the principle sites of action for cocaine (1, Fig. 1) and other stimulants.1-5 Compounds with high affinity and selectivity for the dopamine, serotonin, and norepinephrine transporters (DAT, 5-HTT, and NET, respectively) are all of interest. In the brain, cocaine binds to the DAT, 5-HTT, and NET, and inhibits presynaptic reuptake of the respective neurotransmitters. It also has effects on the cholinergic, muscarinic, and σ receptors as well as sodium channels.6-9 The behavioural and reinforcing effects of cocaine result primarily from inhibition of the DAT and subsequent increases in extracellular levels of dopamine (DA), which in turn stimulate postsynaptic DA receptors.4, 7, 10-15 Hence, the discovery and development of potent and selective DAT inhibitors represents one of the promising approaches for the treatment of cocaine abuse. Several lines of evidence have suggested that inhibition of the 5-HTT can also modulate the reinforcing properties of cocaine.14, 16-18 Animal behaviour studies demonstrated that selective serotonin (5-HT) uptake inhibitors as well as dopamine uptake inhibitors can attenuate cocaine-induced stimulant and reinforcing effects.19, 20 Accordingly, the mixed action inhibitors of DAT and 5-HTT have received much attention as potential pharmacotherapies for treating cocaine abuse.21-24
Figure 1.
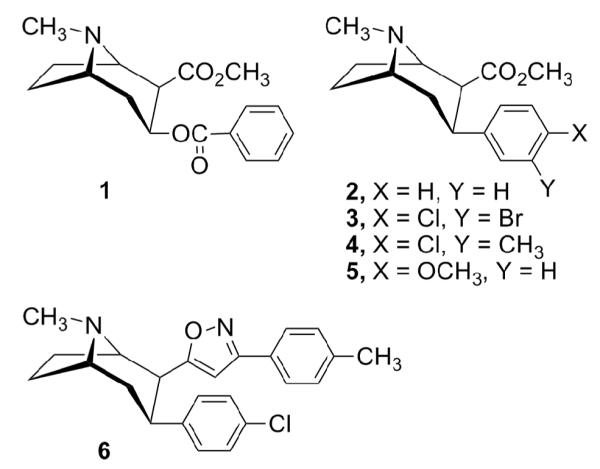
Structures of cocaine (1) and 3β-phenyltropanes
The exploration of potent and selective monoamine transporter inhibitors for cocaine abuse therapy depends on an understanding of the mechanisms of action and their structure-activity relationships (SAR). This has been accomplished experimentally by correlation of the structural variation with inhibition of [3H]WIN35,428, [3H]paroxetine, and [3H]nisoxetine binding at the DAT, 5-HTT, and NET, respectively.25 The information was used as an indication of the compound’s potential ability to inhibit uptake of the DA, 5-HT, and norepinephrine (NE) neurotransmitters. Over the last decade, we and others have synthesized a large number of 3-phenyltropane analogues with 3β-phenyltropane-2β-carboxylic acid methyl ester (WIN35,065-2, 2) as the lead compound, and evaluated them for binding at monoamine transporters in order to identify the key structural features required for potent and selective monoamine transporter inhibitors.6, 25-34 Generally, the overall tropane configuration, the substituents and substitution pattern on the 3-aromatic ring, the nature of the 2β-subsitituents, and the N-substitution are all important for the ligand recognition site interaction. One of the most studied compounds, the DAT selective inhibitor 3β-(4-chlorophenyl)-2β-[3-(4′-methylphenyl)isoxazol-5-yl]tropane (RTI-336, 6) is currently in advanced preclinical development.35-37
In order to gain a better understanding of the molecular mechanisms of cocaine actions in the brain and find highly potent and selective monoamine uptake inhibitors, we have continued our SAR studies of 2. In a recent study, we reported that 3β-(4-methoxyphenyl)tropane-2β-carboxylic acid methyl ester (5) possessed high affinity at the DAT (IC50 = 6.5 nM) and 5-HTT (Ki = 4.3 nM), while having much less affinity at the NET (Ki = 1110 nM).23 Thus, 5 is a promising lead compound to further develop ligands with high affinities for both the DAT and 5-HTT, and low affinity for the NET. In this study, we replaced the 4′-methoxy group of the 3β-phenyl ring in 5 with the corresponding bioisosteric 4′-methylthio group and further characterized the SAR of this type of compound for binding at the DAT, 5-HTT, and NET. We report herein the synthesis and monoamine transporter binding properties of novel 3β-(4-alkylthiophenyl)tropanes 7a-e, 3β-(4-methylsulfinylphenyl) and 3β-(4-methylsulfonylphenyl)tropane analogues 7f-h, and 3β-(4-alkylthiophenyl)nortropane derivatives 8-11 (Fig. 2).
Figure 2.

Structures of 3β-(4-alkylthio, -methylsulfinyl, and -methylsulfonylphenyl)tropane and 3β-(4-alkylthiophenyl)nortropane derivatives
2. Chemistry
All the 3-phenyltropane compounds described herein were prepared starting from natural (—)-cocaine, and therefore, they are optically active and possess the same absolute configuration as (—)-cocaine. The synthesis of 3β-(4-alkylthiophenyl)tropanes 7a-e, and 3β-(4-methylsulfinylphenyl) and 3β-(4-methylsulfonylphenyl)tropane analogues 7f-h is outlined in Schemes 1 and 2. Conjugate addition38 of the appropriate Grignard reagent to anhydroecgonine methyl ester (12) at −45 °C in ethyl ether followed by the addition of trifluoroacetic acid (TFA) afforded 7a-c in good yields ranging from 61% to 86% (Scheme 1). The addition of 4-trifluoromethylthiophenyl magnesium bromide to 12 gave only 7% yield (40% based on the recovered 12) of 7d. Bromination of 3β-(4-methylthiophenyl)tropane-2β-carboxylic acid methyl ester (7a) with 2 equivalents of bromine in acetic acid afforded the desired 3β-(3-bromo-4-methylthiophenyl)tropane 7e in 13% yield, as well as oxidation products 3β-(3-bromo-4-methylsulfinylphenyl)tropane 7f and 3β-(4-methylsulfinylphenyl)tropane 7g in 15% and 34% yields, respectively (Scheme 2). Use of 1 equivalent of bromine didn’t improve the bromination reaction as judged by TLC analysis. Oxidation of 7a using 30% hydrogen peroxide in acetic acid provided the sulfone analogue 7h in 84% yield.
Scheme 1.
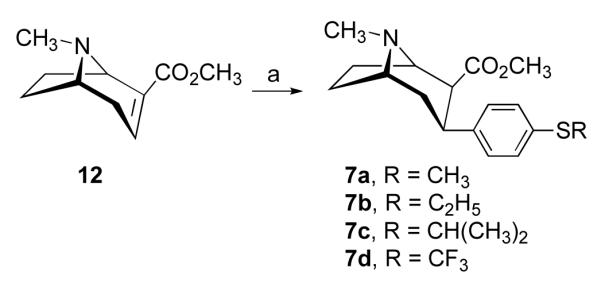
Reagents and conditions: (a) Grignard reagent, −45 °C, 2 h, then −78 °C, TFA.
Scheme 2.

Reagents and conditions: (a) Br2, AcOH, 0 °C; (b) 30% H2O2, AcOH, room temperature.
The synthesis of 3β-(4-alkylthiophenyl)nortropane analogues 8-11 is presented in Scheme 3. Treatment of 3-phenyltropanes 7a,b with 1-chloroethyl chloroformate afforded the corresponding (1-chloroethyl)urethane intermediates, which were converted to the N-nortropanes 8a,b in 86% and 87% yields, respectively, by solvolysis with methanol.39 N-Alkylation of 8a,b with 1-bromo-3-fluoropropane using potassium carbonate in acetonitrile afforded N-(3-fluoropropyl)nortropane analogues 9a,b in 94% and 87% yields, respectively. Using similar conditions, 8a,b were alkylated with allyl bromide in the presence of catalytic potassium iodide to provide 10a,b in 79% and 83% yields, respectively. Finally, the N-propylnortropanes 11a,b were prepared in 60-88% yield by hydrogenation of the corresponding N-ally analogues 10a,b over 10% palladium on carbon. The 1H NMR data of the target compounds are in agreement with the assigned structures. The chemical shift and coupling pattern of the C(2)-H and C(3)-H are consistent with previously reported compounds that possess the 2β,3β-stereochemistry.40-42
Scheme 3.

Reagents and conditions: (a) 1-chloroethyl chloroformate, 1,2-dichloroethane, reflux; (b) CH3OH, reflux; (c) 1-bromo-3-fluoropropane, K2CO3, CH3CN, reflux; (d) allyl bromide, KI, K2CO3, CH3CN, room temperature; (e) 10% Pd/C, H2, EtOH.
3. Biology
Binding affinities at the DAT, 5-HTT, and NET represent inhibition of 0.5 nM [3H]WIN35,428, 0.2 nM [3H]paroxetine, and 0.5 nM [3H]nisoxetine binding, respectively, determined as previously reported.41,42 The results of the binding studies, along with binding data of cocaine and 523 for comparison are listed in Tables 1 and 2. The DAT has two binding sites, and thus IC50 values are reported. Since the 5-HTT and NET have only one binding site, Ki values were calculated for inhibition of binding at these two transporters.
Table 1.
Monoamine transporter binding 3β-(4-alkylthio, -methylsulfinyl, and -methylsulfinyl, and -methylsulfonylphenyl)tropane derivatives
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compda | R | X | n | DAT, IC50b (nM) [3H]WIN35,428 |
5-HTT, Kib (nM) [3H]paroxetine |
NET, Kib (nM) [3H]nisoxetine |
NET/DAT Ratio |
NET/5-HTT Ratio |
| Cocainec | 89.1 | 95 | 1990 | 22 | 21 | |||
| 5c | 6.5 ± 1.3 | 4.3 ± 0.5 | 1110 ± 64 | 171 | 258 | |||
| 7a | CH3 | H | 0 | 9 ± 3 | 0.7 ± 0.2 | 220 ± 10 | 24 | 314 |
| 7b | C2H5 | H | 0 | 232 ± 34 | 4.5 ± 0.5 | 1170 ± 300 | 5 | 260 |
| 7c | CH(CH3)2 | H | 0 | 16 ± 2 | 23 ± 2 | 129 ± 2 | 8 | 7 |
| 7d | CF3 | H | 0 | 200 ± 70 | 8 ± 2 | 1900 ± 300 | 10 | 238 |
| 7e | CH3 | Br | 0 | 10.1 ± 1 | 0.6 ± 0.2 | 121 ± 12 | 12 | 202 |
| 7f | CH3 | Br | 1 | 76 ± 18 | 3.2 ± 0.4 | 690 ± 80 | 9 | 216 |
| 7g | CH3 | H | 1 | 91 ± 16 | 4.3 ± 0.6 | 515 ± 60 | 6 | 120 |
| 7h | CH3 | H | 2 | >10000 | 208 ± 45 | >10000 | 1 | 48 |
All compounds were tested as the HCl salt.
All values are means ± standard error of three or four experiments performed in triplicate.
Data taken from Ref. 23.
Table 2.
Monoamine transporter binding properties of 3β-(4-alkylthiophenyl)nortropane derivatives
 | |||||||
|---|---|---|---|---|---|---|---|
| Compda | R1 | R2 | DAT, IC50c (nM) [3H]WIN35,428 |
5-HTT, Kic (nM) [3H]paroxetine |
NET, Kic (nM) [3H]nisoxetine |
NET/DAT Ratio |
NET/5-HTT Ratio |
| 7a | CH3 | CH3 | 9 ± 3 | 0.7 ± 0.2 | 220 ± 10 | 24 | 314 |
| 8ab | CH3 | H | 28 ± 6 | 0.19 ± 0.01 | 21 ± 6 | 0.8 | 110 |
| 9a | CH3 | FCH2CH2CH2 | 112 ± 2 | 3 ± 1 | 960 ± 100 | 9 | 320 |
| 10a | CH3 | CH2=CHCH2 | 71 ± 25 | 5.5 ± 0.8 | 2000 ± 500 | 28 | 364 |
| 11a | CH3 | CH3CH2CH2 | 74 ± 20 | 5.7 ± 0.6 | 1200 ± 140 | 16 | 211 |
| 7b | C2H5 | CH3 | 232 ± 34 | 4.5 ± 0.5 | 1170 ± 300 | 5 | 260 |
| 8bb | C2H5 | H | 177 ± 62 | 1.26 ± 0.05 | 118 ± 13 | 0.7 | 94 |
| 9b | C2H5 | FCH2CH2CH2 | 1200 ± 200 | 27 ± 2 | >2000 | 2 | 74 |
| 10b | C2H5 | CH2=CHCH2 | 1100 ± 100 | 47 ± 3 | >2000 | 2 | 43 |
| 11b | C2H5 | CH3CH2CH2 | 900 ± 300 | 49 ± 6 | >2000 | 2 | 41 |
Compounds were tested as the HCl salt.
Compounds were tested as the tartrate salt.
All values are means ± standard error of three or four experiments performed in triplicate.
4. Results and Discussion
SAR studies of the 3-phenyltropane class of monoamine uptake inhibitors from our laboratory, as well as from others, have addressed the key structural features required for binding to the DAT, as well as the 5-HTT and NET.25, 31, 43 The binding affinity and selectivity of compounds for the monoamine transporter is highly dependent on the nature and position of the substituents on the 3β-phenyl ring.25, 31, 44 Recently, we reported that the 4′-methoxy analogue 5 is selective for both the DAT (IC50 = 6.5 nM) and 5-HTT (Ki = 4.3 nM) relative to the NET (Ki = 1110 nM).23 In this study, we replaced the 4′-methoxy group of 5 with the bioisosteric 4′-methylthio group to give 3β-(4-methylthiophenyl)tropane-2β-carboxylic acid methyl ester (7a). We also synthesized a number of 3β-(4-alkylthiophenyl) analogues of 7a and expanded the 3β-(4-methylthiophenyl)tropane to the corresponding 3β-(4-methylsulfinylphenyl) and 3β-(4-methylsulfonyl-phenyl)tropanes as well as 3β-(4-alkylthiophenyl)nortropanes with variants of the N-substituents to further characterize the SAR of this type of compound. The ratio of Ki/IC50 values of NET/DAT and the ratio of Ki values of NET/5-HTT were calculated as a measure of the in vitro selectivity of the compounds for the DAT relative to the NET and the 5-HTT relative to the NET, respectively. As shown in Table 1, replacement of the 4′-methoxy group of 5 with the 4′-methylthio group afforded 7a with a comparable DAT affinity as that of 5 (9 nM vs. 6.5 nM) and enhanced affinities for both the 5-HTT and NET. The magnitude of increase at the 5-HTT is more than that of the NET, leading to increased selectivity for the 5-HTT relative to the NET (NET/5-HTT ratio of 314 of 7a vs. 258 of 5). Thus, 7a is more potent at the 5-HTT and more selective for the 5-HTT over the NET than 5. Changing the 4′-methylthio group of 7a with a slightly larger 4′-ethylthio group resulted in 7b, which had less potency for all three monoamine transporters, with the biggest loss of affinity at the DAT (232 nM vs. 9 nM of 7a). Somewhat surprisingly, the 4′-isopropylthio analogue 7c possessed a similar affinity at the DAT (16 nM vs. 9 nM of 7a) and slightly enhanced affinity at the 5-HTT (129 nM vs. 220 nM of 7a) relative to 7a. Compound 7e with an electron-withdrawing trifluoromethyl group also had loss of potency compared to 7a. We previously found that a combination of 3′-halogen atoms with 4′-substituents on the 3β-phenyl ring usually led to enhanced affinity at the DAT.25 The addition of a bromo group ortho to the 4′-methylthio group of 7a, however, had almost no effect on binding affinity at the DAT, 5-HTT, and NET. The 4′-methylsulfinyl compounds 7f and 7g were less potent than the corresponding 4′-methylthio analogues 7e and 7a, respectively, while still having reasonable affinities at all three transporters. The 4′-methylsulfonyl analogue 7h had much less affinity, in particular for the DAT and NET with IC50 and Ki values >10 μM, respectively. With the exception of 7c, all the 4′-alkylthiol and 4′-methylsulfinyl tropanes 7a, 7b, and 7d-g possessed nanomolar or subnanomolar affinity at the 5-HTT (Ki = 0.6-4.5 nM) and good selectivity for the 5-HTT over the NET (NET/5-HTT = 120-314). However, none of these compounds had selectivity similar to 5 for both the DAT and 5-HTT relative to the NET.
The tertiary amino nitrogen of cocaine and its 3-phenyltropane analogues may contribute to the electrostatic or hydrogen-binding interactions between the ligand and transporter binding site.25 Consequently, N-substituents that change electron density at the nitrogen atom should affect their binding properties, possibly with gains in selectivity for specific transporters. In our SAR studies, we noticed that removing the N-methyl group from 3β-phenyltropanes resulted in enhanced affinity for binding at the 5-HTT and NET with virtually no change in binding affinity at the DAT.25 In addition, studies from other groups showed that N-substituted analogues of 3β-(4-iodophenyl)tropane yielded higher 5-HTT affinity.45, 46 Replacement of the N-methyl group by hydrogen in 3β-(4-methylthiophenyl)tropane 7a to give N-nortropane 8a exhibited approximately 4- and 10-fold increases at the 5-HTT and NET, respectively, but had 3-fold less affinity at the DAT (Table 2). Compound 8a with an IC50 value of 28 nM at the DAT, and Ki values of 0.19 nM and 21 nM at 5-HTT and NET, respectively, is a potent and nonselective monoamine uptake inhibitor. N-substituents of 3-fluoropropyl, allyl, and propyl groups in 9a, 10a, and 11a, respectively, showed less affinities at all three transporters than the N-methyl substituted 7a, though they still possessed appreciable affinities at the 5-HTT with Ki values ranging from 3 nM to 5.7 nM. The similar affinities of 10a and 11a at the DAT and 5-HTT suggest that electronic as well as steric factors of N-substituents may not play an important role in receptor binding of these compounds. An analogous effect was observed upon replacement of the N-methyl group in 3β-(4-ethylthiophenyl)tropane 7b by hydrogen or alkyl groups to give 8b-11b, while in this case, they all possessed less affinities than the corresponding 4′-methylthio analogues.
In summary, a new series of 3β-(4-alkylthio, 4-methylsulfinyl, and 4-methylsulfonylphenyl)tropane and 3β-(4-alkylthiophenyl)nortropane derivatives were synthesized and evaluated for their monoamine transporter binding affinities. With exception of the 4′-methylsulfonyl analogue 7h, all the tested compounds exhibited high binding affinities at the 5-HTT with Ki values ranging from 0.19 nM to 49 nM. The 3β-(4-methylthiophenyl)nortropane 8a had a Ki value of 0.19 nM at the 5-HTT and also appreciable affinity at the DAT and NET (IC50 = 28 nM and Ki = 21 nM, respectively), and thus is the most potent compound for all three transporters in the series. Compound 7a and its N-(3-fluoropropyl) analogue 9a and N-allyl analogue 10a are the most selective compounds for the 5-HTT relative to the NET. However, none of the tested compounds showed good selectivity for the DAT over the NET. This study provided useful SAR information for further design of potent and selective monoamine uptake inhibitors.
5. Experimental
Melting points were determined using a MEL-TEMP II capillary melting point apparatus and are uncorrected. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained on a Bruker Avance DPX-300 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) with reference to internal solvent. Mass spectra (MS) were run on a Perkin-Elmer Sciex API 150 EX mass spectrometer equipped with ESI (turbospray) source or on a Hewlett Packard 5989A instrument by electron impact. Elemental analyses were performed by Atlantic Microlab Inc., Atlanta, GA. Optical rotations were measured on an AutoPol III polarimeter, purchased from Rudolf Research. Analytical thin-layer chromatography (TLC) was carried out using EMD silica gel 60 F254 TLC plates. TLC visualization was achieved with a UV lamp or in an iodine chamber. Flash column chromatography was done on a CombiFlash Companion system using Isco prepacked silica gel columns or using EM Science silica gel 60Å (230–400 mesh). Unless otherwise stated, reagent-grade chemicals were obtained from commercial sources and were used without further purification. All moisture- and air-sensitive reactions and reagent transfers were carried out under dry nitrogen.
5.1. 3β-(4-Methylthiophenyl)tropane-2β-carboxylic acid methyl ester (7a)
Magnesium (0.72 g, 0.03 mol) was weighed into a 100 mL round bottom flask. A single crystal of iodine was added and the system was flushed with nitrogen and flame dried. After cooling to room temperature, anhydrous Et2O (2 mL) was added. A solution of 4-bromothioanisole (5.08 g, 0.025 mol) in anhydrous Et2O (20 mL) was prepared and 2 mL was added. After addition of a catalytic amount of 1,2-dibromoethane, the orange iodine color disappeared which indicated the initiation was successful. The rest of the 4-bromothioanisole solution was added slowly over 30 min while the solution was kept refluxing. After addition, the reaction mixture was refluxed for 1 h. The fleshly prepared Grignard solution was then diluted with anhydrous Et2O (76 mL) and cooled to −45 °C. A solution of anhydroecgonine methyl ester (12) (1.80 g, 0.01 mol) in 1:1 mixture of CH2Cl2-Et2O (15 mL) was added slowly and the reaction mixture was stirred at −45 °C for another 2 h. After cooling to −78 °C, the reaction was quenched by slow addition of a solution of TFA (4.60 mL, 60.0 mmol) in Et2O (6 mL). The mixture was warmed to room temperature and 6 N HCl (40 mL) was added. The aqueous layer was separated, made basic to pH 11 using NH4OH, and extracted with EtOAc (3 × 100 mL). The combined EtOAc extracts were washed with brine (3 × 50 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 20% Et2O in hexane with the addition of 5% Et3N afforded 7a (2.60 g, 87%) as an oil. 1H NMR (300 MHz; CDCl3) δ 7.18 (s, 4H), 3.60–3.51 (m, 1H), 3.50 (s, 3H), 3.42–3.34 (m, 1H), 2.96 (ddd, J = 12.8, 5.1, 5.1 Hz, 1H), 2.90–2.84 (m, 1H), 2.57 (ddd, J = 12.6, 12.8, 3.0 Hz, 1H), 2.45 (s, 3H), 2.30–2.01 (m, 5H), 1.78–1.54 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 171.6, 139.9, 135.0, 127.6, 126.3, 65.0, 62.0, 52.4, 50.7, 41.7, 33.8, 33.1, 25.6, 24.9, 15.8; MS (EI) m/z 305 (M+). The free base was converted to the hydrochloride salt: mp 157–158 °C; [α]D20 −128° (c 0.29, CH3OH); Anal. Calcd for C17H23NO2S·HCl·1.75H2O: C, 54.68; H, 7.42; N, 3.75. Found: C, 54.79; H, 7.50; N, 3.77.
5.2. 3β-(4-Ethylthiophenyl)tropane-2β-carboxylic acid methyl ester (7b)
The procedure for 7a was followed using 1.80 g (0.01 mol) of 12 to give 1.98 g (62%) of 7b as a solid: mp 55–56 °C; 1H NMR (300 MHz; CDCl3) δ 7.30–7.17 (m, 4H), 3.63–3.53 (m, 1H), 3.49 (s, 3H), 3.40–3.32 (m, 1H), 3.05–2.85 (m, 4H), 2.57 (ddd, J = 12.6, 12.6, 3.0 Hz, 1H), 2.28–2.00 (m, 5H), 1.80–1.55 (m, 3H), 1.28 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 171.6, 140.7, 133.1, 128.7, 127.6, 65.0, 62.0, 52.4, 50.7, 41.7, 33.7, 33.2, 27.6, 25.6, 24.9, 14.2; MS (ESI) m/z 320.2 (M+1). The free base was converted to the hydrochloride salt: mp 155–157 °C; [α]D20 −120° (c 0.35, CH3OH); Anal. Calcd for C18H25NO4S·HCl·1.5H2O: C, 56.46; H, 7.63; N, 3.66. Found: C, 56.70; H, 7.61; N, 3.65.
5.3. 3β-(4-Isopropylthiophenyl)tropane-2β-carboxylic acid methyl ester (7c)
The procedure for 7a was followed using 850 mg (4.68 mmol) of 12 to give 0.96 g (61%) of 7c as a solid: mp 103–105 °C; 1H NMR (300 MHz; CDCl3) δ 7.30 (d, J = 8.3 Hz, 2H), 7.18 (d, J = 8.3 Hz, 2H), 3.62–3.53 (m, 1H), 3.48 (s, 3H), 3.42–3.22 (m, 2H), 2.97 (ddd, J = 12.5, 5.1, 5.1 Hz, 1H), 2.92–2.87 (m, 1H), 2.58 (ddd, J = 12.5, 12.6, 2.4 Hz, 1H), 2.30–2.04 (m, 5H), 1.78–1.54 (m, 3H), 1.26 (d, J = 6.0 Hz, 6H); 13C NMR (75 MHz; CDCl3) δ 172.2, 142.2, 132.5, 132.2, 128.0, 65.5, 62.5, 53.0, 51.3, 42.1, 38.6, 34.2, 33.7, 26.1, 25.3, 23.4; MS (ESI) m/z 334.4 (M+1). The free base was converted to the hydrochloride salt: mp 165–166 °C; [α]D20 −116° (c 0.38, CH3OH); Anal. Calcd for C19H27NO2S·HCl·0.75H2O: C, 59.51; H, 7.75; N, 3.65. Found: C, 59.62; H, 7.89; N, 3.37.
5.4. 3β-(4-Trifluoromethylthiophenyl)tropane-2β-carboxylic acid methyl ester (7d)
The procedure for 7a was followed using 900 mg (5.00 mmol) of 12 to give 0.12 g (7%) of 7d as a solid: mp 64–65 °C; 1H NMR (300 MHz; CDCl3) δ 7.60–7.51 (m, 2H), 7.37–6.27 (m, 2H), 3.65–3.55 (m, 1H), 3.49 (s, 3H), 3.41–3.32 (m, 1H), 3.02 (ddd, J = 12.5, 5.1, 5.1 Hz, 1H), 2.97–2.88 (m, 1H), 2.56 (ddd, J = 12.6, 12.5, 2.7 Hz, 1H), 2.33–2.05 (m, 5H), 1.80–1.54 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 172.0, 146.9, 136.1, 129.9 (q, JC,F = 306 Hz), 128.7, 121.4 (q, JC,F = 1.9 Hz), 65.5, 62.3, 52.8, 51.3, 42.1, 34.0, 33.9, 26.0, 25.4; MS (EI) m/z 359 (M+). The free base was converted to the hydrochloride salt: mp 169-171 °C; [α]D20 −113° (c 0.35, CH3OH); Anal. Calcd for C17H20F3NO2S·HCl·0.25H2O: C, 51.00; H, 5.41; N, 3.50. Found: C, 50.87; H, 5.52; N, 3.37.
5.5. 3β-(3-Bromo-4-methylthiophenyl)tropane-2β-carboxylic acid methyl ester (7e), 3β-(3-bromo-4-methylsulfinylphenyl)tropane-2β-carboxylic acid methyl ester (7f), and 3β-(4-methylsulfinylphenyl)tropane-2β-carboxylic acid methyl ester (7g)
To a stirred solution of 7a (153 mg, 0.50 mmol) in AcOH (2 mL) at room temperature under nitrogen was added Br2 (0.056 mL, 1.00 mmol). After stirring for 1 h, the reaction mixture was poured into a mixture of NaHCO3 and ice. The resultant solution was extracted with CH2Cl2 (3 × 30 mL). The combined CH2Cl2 extracts were washed with 20% Na2S2O3 (30 mL), brine (30 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 5% Et2O in hexane with the addition of 5% Et3N followed by 0 → 30% CH3OH in CH2Cl2 with the addition of 1% NH4OH afforded 7e (25.0 mg, 13%), 7f (30.0 mg, 15%), and 7g (55.0 mg, 34%).
Compound 7e: oil; 1H NMR (300 MHz; CDCl3) δ 7.39 (d, J = 1.5 Hz, 1H), 7.23 (dd, J = 8.1, 1.5 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 3.64–3.54 (m, 1H), 3.53 (s, 3H), 3.42–3.33 (m, 1H), 2.94 (ddd, J = 12.4, 4.8, 4.8 Hz, 1H), 2.88–2.81 (m, 1H), 2.53 (ddd, J = 12.4, 12.3, 2.7 Hz, 1H), 2.44 (s, 3H), 2.30–2.02 (m, 5H), 1.80–1.57 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 172.1, 141.7, 136.6, 132.0, 127.1, 125.6, 121.8, 65.5, 62.4, 52.8, 51.4, 42.1, 34.2, 33.4, 26.1, 25.4, 16.1; MS (ESI) m/z 384.3 (M+1) (79Br), 386.2 (M+1) (81Br). The free base was converted to the hydrochloride salt: mp 148-150 °C; [α]D20 −90° (c 0.30, CH3OH); Anal. Calcd for C17H22BrNO2S·HCl·0.5H2O: C, 47.51; H, 5.63; N, 3.26. Found: C, 47.64; H, 5.99; N, 3.00.
Compound 7f: solid; mp 63–66 °C; 1H NMR (300 MHz; CDCl3) δ 7.82 (dd, J = 8.7, 2.4 Hz, 1H), 7.50–7.40 (m, 2H), 3.68–3.58 (m, 1H), 3.54 (s, 3H), 3.47–3.37 (m, 1H), 3.10–2.90 (m, 2H), 2.79 (s, 3H), 2.68-2.52 (m, 1H), 2.32–2.03 (m, 5H), 1.80–1.55 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 171.8, 148.8, 142.4, 132.0, 127.7, 125.2, 118.2, 65.3, 62.2, 52.5, 51.4, 42.1, 41.9, 33.9, 33.7, 25.8, 25.2; MS (ESI) m/z 400.3 (M+1) (79Br), 402.2 (M+1) (81Br). The free base was converted to the hydrochloride salt: mp 148-150 °C; [α]D20 −91° (c 0.28, CH3OH); Anal. Calcd for C17H22BrNO3S·HCl·1.5H2O: C, 44.02; H, 5.65; N, 3.02. Found: C, 43.75; H, 5.65; N, 2.99.
Compound 7g: solid; mp 94–97 °C; 1H NMR (300 MHz; CDCl3) δ 7.55 (d, J = 7.6 Hz, 2H), 7.42 (d, J = 7.6 Hz, 2H), 3.70–3.60 (m, 1H), 3.50 (s, 3H), 3.47–3.37 (m, 1H), 3.10-3.02 (m, 1H), 2.98-2.90 (m, 1H), 2.70 (s, 3H), 2.69–2.52 (m, 1H), 2.32–2.05 (m, 5H), 1.86–1.60 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 171.8, 146.7, 142.7, 128.4, 123.2, 65.2, 62.1, 52.5, 51.2, 43.8, 41.9, 33.8, 25.8, 25.1; MS (ESI) m/z 322.3 (M+1). The free base was converted to the hydrochloride salt: mp 115 °C (fusion); [α]D20 −115° (c 0.31, CH3OH); Anal. Calcd for C17H23NO3S·HCl·1.5H2O: C, 53.05; H, 7.07; N, 3.64. Found: C, 53.30; H, 7.01; N, 3.78.
3β-(4-Methylsulfonylphenyl)tropane-2β-carboxylic acid methyl ester (7h)
To a stirred solution of 7a (500 mg, 1.60 mmol) in AcOH (13 mL) at room temperature was added 30% H2O2 (4.66 mL). After stirring at room temperature for 20 h, the reaction mixture was poured into a mixture of NaHCO3 and Na2SO3 in water, and extracted with EtOAc (3 × 50 mL). The combined EtOAc extracts were washed with brine (3 × 50 mL) and dried (Na2SO4). Removal of the solvent under reduced pressure afforded 7h (470 mg, 84%) as a solid: mp 123–124 °C; 1H NMR (300 MHz; CDCl3) δ 7.89–7.80 (m, 2H), 7.50–7.42 (m, 2H), 3.67–3.58 (m, 1H), 3.52 (s, 3H), 3.43–3.34 (m, 1H), 3.02–2.92 (m, 5H), 2.61 (ddd, J = 12.3, 12.3, 2.7 Hz, 1H), 2.30–2.07 (m, 5H), 1.80–1.56 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 171.6, 150.1, 137.7, 128.1, 126.8, 65.2, 62.0, 52.4, 51.1, 44.4, 41.8, 33.8, 33.6, 25.6, 25.1; MS (EI) m/z 337 (M+). The free base was converted to the hydrochloride salt: mp 159–162 °C; [α]D20 −106° (c 0.74, CH3OH); Anal. Calcd for C17H23NO4S·HCl·1.25H2O: C, 51.51; H, 6.74; N, 3.53. Found: C, 51.65; H, 6.58; N, 3.42.
3β-(4-Methylthiophenyl)nortropane-2β-carboxylic acid methyl ester (8a)
A mixture of 7a (310 mg, 1.00 mmol) and 1-chloroethyl chloroformate (0.43 mL, 4.00 mmol) in 1,2-dichloroethane (3 mL) was refluxed under nitrogen for 20 h. The reaction mixture was concentrated under reduced pressure and the residue was dissolved in MeOH (10 mL). After refluxing for 5 h, the mixture was cooled to room temperature and concentrated under reduced pressure. The residue was made basic using NaHCO3 and extracted with CH2Cl2 (3 × 50 mL). The combined CH2Cl2 extracts were dried (Na2SO4) and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 5% CH3OH in CH2Cl2 with the addition of 1% NH4OH afforded 8a (250 mg, 86%) as a solid: mp 48–49 °C; 1H NMR (300 MHz; CDCl3) δ 7.25–7.09 (m, 4H), 3.78–3.68 (m, 2H), 3.38 (s, 3H), 3.20 (ddd, J = 12.3, 5.4, 5.4 Hz, 1H), 2.73 (s, 2H), 2.52–2.33 (m, 4H), 2.20–1.97 (m, 2H), 1.85–1.55 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 173.8, 139.4, 136.2, 127.9, 126.7, 56.4, 53.7, 51.2, 35.3, 33.8, 29.1, 27.7, 16.0; MS (ESI) m/z 292.1 (M+1). The free base was converted to the tartrate salt: mp 118–121 °C; [α]D20 −103° (c 0.26, CH3OH); Anal. Calcd for C20H27NO8S: C, 54.41; H, 6.16; N, 3.17. Found: C, 54.27; H, 6.15; N, 3.09.
3β-(4-Ethylthiophenyl)nortropane-2β-carboxylic acid methyl ester (8b)
The procedure for 8a was followed using 640 mg (2.00 mmol) of 7b to give 530 mg (87%) of 8b as a solid: mp 49–51 °C; 1H NMR (300 MHz; CDCl3) δ 7.24 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H), 3.80–3.68 (m, 2H), 3.37 (s, 3H), 3.34–3.15 (m, 2H), 2.90 (q, J = 7.3 Hz, 2H), 2.80–2.70 (m, 1H), 2.40 (ddd, J = 12.9, 12.9, 2.4 Hz, 1H), 2.25–1.94 (m, 2H), 1.87–1.56 (m, 3H), 1.27 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 173.7, 140.0, 134.4, 129.2, 127.8, 56.3, 53.7, 51.1, 51.0, 35.2, 33.6, 29.0, 27.8, 27.6, 14.3; MS (ESI) m/z 306.1 (M+1). The free base was converted to the tartrate salt: mp 108–110 °C; [α]D20 −79° (c 0.21, CH3OH); Anal. Calcd for C21H29NO8S·0.25H2O: C, 54.83; H, 6.46; N, 3.04. Found: C, 54.69; H, 6.45; N, 3.02.
3β-(4-Methylthiophenyl)-8-(3-fluoropropyl)nortropane-2β-carboxylic acid methyl ester (9a)
To a stirred mixture of 8a (146 mg, 0.50 mmol) and K2CO3 (138 mg, 1.00 mmol) in CH3CN (5 mL) at room temperature under nitrogen was added 1-bromo-3-fluoro-propane (93.0 mg, 0.60 mmol). After refluxing for 2 h, the reaction mixture was cooled to room temperature and diluted with EtOAc (50 mL). The mixture was washed with brine (3 × 30 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 5% Et2O in hexane with the addition of 5% Et3N afforded 9a (165 mg, 94%) as an oil: 1H NMR (300 MHz; CDCl3) δ 7.18 (s, 4H), 4.60 (t, J = 6.0 Hz, 1H), 4.44 (t, J = 6.0 Hz, 1H), 3.71–3.64 (m, 1H), 3.49 (s, 3H), 3.43–3.37 (m, 1H), 2.98 (ddd, J = 12.6, 4.8, 4.8 Hz, 1H), 2.93–2.87 (m, 1H), 2.56 (ddd, J = 12.6, 12.6, 2.7 Hz, 1H), 2.45 (s, 3H), 2.43–2.30 (m, 2H), 2.18–1.92 (m, 2H), 1.86–1.58 (m, 5H); 13C NMR (75 MHz; CDCl3) δ 172.0, 140.5, 135.3, 128.0, 126.7, 82.4 (d, JC,F = 162 Hz), 63.4, 61.6, 52.9, 51.0, 49.4 (JC,F = 5.7 Hz), 34.1 (d, JC,F = 18.5 Hz), 30.4, 30.1, 26.2, 26.1, 16.3; MS (ESI) m/z 352.6 (M+1). The free base was converted to the hydrochloride salt: mp 85–87 °C; [α]D20 −120° (c 0.53, CH3OH); Anal. Calcd for C19H26FNO2S·HCl·1.5 H2O: C, 54.99; H, 7.29; N, 3.38. Found: C, 55.09; H, 7.14; N, 3.43.
3β-(4-Ethylthiophenyl)-8-(3-fluoropropyl)nortropane-2β-carboxylic acid methyl ester (9b)
The procedure for 9a was followed using 100 mg (0.33 mmol) of 8b to give 105 mg (87%) of 9b as an oil: 1H NMR (300 MHz; CDCl3) δ 7.26–7.14 (m, 4H), 4.60 (t, J = 6.0 Hz, 1H), 4.44 (t, J = 6.0 Hz, 1H), 3.71–3.63 (m, 1H), 3.48 (s, 3H), 3.43–3.38 (m, 1H), 3.02–2.85 (m, 4H), 2.57 (ddd, J = 12.6, 12.6, 2.7 Hz, 1H), 2.44–2.31 (m, 2H), 2.26–1.92 (m, 2H), 1.86–1.57 (m, 5H), 1.28 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 172.0, 141.3, 133.5, 129.3, 128.1, 82.4 (d, JC,F = 162 Hz), 63.4, 61.6, 53.0, 51.0, 49.4 (d, JC,F = 5.7 Hz), 34.1 (d, JC,F = 14.6 Hz), 30.4, 30.1, 28.2, 26.2, 26.1, 14.6; MS (ESI) m/z 366.5 (M+1); The free base was converted to the hydrochloride salt: mp 165–167 °C; [α]D20 −118° (c 0.28, CH3OH); Anal. Calcd for C20H28FNO2S·HCl: C, 59.76; H, 7.27; N, 3.48. Found: C, 59.76; H, 7.23; N, 3.26.
3β-(4-Methylthiophenyl)-8-allylnortropane-2β-carboxylic acid methyl ester (10a)
To a stirred mixture of 8a (582 mg, 2.00 mmol), K2CO3 (552 mg, 4.00 mmol), and KI (10.0 mg) in CH3CN (10 mL) at room temperature under nitrogen was added allyl bromide (0.21 mL, 2.40 mmol). After stirring at room temperature for 3 h, the reaction mixture was diluted with EtOAc (50 mL). The mixture was washed with brine (3 × 30 mL), dried (Na2SO4), and concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 5% Et2O in hexane with the addition of 5% Et3N afforded 10a (520 mg, 79%) as a solid: mp 46–48 °C; 1H NMR (300 MHz; CDCl3) δ 7.21–7.15 (m, 4H), 5.86–5.68 (m, 1H), 5.18–4.94 (m, 2H), 3.71–3.64 (m, 1H), 3.49 (s, 3H), 3.47–3.41 (m, 1H), 3.08–2.96 (m, 2H), 2.92–2.78 (m, 2H), 2.60 (ddd, J = 12.6, 12.6, 2.7 Hz, 1H), 2.45 (s, 3H), 2.18–1.97 (m, 2H), 1.82–1.58 (m, 3H); 13C NMR (75 MHz; CDCl3) δ 171.8, 140.2, 136.6, 135.2, 127.9, 126.5, 116.2, 61.9, 61.1, 56.6, 52.7, 50.8, 34.0, 33.8, 26.0, 25.8, 16.0; MS (ESI) m/z 332.5 (M+1). The free base was converted to the hydrochloride salt: mp 82–84 °C; [α]D20 −78° (c 0.29, CH3OH); Anal. Calcd for C19H25NO2S·HCl·0.5H2O: C, 60.54; H, 7.22; N, 3.72. Found: C, 60.36; H, 7.32; N, 3.44.
3β-(4-Ethylthiophenyl)-8-allylnortropane-2β-carboxylic acid methyl ester (10b)
The procedure for 10a was followed using 270 mg (0.89 mmol) of 8b to give 255 mg (83%) of 10b as a solid: mp 71–72 °C; 1H NMR (300 MHz; CDCl3) δ 7.29–7.14 (m, 4H), 5.85–5.68 (m, 1H), 5.18–4.95 (m, 2H), 3.73–3.66 (m, 1H), 3.49 (s, 3H), 3.46–3.40 (m, 1H), 3.08–2.78 (m, 6H), 2.60 (ddd, J = 12.6, 12.6, 2.7 Hz, 1H), 2.19–2.04 (m, 2H), 1.80–1.55 (m, 3H), 1.28 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 171.9, 141.1, 136.7, 133.4, 129.1, 128.0, 116.3, 62.1, 61.2, 56.7, 52.8, 51.0, 34.1, 34.0, 28.0, 26.1, 25.9, 14.5; MS (ESI) m/z 346.3 (M+1). The free base was converted to the hydrochloride salt: mp 71–73 °C; [α]D20 −72° (c 0.29, CH3OH); Anal. Calcd for C20H27NO2S·HCl·0.25H2O: C, 62.16; H, 7.43; N, 3.62. Found: C, 62.28; H, 7.65; N, 3.49.
3β-(4-Methylthiophenyl)-8-propylnortropane-2β-carboxylic acid methyl ester (11a)
A mixture of 10a (130 mg, 0.40 mmol) and 10 wt% Pd/C (26.0 mg) in EtOH was hydrogenated at 1 atm for 16 h. The mixture was filtered through a short pad of Celite and the filtrate was concentrated under reduced pressure. Flash column chromatography of the crude product on silica gel using 0 → 10% Et2O in hexane with the addition of 3% Et3N afforded 11a (115 mg, 88%) as an oil: 1H NMR (300 MHz; CDCl3) δ 7.21–7.15 (m, 4H), 3.76–3.66 (m, 1H), 3.49 (s, 3H), 3.43–3.34 (m, 1H), 2.97 (ddd, J = 12.5, 5.1, 5.1 Hz, 1H), 2.93–2.87 (m, 1H), 2.57 (ddd, J = 12.5, 12.3, 2.7 Hz, 1H), 2.44 (s, 3H), 2.28–1.94 (m, 4H), 1.80–1.58 (m, 3H), 1.50–1.30 (m, 2H), 0.87 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 172.2, 140.7, 135.3, 128.1, 126.8, 62.8, 62.0, 55.8, 53.0, 51.1, 34.3, 34.1, 26.3, 26.0, 22.4, 16.3, 11.9; MS (ESI) m/z 334.3 (M+1). The free base was converted to the hydrochloride salt: mp 83–85 °C; [α]D20 −115° (c 0.34, CH3OH); Anal. Calcd for C19H27NO2S·HCl·0.75H2O: C, 59.51; H, 7.75; N, 3.65. Found: C, 59.78; H, 8.03; N, 3.27.
3β-(4-Ethylthiophenyl)-8-propylnortropane-2β-carboxylic acid methyl ester (11b)
The procedure for 11a was followed using 140 mg (0.41 mmol) of 10b to give 85.0 mg (60%) of 11b as an oil: 1H NMR (300 MHz; CDCl3) δ 7.25–7.05 (m, 4H), 3.68–3.55 (m, 1H), 3.41 (s, 3H), 3.34–3.23 (m, 1H), 2.94–2.75 (m, 4H), 2.50 (ddd, J = 12.5, 12.5, 2.7 Hz, 1H), 2.22–1.82 (m, 4H), 1.70–1.43 (m, 3H), 1.42–1.20 (m, 2H), 1.20 (t, J = 7.4 Hz, 3H), 0.79 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz; CDCl3) δ 172.1, 141.5, 133.4, 129.3, 128.1, 62.8, 61.9, 55.8, 53.0, 51.0, 34.3, 34.1, 28.2, 26.3, 26.0, 22.4, 14.6, 11.9; MS (ESI) m/z 348.0 (M+1). The free base was converted to the hydrochloride salt: mp 78–80 °C; [α]D20 −124° (c 0.39, CH3OH); Anal. Calcd for C20H29NO2S·HCl·1.25H2O: C, 59.09; H, 8.06; N, 3.45. Found: C, 59.11; H, 8.15; N, 3.49.
Acknowledgements
This research was supported by the National Institute on Drug Abuse, Grant No. DA05477.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reith MEA, Meisler BE, Sershen H, Lajtha A. Biochem. Pharmacol. 1986;35:1123–1129. doi: 10.1016/0006-2952(86)90148-6. [DOI] [PubMed] [Google Scholar]
- 2.Riddle EL, Fleckenstein AE, Hanson GR. The AAPS Journal. 2005;7:E847–E851. doi: 10.1208/aapsj070481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heikkila RE, Manzino L. Eur. J. Pharmacol. 1984;103:241–248. doi: 10.1016/0014-2999(84)90483-7. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Reith MEA. CNS Neurol. Disord. Drug Targets. 2008;7:393–409. doi: 10.2174/187152708786927877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xi ZX, Gardner EL. Curr. Drug Abuse Rev. 2008;1:303–327. doi: 10.2174/1874473710801030303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll FI, Lewin AH, Boja JW, Kuhar MJ. J. Med. Chem. 1992;35:969–981. doi: 10.1021/jm00084a001. [DOI] [PubMed] [Google Scholar]
- 7.Kuhar MJ, Ritz MC, Boja JW. Trends Neurosci. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 8.Kalivas PW. Am. J. Addict. 2007;16:71–78. doi: 10.1080/10550490601184142. [DOI] [PubMed] [Google Scholar]
- 9.Howell LL, Kimmel HL. Biochem. Pharmacol. 2008;75:196–217. doi: 10.1016/j.bcp.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B, Miller GW, Caron MG. Nat. Neurosci. 1998;1:132–137. doi: 10.1038/381. [DOI] [PubMed] [Google Scholar]
- 11.Wilcox KM, Rowlett JK, Paul IA, Ordway GA, Woolverton WL. Psychopharmacology (Berl) 2000;153:139–147. doi: 10.1007/s002130000457. [DOI] [PubMed] [Google Scholar]
- 12.Wise RA, Leeb K, Pocock D, Newton P, Burnette B, Justice JB. Psychopharmacology (Berl) 1995;120:10–20. doi: 10.1007/BF02246140. [DOI] [PubMed] [Google Scholar]
- 13.Volkow ND, Wang G-J, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, Hitzemann R, Shea CE. Nature. 1997;386:827–833. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- 14.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 15.Bergman J, Madras BK, Johnson SE, Spealman RD. J. Pharmacol. Exp. Ther. 1989;251:150–155. [PubMed] [Google Scholar]
- 16.Carroll FI, Howell LL, Kuhar MJ. J. Med. Chem. 1999;42:2721–2736. doi: 10.1021/jm9706729. [DOI] [PubMed] [Google Scholar]
- 17.Howell LL, Carroll FI, Votaw JR, Goodman MM, Kimmel HL. J. Pharmacol. Exp. Ther. 2007;320:757–765. doi: 10.1124/jpet.106.108324. [DOI] [PubMed] [Google Scholar]
- 18.Ritz MC, Kuhar MJ. J. Pharmacol. Exp. Ther. 1989;248:1010–1017. [PubMed] [Google Scholar]
- 19.Howell LL, Byrd LD. J. Pharmacol. Exp. Ther. 1995;275:1551–1559. [PubMed] [Google Scholar]
- 20.Spealman RD. Psychopharmacology (Berl) 1993;112:93–99. doi: 10.1007/BF02247368. [DOI] [PubMed] [Google Scholar]
- 21.Blough Bruce E., Abraham P, Lewin Anita H., Kuhar Michael J., Boja John W., Carroll FI. J. Med. Chem. 1996;39:4027–4035. doi: 10.1021/jm960409s. [DOI] [PubMed] [Google Scholar]
- 22.Blough BE, Abraham P, Mills AC, Lewin AH, Boja JW, Scheffel U, Kuhar MJ, Carroll FI. J. Med. Chem. 1997;40:3861–3864. doi: 10.1021/jm970492z. [DOI] [PubMed] [Google Scholar]
- 23.Jin C, Navarro HA, Carroll FI. J. Med. Chem. 2008;51:8048–8056. doi: 10.1021/jm801162z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carey RJ, Huston JP, Müller CP. Prog. Brain Res. 2008;172:347–360. doi: 10.1016/S0079-6123(08)00917-5. [DOI] [PubMed] [Google Scholar]
- 25.Carroll FI. J. Med. Chem. 2003;46:1775–1794. doi: 10.1021/jm030092d. [DOI] [PubMed] [Google Scholar]
- 26.Davies HML, Saikali E, Huby NJS, Gilliat VJ, Matasi JJ, Sexton T, Childers SR. J. Med. Chem. 1994;37:1262–1268. doi: 10.1021/jm00035a005. [DOI] [PubMed] [Google Scholar]
- 27.Xu L, Kelkar SV, Lomenzo SA, Izenwasser S, Katz JL, Kline RH, Trudell ML. J. Med. Chem. 1997;40:858–863. doi: 10.1021/jm960739c. [DOI] [PubMed] [Google Scholar]
- 28.Carroll FI, Lewin AH, Mascarella SW. In: Neurotransmitter Transporters: Structure, Function, and Regulation. 2nd Edition Reith MEA, editor. Humana Press; Totowa, NJ: 2001. pp. 381–432. [Google Scholar]
- 29.Xu L, Kulkarni SS, Izenwasser S, Katz JL, Kopajtic T, Lomenzo SA, Newman AH, Trudell ML. J Med Chem. 2004;47:1676–1682. doi: 10.1021/jm030430a. [DOI] [PubMed] [Google Scholar]
- 30.Runyon SP, Carroll FI. Curr. Top. Med. Chem. 2006;6:1825–1843. doi: 10.2174/156802606778249775. [DOI] [PubMed] [Google Scholar]
- 31.Runyon SP, Carroll FI. In: Dopamine Transporters, Chemistry, Biology and Pharmacology. Trudell ML, Izenwasser S, editors. Wiley; 2007. [Google Scholar]
- 32.Kozikowski AP, Araldi GL, Prakash KR, Zhang M, Johnson KM. J. Med. Chem. 1998;41:4973–4982. doi: 10.1021/jm9802564. [DOI] [PubMed] [Google Scholar]
- 33.Jin C, Navarro HA, Carroll FI. Bioorg. Med. Chem. 2008;16:5529–5535. doi: 10.1016/j.bmc.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin C, Navarro HA, Page K, Carroll FI. Bioorg. Med. Chem. 2008;16:6682–6688. doi: 10.1016/j.bmc.2008.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carroll FI, Pawlush N, Kuhar MJ, Pollard GT, Howard JL. J. Med. Chem. 2004;47:296–302. doi: 10.1021/jm030453p. [DOI] [PubMed] [Google Scholar]
- 36.Carroll FI, Howard JL, Howell LL, Fox BS, Kuhar MJ. AAPS J. 2006;8:E196–E203. doi: 10.1208/aapsj080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll FI, Fox BS, Kuhar MJ, Howard JL, Pollard GT, Schenk S. Eur. J. Pharmacol. 2006;553:149–156. doi: 10.1016/j.ejphar.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 38.Carroll FI, Mascarella SW, Kuzemko MA, Gao Y, Abraham P, Lewin AH, Boja JW, Kuhar MJ. J. Med. Chem. 1994;37:2865–2873. doi: 10.1021/jm00044a007. [DOI] [PubMed] [Google Scholar]
- 39.Boja JW, Kuhar MJ, Kopajtic T, Yang E, Abraham P, Lewin AH, Carroll FI. J. Med. Chem. 1994;37:1220–1223. doi: 10.1021/jm00034a021. [DOI] [PubMed] [Google Scholar]
- 40.Carroll FI, Abraham P, Lewin AH, Parham KA, Boja JW, Kuhar MJ. J. Med. Chem. 1992;35:2497–2500. doi: 10.1021/jm00091a019. [DOI] [PubMed] [Google Scholar]
- 41.Carroll FI, Gao Y, Abraham P, Lewin AH, Lew R, Patel A, Boja JW, Kuhar MJ. J. Med. Chem. 1992;35:1813–1817. doi: 10.1021/jm00088a017. [DOI] [PubMed] [Google Scholar]
- 42.Carroll FI, Gao Y, Rahman MA, Abraham P, Parham K, Lewin AH, Boja JW, Kuhar MJ. J. Med. Chem. 1991;34:2719–2925. doi: 10.1021/jm00113a008. [DOI] [PubMed] [Google Scholar]
- 43.Singh S. Chem. Rev. 2000;100:925–1024. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]
- 44.Carroll FI, Blough BE, Nie Z, Kuhar MJ, Howell LL, Navarro HA. J. Med. Chem. 2005;48:2767–2771. doi: 10.1021/jm040185a. [DOI] [PubMed] [Google Scholar]
- 45.Neumeyer JL, Tamagnan G, Wang S, Gao Y, Milius RA, Kula NS, Baldessarini RJ. J. Med. Chem. 1996;39:543–548. doi: 10.1021/jm9505324. [DOI] [PubMed] [Google Scholar]
- 46.Tamagnan G, Neumeyer JL, Gao Y, Wang S, Kula NS, Baldessarini RJ. Bioorg. Med. Chem. Lett. 1997;7:337–340. [Google Scholar]