

Abstract
The contribution of a dysfunctional TGF-β type II receptor (TGFβRII) to prostate cancer initiation and progression was investigated in an in vivo mouse model. Transgenic mice harboring the dominant-negative mutant TGF-β type II receptor (DNTGFβRII) in mouse epithelial cell were crossed with the TRAMP prostate cancer transgenic mouse to characterize the in vivo consequences of inactivated TGF-β signaling on prostate tumor initiation and progression. Histopathological diagnosis of prostate specimens from the TRAMP+/DNTGFβRII double transgenic mice, revealed the appearance of early malignant changes and subsequently highly aggressive prostate tumors at a younger age, compared to littermates TRAMP+/Wt TGFβRII mice. Immunohistochemical and western blotting analysis revealed significantly increased proliferative and apoptotic activities, as well as vascularity and macrophage infiltration that correlated with an elevated VEGF and MCP-1 protein levels in prostates from TRAMP+/DNTGFβRII+ mice. An epithelial-mesenchymal transition (EMT)-effect was also detected in prostates of TRAMP+/DNTGFβRII mice, as documented by the loss of epithelial markers (E-cadherin and β-catenin) and upregulation of mesenchymal markers (N-cadherin) and EMT-transcription factor Snail. A significant increase in the androgen receptor (AR) mRNA and protein levels was associated with the early onset of prostate tumorigenesis in TRAMP+/DNTGFβRII mice. Our results indicate that in vivo disruption of TGF-β signaling accelerates the pathological malignant changes in the prostate by altering the kinetics of prostate growth and inducing EMT. The study also suggests that a dysfunctional TGFβRII augments AR expression and promotes inflammation in early stage tumor growth thus conferring a significant contribution by TGF-β to prostate cancer progression.
Keywords: Apoptosis, Cell Proliferation, TGF-β Signaling, Membrane Receptors, Transgenic Mouse Model, Prostate Cancer, Metastasis
INTRODUCTION
Approximately 30,000 American men lose their lives due to prostate cancer every year (1). Prostate cancer is a heterogeneous cancer with a natural history of progression from prostatic intraepithelial neoplasia (PIN) to locally invasive androgen-dependent to androgen-independent metastatic disease which is associated with increased patient mortality (2). Overcoming the androgen-independence of prostate tumors is considered the most critical therapeutic endpoint for improving patient survival (3). Evidence from in vitro studies supports a dynamic contribution of androgen receptor (AR) cross-talk with transforming growth factor-β (TGF-β) signaling towards the emergence of androgen-resistant prostate cancer (4). AR overexpression is an effective way of hormone refractory prostate tumors to overcome the growth inhibition effects of elevated serum TGF-β levels, even in the absence of DHT (4).
TGF-β is a ubiquitous growth factor that elicits diverse cellular responses in cell type-dependent context during different stages of normal development and tumor growth. Disruption of TGF-β signaling correlates with pathological manifestation of PIN and prostate cancer development (5, 6). Emergence of androgen-independent and metastatic tumors, is associated with modulations in activity and/or deregulation of expression of key apoptosis, proliferation and migration proteins, such as AR, bcl-2 and β-catenin (7–10). AR modulates TGF-β/Smad signaling in androgen-independent prostate tumors by directly antagonizing the growth inhibitory function of TGF-β /Smad signaling (11–13), while TGF-β may inhibit AR action via targeting binding of Smad proteins to A R (14, 15). TGF-β signaling proceeds via transmembrane heterotetrameric complexes from two types of serine threonine kinase receptors, type I (TGFβRI) and type II (TGFβRII) (5). Upon ligand binding, TGFβRII receptor phosphorylates and activates TGFβRI receptor, which initiates the downstream signaling cascade by phosphorylating the receptor-regulated Smads (R-Smads) (16). Deregulation of TGF-β signaling plays a key role in the pathogenesis of cancer and other diseases, due to either loss of expression or mutational inactivation of its membrane receptors or intracellular effectors, the Smads (6). Loss of responsiveness to TGF-β by introducing a dominant negative mutant TGβRII receptor induces malignant transformation of non-tumorigenic rat prostate epithelial cells (17). Overexpression of TGFβRII in prostate cancer cells restores TGF-β sensitivity and suppresses prostate tumorigenic growth (18, 19).
The transgenic adenocarcinoma of mouse prostate (TRAMP) model is transgenic for the SV40 large T antigen (TAg) under the control of the rat probasin promoter (20, 21). TAg transgene expression occurs with sexual maturity and results in prostate cancer development and progression in a pattern resembling the clinical progression of human prostate cancer from androgen independence to metastasis (22, 23). To determine the consequences of a dysfunctional TGF-β signaling via loss of TGFβRII on prostate cancer progression, the dominant negative TGFβRII (DNTGFβRII) transgenic mouse model (24) in which TGFβ signaling is impaired via the expression of a dominant negative TGFβRII in epithelial cells, was crossed with the TRAMP mouse. The truncated version of human TGFβRII dimerizes with TGFβRI, but prevents its activation, since it lacks the C-terminus kinase domain resulting in an inactive TGF-β signaling (24). Here we generated and characterized a double transgenic TRAMP+/DNTGFβRII mouse model. We found that TGFβRII inactivation in prostate epithelial cells in vivo, causes an earlier onset of prostate cancer consequential to enhanced growth, vascularity and inflammation of the prostate.
MATERIALS AND METHODS
Mice
Animals were maintained under environmentally controlled conditions and subject to a 12-h light/dark cycle with food and water ad libitum. Animals (6–8 mice per group) were divided into the following experimental groups: (1) TRAMP+/DNTGFβRII+; (2) TRAMP+/WtTGFβRII; (3) TRAMP-/DNTGFβRII+; (4) TRAMP-/WtTGFβRII. For immunohistochemical analysis, prostate tissue specimens were fixed in 10% (v/v) formalin (Sigma-Aldrich, St. Louis, MO) and formalin-fixed paraffin-embedded tissue specimens were sectioned (6µm) using a Finesse microtome (Thermo Shandon Inc., Pittsburg, PA).
Characterization of DNTGF-β RII transgenic conditional knockout mice
The DNTGFβRII mice were obtained from Dr. Lalage Wakefield (National Cancer Institute, Bethesda). The DNTGFβRII transgene contains three different integrations of a construct that has a truncated version of the human TGFβRII expressed under the control of a mouse metallothionein MT1 (induced by ZnSO4 in drinking water) and MT locus control regions (25). Metallothioneins (MTs) are low molecular weight, cysteine-rich proteins which expression is rapidly induced in response to cadmium and zinc. The dominant-negative TGFβRII transgene mouse is in the FVB/N genetic background (24). By crossing the original DNTGFβRII+/+ transgenic female mice (Background FVB) with wild type C57BL/6 males mice for six generations, DNTGFβRII+/− in the C57BL/6 background mice have been generated. Transgenic mice were identified by PCR. For DNTGFβRII genotyping PCR conditions were: 30 cycles, 60s, 94°C, 60s, 56°C; 60s 72°C. Forward primer:5’-AAGATGATGTTGTCATTGCACTC-3’;Reverse primer 5’ -TGGAGAAAGAATGACGAGAACA-3’ yield a 183-bp PCR product.
Generation of TRAMP/DNTGFβRII transgenic mice
The TRAMP mouse model (C57BL/6-Tg –TRAMP-8247Ng; Jackson Laboratory, Bar Harbor, ME) is a well-established model of prostate cancer progression to advanced disease. The TRAMP transgene is in the C57BL/6 genetic background. TRAMP was detected by PCR using the following specific primers: TRAMP forward primer -5’ CAGAGCAGAATTGTGGAGTGG-3’; TRAMP reverse primer 5’ GGACAAACCACAACTAGAATGCAGTG-3’, a 489-bp product. PCR conditions: 25 cycles 60 secs; at 95°C, 60 secs at 55°C, and 60 secs at 72°C. DNTGFβRII heterozygous males in the C57BL/6 background, were crossed to TRAMP heterozygous females (of same background). Male mice (C57BL/6) of the following genotypes were thus generated: TRAMP+/DNTGFβRII+, littermates TRAMP+/WtTGFβRII; TRAMP-/DNTGFβRII+, and TRAMP-/WtTGFβRII to serve as controls.
Truncated version of human DNTGFβRII mRNA and protein expression induced by ZnSO4
RT–PCR was used to determine dominant-negative TGFβRII receptor mRNA levels in TRAMP+/DNTGFβRII+ and littermates TRAMP+/WtTGFβRII; TRAMP-/DNTGFβRII+; TRAMP-/WtTGFβRII transgenic mice (with and without 25mM ZnSO4 in drinking water). Total RNA (TRIzol, Invitrogen, Carlsbod, CA), was reverse-transcribed using Promega Corporation kit (Madison, WI). Transcription conditions were as follows: 25 °C for 10mins, 42 °C for 60mins, and 95 °C for 5mins. Primer forward: 5-CAC TGA CAA CAA CGG TGC AGT C-3. Primer reverse: 5-GCA ACA AGT CAG GAT TGC TGG TG-3. The expected PCR product was a 372-bp fragment. Quantum mRNA 18s (Ambion Inc., Austin, TX) was used as an internal control. Thermocycling conditions were as follows: 94°C, 1min before the first cycle, 94°C, 30s, 56°C, 30s, and 72°C, 30s, for 36 times and followed by a final extension at 72°C, 7mins. PCR products were separated in 2% agarose gels, stained with ethidium bromide (Invitrogen) and visualized using UVP Bioimaging system (UVP LCC, Upland, CA). The band intensity was quantified using Scion image analysis software (Scion Corporation, Frederick, MD).
Paraffin sections (6µm) were deparaffinized, rehydrated and stained with hematoxylin-and-eosin and were subjected to pathological evaluation by the pathologist (BM). To examine DNTGFβRII protein expression, formalin-fixed paraffin-embedded sections of mouse prostates, were deparaffinized and subjected to antigen retrieval in citrate buffer (pH 6) (Dako, Carpinteria, CA); slides were rinsed with TBS-T [50 mM Tris, 150mM NaCl, pH 7.6 and 0.1% (v/v) Triton X-100], and subsequently exposed to the rabbit polyclonal antibody against human TGFβRII (Catalogue No: 06–227; Upstate, Lake Placid, NY.) (4°C; overnight). Negative controls consisted of exposing tissue sections to rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Slides were incubated with biotinylated goat anti-rabbit IgG (Chemicon, Billerica, MA) (1hr at room temperature) and subsequently exposed to streptavidin-conjugated Texas Red (Chemicon). Slides were examined under a Nikon fluorescence microscope (Nikon Inc., Melville, NY).
Apoptosis detection
The incidence of apoptosis was examined in situ using the terminal deoxynucleotidyl transferase–mediated dUTP-biotin nick end labeling (TUNEL) assay (Chemicon International, Temecula CA). Sections were counterstained with methyl green and TUNEL-positive cells were counted (26). The apoptotic index was expressed as the percentage of TUNEL positive cells over the total number of cells in three random fields (40X).
Evaluation of cell proliferation, vascularity, inflammation and EMT
Cell proliferation, tissue vascularity, macrophage recruitment and EMT markers, as well as AR expression were determined by immunohistochemical and Western blot analysis.
Immunohistochemical analysis
The following antibodies were used: the rabbit polyclonal antibody against mouse E-cadherin (Catalogue No: 3195, Cell Signaling Technology, Inc. Danvers, MA), N-cadherin, AR and mouse TGFβRII (Catalogue No: sc-7939, sc815 and sc33931, respectively, Santa Cruz Biotechnology, Santa Cruz, CA); the antibody against nuclear antigen Ki-67 (Catalogue No; ab15580-100, Abcam Inc, Cambidge, MA) was used as a marker of proliferation; the rabbit polyclonal antibody against mouse von Willebrand Factor (vWF) (Catalogue No: A0082, Dako, Carpinteria, CA) was used to evaluate microvessel density as an indicator of tissue vascularity; the rat anti-mouse CD68 (Catalogue No: MCA1957, Abd Serotec, Oxford, UK) was used to detect macrophage infiltration. Citrate buffer and proteinase K solution (20µg/ml), was used for vWF antigen retrieval. Serial sections were exposed to Ki-67, AR, von Willebrand Factor and CD68 antibodies, overnight at 4°C (negative controls consisted of incubation with IgG from the host of primary antibody). Sections were subsequently exposed to biotinylated goat anti-rabbit IgG and horseradish peroxidase-streptavidin conjugate (Chemicon, Billerica, MA). Color development was accomplished using a FAST 3 , 3′-diaminobenzidine (DAB)-based kit (Sigma-Aldrich, Louis, MO) and counterstained with hematoxylin. Images were captured using an Olympus BX51 microscope system (Olympus America, Lake Success, NY). The number of Ki-67 positive cells over total number of prostate epithelial cells (300–500) was counted two independent observers.
Western blot analysis
Prostate tissue was homogenized in TRIzol Reagent and protein was extracted following the manufacturer’s instructions. Protein expression was determined by immunoblotting using the following specific antibodies: anti-VEGF (Catalogue No; sc7269, Santa Cruz Biotechnology), anti MCP-1 and anti-Snail (Catalogue No: 2026 and 3879, Cell Signaling Technology). Anti E-cadherin, anti N-cadherin, anti-mouse TGFβRII (same as described above for immunohistochemical analysis), anti β-catenin (Catalogue No: 9582, Cell Signaling Technology). Protein levels were normalized to α-actin expression, using the α-actin antibody (Oncogene Research Products, La Jolla, CA). Protein content was quantified using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL) and protein samples (30µg) were subjected to SDS-PAGE and transferred to Hybond-C membranes (Amersham Pharmacia Biotech, Piscataway, NJ). Membranes were blocked in 5% milk in TBS-T (TBS containing 0.05% Tween 20) and following incubation with the respective primary antibody (overnight at 4°C), membranes were exposed to species-specific horseradish peroxidase–labeled secondary antibody. Signal detection was achieved with SuperSignal West Dura Extended Duration Substrate (Pierce) and visualized using a UVP Imaging System. Fold change was determined based on α-actin expression as a control.
Real time PCR Analysis
Real-time RT–PCR was used to determine TGF-β, AR, Bax, Bad, p21 and mouse TGFβRII mRNA expression. Total RNA was extracted from prostate tissue using the TRIzol Reagent (Invitrogen), and 1µg RNA was reverse-transcribed using a Promega kit according to the following conditions: 25°C for 10min, 42°C for 60min, and 95°C for 5min. Inventoried Primer pairs and TaqMan probes (Applied Biosystems, Foster City, CA, Catalogue No: Mm 03024053_m1, Mm 00442688_m1, Mm 00432050_m1, Mm 00432042_m1, Mm 1303209 _m1, Mm 03024091_m1) were used to determine TGF-β and AR mRNA levels. Real-time PCR was conducted on an ABI Prism 7300 system, in TaqMan Universal PCR Master Mix, primers (95°C for 10min, followed by 95°C, 15s and 60°C, 60s). Numerical data (normalized to 18SmRNA levels) indicate mean values ±SEM, n=6–8.
Statistical analysis
The data presented on Table 1 were analyzed for statistical significance using the unpaired t-test. Results from all other experiments were analyzed by one-way ANOVA analysis of variance followed by Tukey's and Dunn’s test using the SigmaStat 2.03 program (SPSS, Chicago, IL). Numerical values are expressed as the mean ± SEM. Statistical differences were considered significant at p < 0.05.
Table 1.
| Table 1 (a): Tumor grade evaluation of prostate TRAMP tumors with dominant negative TGFβRII versus TRAMP mice with wild type TGFβRII littermates. Numerical values are the mean ±S.D. (N=6–8 mice/group). | |||||
|---|---|---|---|---|---|
| 12 Weeks | 16 Weeks | 20 Weeks | 24 Weeks | Control ( 20 Weeks TRAMP -) |
|
| TRAMP+/Wt | TRAMP+/Wt | TRAMP+/Wt | TRAMP+/Wt | TRAMP−/Wt | |
| TRAMP+/DN | TRAMP+/DN | TRAMP+/DN | TRAMP+/DN | TRAMP−/DN | |
| Anterior | 3.00 ± 0.00 | 3.33 ± 0.29 | 3.67± 1.16 | 3.75 ± 0.35 | 1.83 ± 0.50 |
| 3.50 ± 0.41 | 4.17 ± 0.29 | 5.33± 0.76* | 4.50 ± 1.32 | 2.17 ± 0.29 | |
| Dorsal+Lateral | 3.17 ± 0.29 | 3.67 ± 0.58 | 4.33± 0.58 | 4.67± 0.76 | 1.83 ± 0.58 |
| 3.57 ±0.29 | 4.67 ± 0.58 | 5.00 ± 0.71 | 5.33± 0.58 | 2.16± 0.29 | |
| Ventral | 2.00± 0.00 | 3.00 ± 0.00 | 3.17± 0.29 | 2.67 ± 0.29 | 1.50±0.50 |
| 2.17±0.29 | 3.33 0.58 | 3.33± 0.58 | 3.00 ± 0.50 | 2.17 ± 0.29 | |
| Table 1(b): Quantitative analysis of prostate apoptosis, proliferatiion, vascularity and inflammation. Values shown are the mean ±SEM (N=6–8 mice/group). The prostates from TRAMP negative control littermates exhibited occasional Ki67, apoptosis and macrophage-positive cells (Supplemental Table 1) | |||||
|---|---|---|---|---|---|
| Ki67 staining | Apoptosis | Vessel | Macrophage | ||
| 12 Weeks | TRAMP+/Wt | 24.84 ±2.89 | 4.52 ± 0.67 | 2.00±1.00 | 5.00 ± 0.58 |
| TRAMP+/DN | 28.81± 2.09 | 8.96 ± 0.64 * | 2.33±1.15 | 5.60 ± 0.67 | |
| 16 Weeks | TRAMP+/Wt | 44.73 ± 6.65 | 5.37 ± 0.50 | 3.00±2.00 | 12.33± 1.45 |
| TRAMP+/DN | 52.22 ± 6.97 | 7.35 ± 1.00 | 2.70±1.50 | 25.33± 5.04 * | |
| 20 Weeks | TRAMP+/Wt | 38.94± 5.05 | 5.55 ± 1.89 | 3.33 ± 0.58 | 18.67±1.45 |
| TRAMP+/DN | 53.86 ±2.61 * | 14.88 ± 1.23 * | 3.66± 0.58 | 36.67±4.63 * | |
| 24 Weeks | TRAMP+/Wt | 42.56±4.89 | 8.56 ± 2.19 | 3.70±1.53 | 55.67±4.98 |
| TRAMP+/DN | 43.99± 3.23 | 13.92 ± 2.33 | 4.00±1.00 | 88.33±8.41 * | |
indicates a significant difference between the TRAMP+/DNTGFβRII+ and TRAMP+/WtTGFβRII in 20 wks mice (p<0.05).
indicates a significant difference between the TRAMP+/DNTGFβRII+ and TRAMP+/WtTGFβRII (p<0.05).
RESULTS
DNTGFβRII expression in prostates from double transgenic TRAMP+/DNTGβRII mice
By crossing heterozygous DNTGFβRII+/− mice in the C57BL/6 background with heterozygous TRAMP mice in the same background, we generated the following transgenic mice: TRAMP+/DNTGFβ RII (Fig. 1, [I], lanes 4,7), TRAMP+/WtTGFβ RII (Fig. 1, [I], lanes 6, 9) and their control litters: TRAMP-/DNTGFβRII (Fig. 1, [I], lanes 1, 2, 3, 5, 11) and TRAMP-/WtTGFβRII (Fig. 1, [I], lanes 8,10). RT-PCR revealed that DNTGFβRII gene expression is successfully turned on in the prostate of TRAMP+/DNTGFβRII (Fig. 1, [II], Lane 2) and TRAMP-/DNTGFβRII mice (Supplemental Fig. 2 [I]) after 29 days on 25mM ZnSO4. Lower levels of mRNA DNTGFβRII were detected in the prostates of TRAMP+/DNTGFβRII (Fig. 1, [II], Lane 3) and TRAMP-/DNTGFβRII (Supplemental Fig . 2 [I]) without ZnSO4. Prostate mRNA DNTGFβRII expression in TRAMP+/DNTGFβRII was higher under zinc-induction (Fig. 1, [III]). DNTGFβRII protein was detected using immunohistochemical staining (24). As shown on Figure 1 [IV], cell membrane DNTGFβRII expression was localized in prostate epithelial cells from TRAMP+/DNTGFβRII mice (upper panel).
Figure 1.
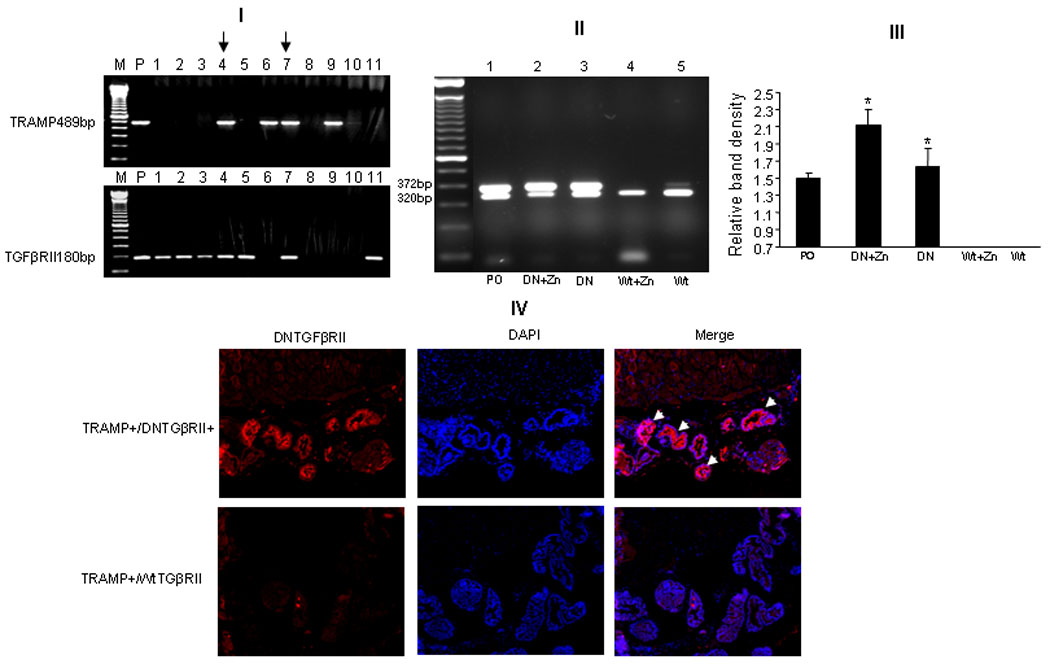
[I] : Genotype characterization of TRAMP+/DNTGFβRII transgenic mice. Lane P, positive control; Lanes 4 and 7, TRAMP+/ DNTGFβRII mice; Lanes 6 and 9 , TRAMP+/WtTGFβRII mice and control litters: TRAMP-/DNTGFβRII (Lanes 1, 2, 3, 5, 11) and TRAMP-/WtTGFβRII (Lanes 8, 10). [II]: RT-PCR of DNTGFβRII transgenic gene expression in the prostate. Lane 1, Positive control. Lanes 2 and 3, prostate mRNA from 12 wk-old TRAMP+/DNTGFβRII mice in the presence and absence of 25mM ZnSO4. Lanes 4 and 5, prostates from 12 wks-old TRAMP+/WtTGFβRII mice. Upper bands: DNTGFβ RII 372bp. Lower bands, comp timer internal control 18S 320bp. [III]: Band intensity for DNTGFβ RII was normalized to 18S. [IV]: DNTGFβRII immunofluorescence using an anti- DNTGFβRII (red) antibody and DAPI (blue) for nuclear staining: Upper panel indicates prostate from a 12wk-TRAMP+/DNTGFβRII mouse in presence of 25mM ZnSO4.
To test the consequences of the exogenous DNTGFβRII on normal TGFRII expression in relation to DNTGFRII, we determined the expression of mouse endogenous wild type TGFβRII and the main TGFβ signaling downstream mediator p21, in control mice (TRAMP-,12-wks), in the absence or presence ZnSO4. Mechanistically the transgenic DNTGFβRII is expected to compete with original wild type TGFβRII, without affecting the expression of the latter in the TRAMP mouse. We found a slight increase in mouse wild type TGFβRII mRNA, but no significant difference in the expression of mouse wild type TGFβRII and DNTGFβRII positive mice at the mRNA level (Supplemental Fig. 1, [I] ). Nor there was a significant difference in p21 mRNA between the DNTGFβRII and wild type TGFβRII mice (Suppl. Fig. 1). The modest increase in wild type TGFβRII protein was observed in DNTGFβRII positive mice (Supplemental Fig.1, [II] and [III]).
Pathological evaluation
The results of histopathological grading of prostatic tumor lesions using a standard grading scale in TRAMP mice (27) are shown in Figure S3 (Supplemental Data). A semiquantitative analysis of lesion distribution in all lobes of prostate is presented in Table 1a. TRAMP negative mice (20wk-old) were used as controls. Flat lesions (grade 1–1.8) were observed in the majority of littermates TRAMP-/Wt TGFβRII mice (Fig. S3, 20 wks, panel A and Table 1a). Flat lesions and focal cell piling (Grade 2–2.5) were detected in the 20 wk-old control TRAMP-/DNTGFβRII+ mice (Fig. S3, panel B), indicating that DNTGFβRII mice have potential for earlier development of hyperplastic lesions. In 12-wks TRAMP mice, histological analysis revealed focal cribriform lesions protruding into the lumen (Grade 3) occurred in the anterior lobes of TRAMP+/DNTGFβRII+ and TRAMP+/WtTGFβRII mice. At 16 wks of age, TRAMP+/DNTGFβRII+ mice developed neuroendocrine type adenocarcinoma with local invasion (Grade 6), in the prostate lateral lobe (Fig. 3S, panel F).
In the ventral prostates, papillary lesions protruding into lumen (grade 2–3) were detected in TRAMP+/WtTGFβRII and TRAMP+/DNTGFβRII+ mice (Table 1a). The anterior prostate lobes in both groups of mice, exhibited regions of focal epithelial hyperplasia (Fig. 5, [I], panels E , F). More aggressive focal epithelial hyperplasia was expressed in TRAMP+/DNTGFβRII+ mice (Fig. 5, [I], panel F). The anterior prostate lobes of TRAMP+/DNTGFβRII+ mice exhibited grades 4–5. (Table 1). Lesions in TRAMP+/DNTGFβRII+ mice were more advanced than in TRAMP+/WtTGFβRII mice, exhibiting distinct grade 5 intraluminal masses, that expanded the acini in dorsal lobes (Fig. S3,,panels C and D). Histological diagnosis indicated that relative to TRAMP mice with intact TGFβRII signaling, prostate tumor lesions in the TRAMP+/DNTGFβRII+ mice are more severe over 12–24 wks in the anterior and dorsal-lateral lobes. Ventral lobes showed reduced tumor progression with aging compared to the other lobes in dysfunctional and intact TGFβRII TRAMP mice (Fig. S3).
Figure 5.
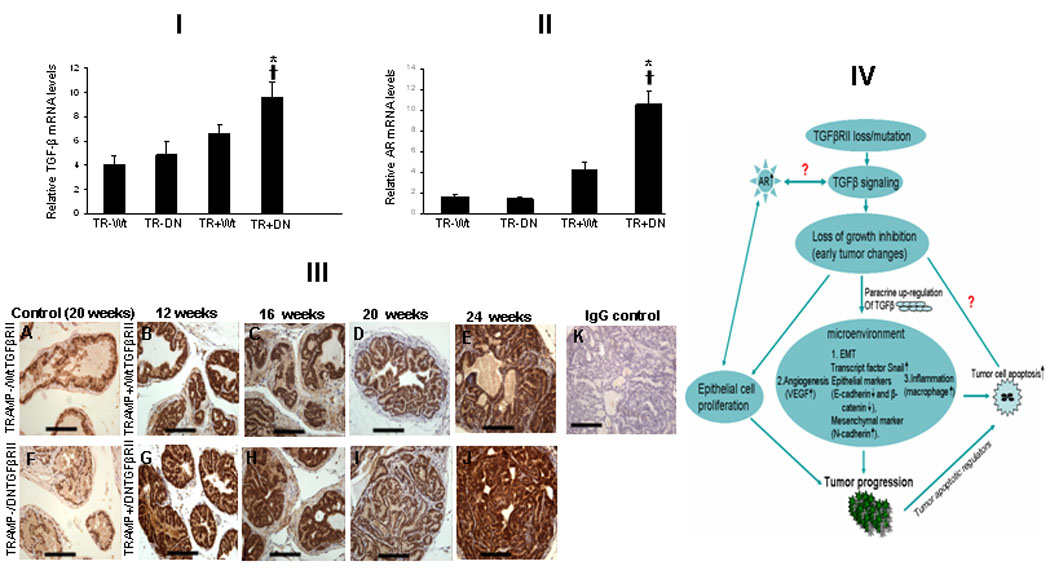
[I] TGF-β and AR expression targeted in prostates from TRAMP+/DNTGFβRII+, TRAMP+/WtTGFβRII mice (20wks). TGF-β ligand mRNA levels were assessed in prostate specimens by real-time RT–PCR. *Significant difference; †Values in TRAMP+/DNTGFβRII+ mice significantly different compared to controls. [II]: AR mRNA expression in prostates from TRAMP+/DNTGFβRII+, TRAMP+/WtTGFβRII mice (*) indicates significant difference. †Values in TRAMP+/DNTGFβRII+ mice significantly different compared to controls. [III] AR immunoreactivity in prostate specimens from 12–24wks-old mice. A and F, controls; G, H, I, J indicate AR expression in aged-matched TRAMP+/DNTGFβRII+ prostates. Panel K, Negative control. [IV]: Impact of TGFβRII signaling loss on prostate growth dynamics. TGFβ dysfunctional signaling has direct consequences on the microenvironment, promoting EMT, inflammation and angiogenesis towards tumor progression.
Consequences of disrupted TGFβRII on prostate cell proliferation and apoptosis in TRAMP mice
The dominant negative TGFβRII led to an increased proliferative index at each stage of progression (12, 16, 20, 24 wks of age) (Fig. 2, [I]). This was paralleled by an increase in the apoptotic cells in TRAMP+/DNTGβRII+, compared to TRAMP+/Wt TGFβRII (Fig. 2, II). The prostate proliferative index in TRAMP+/DNTGFβRII+ mice was higher compared to control TRAMP+/Wt TGFβRII mice at respective ages, 12, 16, 20, 24 wks (Table 1a). The prostate cell apoptotic index was also higher in the TRAMP+/DNTGβRII+ prostates compared to the TRAMP+/WtTGFβRII mice (Table 1b). To dissect this elevated apoptosis, we examined the expression of key pro-apoptotic effector players Bax and Bad. As shown on Figure 2, [III], bax gene expression was up-regulated in 20 wks-old TRAMP+/DNTGβRII, compared to TRAMP+/WtTGFβRII. There was also an increase detected for bad mRNA, but this failed to reach a statistical significant difference (Fig. 2, [III]).
Figure 2.
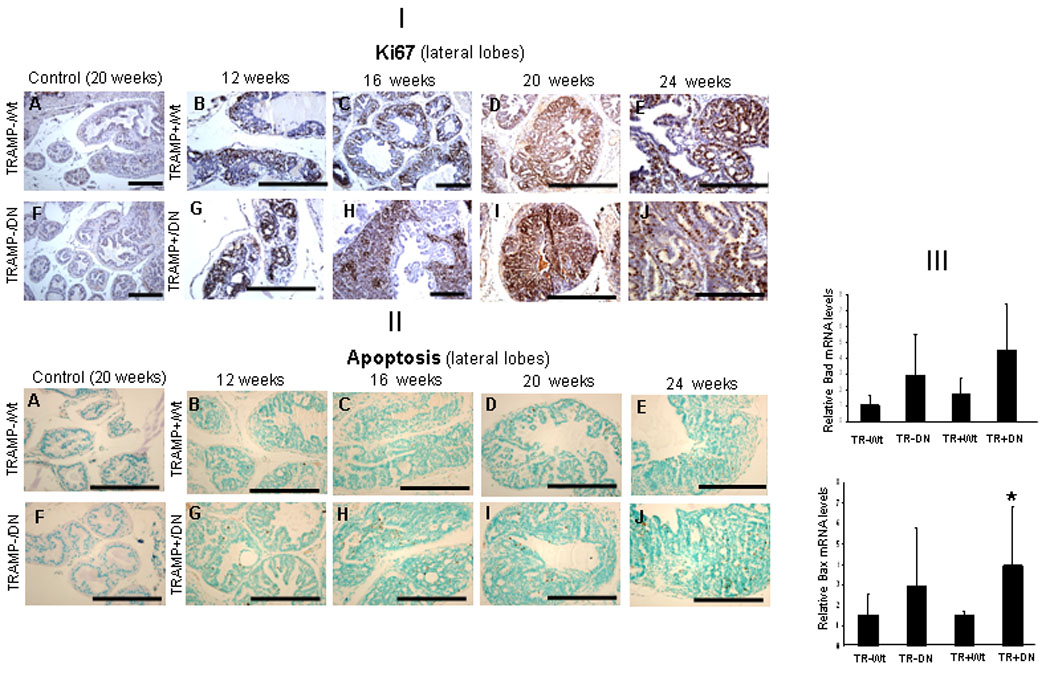
Prostate cell proliferation and apoptosis in TRAMP+/DNTGFβRII, TRAMP+/WtTGFβRII and control littermates. [I]: Ki-6 7 immunostaining and hematoxylin counterstaining (blue) were performed in prostates from 12, 16, 20 and 24wks mice (on ZnSO4 for 45 days). Prostate tissue from controls (20wks) TRAMP-/WtTGFβRII and TRAMP-/DNTGFβRII mice with low Ki-67 staining is shown on panels A and F. Panels B, C, D, E, Ki-67 immunostaining of TRAMP+/WtTGFβRII prostates (12–24wks age). Panels G, H, I, J reveal Ki-67 staining of TRAMP+/DNTGFβRII+ mice of the same age range. [II]: Detection of TUNEL-positive cells; counterstaining with methyl green in prostates from 12–24wks-old mice. Prostates from TRAMP-/WtTGFβRII and TRAMP-/DNTGFβRII+ mice exhibited fewer apoptotic cells (panels A and F) . Panels B, C, D, E reveal TUNEL staining in prostates from TRAMP+/WtTGFβRII mice (12–24wks). Panels G, H, I, J, indicate apoptotic cells. [III]: Real-time RT-PCR was used to analyze Bad and Bax mRNA expression in prostates from TRAMP+/DNTGFβRII+, TRAMP+/WtTGFβRII mice and control littermates (20-wks). (*) Significantly different in TRAMP+/DNTGFβRII+ compared to TRAMP+/WtTGFβRII. p<0.05
Prostate vascularity and inflammation in transgenic DNTGFβRII/TRAMP mice
To investigate whether increased tumorigenicity of TRAMP+/DNTGFβRII+ was associated with increased angiogenesis, vWF-immunostaining was examined (Fig. 3, [I]). There was no significant difference in the vascularity of prostate stroma between TRAMP+/DNTGFβRII+ and TRAMP+/WtTGFβRII (Table 1a) and their littermates mice (Supplemental Fig. S2). However, there was increased intensity of neo-capillary-like vessels observed in prostate tumor regions of TRAMP+/DNTGFβRII (Fig. 3, [I]. panels F, G, H, I, J) compared to controls (Fig. 3, [I], panels, A, B, C, D, E). An increase in VEGF protein levels was detected in TRAMP+/DNTGFβRII prostates (Fig. 3, [III]). There was also an increase in macrophage infiltration into prostate tumors from TRAMP+/ DNTGFβRII+ compared to TRAMP+/WtTGFβRII mice detected at 16, 20 and 24-wks (Fig. 3, [II]). CD68-positive cells were abundant in prostate stroma in TRAMP mice (12–24wks). With advancing age (> 24wks), prostate glands of TRAMP+/DNTGFβRII+ mice exhibited strong luminal CD68-immunoreactivity (Fig. 3, panel J). Moreover, there was a marked increase in the MCP-1 protein levels in prostate tissue from the TRAMP+/DNTGFβRII that directly correlated with high macrophage detection (Fig. 3, [IV]).
Figure 3.
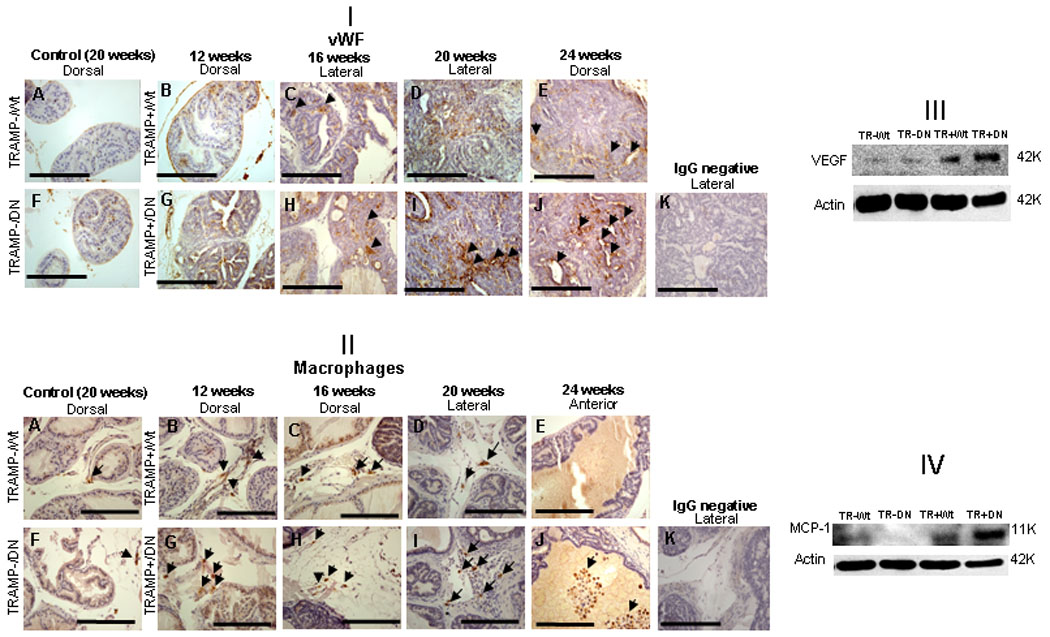
Microvessel density and macrophage infiltration in prostates of TRAMP+/DNTGFβRII+, TRAMP+/WtTGFβRII mice. [I] : Immunostaining with anti-vWF antibody and hematoxylin counterstaining (blue) were performed in prostate tissue from 12–24wks mice. Panels A and F, prostates from controls (at 20wks); Panels B, C, D, E indicate vWF staining of TRAMP+/WtTGFβRII+ mice of increasing ages (12–24wks). Panel G, H, I, J presented vWF staining of TRAMP+/DNTGFβRII+ mice (12–24wks). Panel K, negative control IgG staining. [II]: CD68 staining for macrophages (brown). Prostates from control mice are shown in panels A and F. Panels B, C, D, E indicate CD68 staining in TRAMP+/WtTGFβRII+ prostates. Panels G, H, I, J reveal CD68 immunoreactivity in prostate with impaired TGF-β signaling. Panel K, negative control IgG staining. [III and IV]: VEGF and MCP-1 protein expression in prostates of TRAMP+/DNTGFβRII+ and control mice (20wks) was evaluated by Western blotting.
EMT-like pathological change in transgenic TRAMP/DNTGFβRII mice
Reduced E-cadherin and increased N-cadherin expression, are recognized characteristic features of EMT. Figure 4[I] reveals characteristic E-cadherin and N-cadherin immunoreactivity in prostate from TRAMP+/DNTGFβRII and TRAMP+/WtTGFβRII mice. Reduced E-cadherin immunoreactivity was detected in 12 and 16 wks-old TGFβRII transgenic (Fig. 4, panels B, D) compared to wild type TGFβRII (Fig. 4, panels A and C) TRAMP mice. E-cadherin immunoreactivity was lost (Fig. 4[I]; panels E and F) in 20-wks TRAMP+/DNTGFβRII. Moderately differentiated prostate tumors detected in the lateral prostates of (24-wks) TRAMP+/DNTGFβRII lack E-cadherin, while well-differentiated prostate tumors from age-matched TRAMP+/WtTGFβRII exhibited strong E-cadherin immunoreactivity (Fig. 4, panels, E and F). The higher density of N-cadherin positive myofibroblast -like cells extended into prostate tumor were observed in the TRAMP+/DNTGFβRII mice (Fig.4, [II], Panels B, D, F, H), compared to TRAMP+/WtTGFβRII mice (Fig.4, [II], Panels A, C, E and G). Mechanistic profiling revealed downregulation of epithelial markers, E-cadherin, β-catenin and up-regulation of the mesenchymal marker N-cadherin, and Snail, an EMT player, in TRAMP+/DNTGFβRII prostates (Fig. 4, [III] and [IV]).
Figure 4.
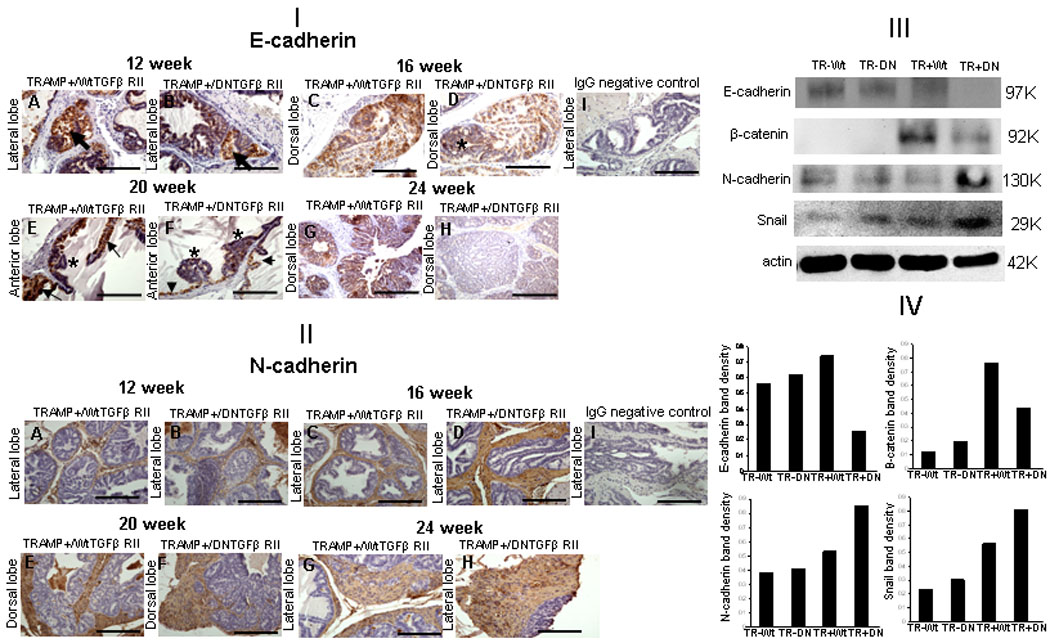
EMT in prostates with impaired TGF-β signaling [I]. Panels A and B: E-cadherin immunoreactivity in the lateral lobes of TRAMP+/WtTGFβRII and TRAMP+/DNTGFβRII+ mice (12-wks), respectively. Panels C and D, E-cadherin immunoreactivity in the dorsal lobes at 16-wks; panels E and F, in anterior prostate lobes of WtTGFβRII and DNTGFβRII+ TRAMP mice at 20-wks, respectively. G and H reveal E-cadherin expression in dorsal lobes. Arrow indicates E-cadherin positive cells; (*), E-cadherin negative. [II]. Panels A and B: N-cadherin immunoreactivity in lateral lobes, TRAMP+/WtTGFβRII and TRAMP+/DNTGFβRII+ mice at 12-wks, respectively. Panels C and D, N-cadherin immunoreactivity in lateral lobes (16-wks). Panels E and F, N-cadherin immunoreactivity pattern in the dorsal prostate lobes of WtTGFβRII and DNTGFβRII of TRAMP mice (20-wks), respectively. Panels G and H, reveal N-cadherin lateral lobe expression. Panels I: IgG negative control. [III]: Prostate E-cadherin, N-cadherin, β-catenin, and Snail expression in TRAMP+/DNTGFβRII+, TRAMP+/WtTGFβRII mice and control littermates (20wks). Prostate lysates were subjected to Western blotting for E-cadherin, N-cadherin, β-catenin, and Snail protein expression. [IV]: Relative band intensity from (III).
Induction of TGFβ ligand and AR expression in DNTGFβRII/TRAMP prostates
As shown on Figure 5 (Panel [I]), the presence of dysfunctional DNTGFβRII receptor resulted in elevated TGFβ ligand mRNA expression. We found a significant uperegulation of AR mRNA in TRAMP+/DNTGFβRII transgenic mice compared to TRAMP+/WtTGFβRII and controls (Fig.5, [II]). AR immunoreactivity was primarily of nuclear localization among the prostate epithelial cells (Fig. 5 [III]). TRAMP-/DNTGFβRII and TRAMP-/WtTGFβRII derived prostates exhibited low AR immunoreactivity, while a strong AR expression was detected in TRAMP+ prostates. Higher AR expression was detected in TRAMP mice with inactivated DNTGFβRII, compared to TRAMP mice with wild-type TGFβRII, a change that correlated with the AR mRNA increase in prostate epithelial cells of TRAMP+/DNTGFβRII (Fig. 5 [II]).
DISCUSSION
Functional defects in TGF-β signaling have been implicated in cancer development and progression (28), but the mechanisms underlying resistance to TGF-β mediated growth inhibition in prostate cancer cells are not fully understood (29), although TGFβRII mutations are more frequent than TGFβRI mutations (30). This study investigated the temporal involvement of mutational inactivation of TGFβRII in prostate epithelial cells in prostate tumorigenesis. Double transgenic TRAMP+/DNTGFβRII+ mice exhibited pathological evidence of PIN and poorly differentiated prostate adenocarcinoma at an early age, compared to TRAMP+/ Wt TGFβRII mice, indicating that TGF-β signaling triggers abnormal growth kinetics at the onset of prostate cancer development. Our results are in accordance with the role of TGF-β a s a tumor suppressor in the early stages of prostate tumorigenesis, and as a tumor promoter in advanced disease (31, 32). Another in vivo study engaging the expression of a dominant negative TGFβRII in mouse skeletal tissue, reported that disruption of TGF-β signaling led to prostate cancer metastasis, challenging the existing knowledge that TGF-β is strong promoter of cancer metastasis (12). Here the dominant negative TGFβRII was expressed in mouse epithelial cells, responsible for giving rise to tumor cells in TRAMP mice. One could argue that TGFβRII loss of function in the epithelial cells may increase TGFβ ligand through paracrine effects. Consequently, increased TGFβ levels stimulate non-malignant stromal cell types of the tumor in angiogenesis, ECM degradation, and EMT (33–35), leading to tumor invasion and metastasis. This evidence is in accord with our results indicating increased intraprostatic TGFβ mRNA levels to correlate with TRAMP+/DNTGFβRII+ mice.
Recurrence of highly aggressive, metastatic prostate tumors (36–38) due to the emergence of androgen-independent prostate cancer cells, results from gene alterations in players regulating androgen-dependent apoptosis of normal prostate epithelium (39). One cannot dismiss the evidence that changes in AR signaling may also become a contributing factor by utilizing low levels of androgens (40), or by co-opting the AR in other signaling pathways (3, 41–43). TGFβRII has been implicated in the androgenic control of TGF-β signaling in prostate epithelial cells (44). The TGF-β promoter contains three distal and three proximal AREs (androgen-response elements)m that physically interact with the DNA-binding domain of AR (45). In our study, disruption of TGFβRII signaling led to a significant increase the proliferative index of prostate cancer cells, a change that was paralleled by a significant upregulation of AR. The present findings resonate with our previous in vitro studies (46), supporting the engagement of TGF-β signaling in anti-androgenic properties of prostate cancer cells, via a dysfunctional TGF-β/AR cross-talk. Interestingly a contextual role of TGF-β in breast cancer was associated with the estrogen receptor (ER) status (47).
In our experimental model, dominant negative TGFβRII led to the early manifestation of the aggressive premalignant pathological characteristics of prostate tumors. TGF-β triggers apoptosis depending on cell-autonomous and environmental factors whose molecular identity remains unknown (48). The increased incidence of apoptosis among the epithelial cells in TRAMP+/DNTGFβRII+ prostates was unexpected and might represent a “feedback” phenomenon under abnormally high proliferative conditions, to which cell growth dynamics react to maintain an appropriate apoptotic rate, counteracting TGF-β signaling. This finding might appear in sharp contrast to the apoptotic role of TGF-β, but nevertheless consistent with the enhanced and frequent apoptosis occurring in hyperplastic lesions of TGFβRII null mammary epithelium (49), as well in high-grade neoplasms (50). Converging oncogenic gene mutations might result in dramatic losses of the normal apoptotic response to TGF-β in vivo. Moreover, the microenvironment and immune system, enjoy a complex dynamic exchange, regulated by inflammatory cells, ECM and degradation and cytokine signaling controlling these processes. Our results indicate that DNTGFβRII expression targets important cellular processes in the prostate epithelial cells, such as leading to enhanced inflammation (via MCP-1 induction and macrophage infiltration), increased vascularity (as demonstrated by increased VEGF) and EMT induction (associated with metastatic progression).
Loss of E-cadherin, expression correlated with an increased proliferative rate and tumor aggressiveness in the TRAMP+/DNTGFβRII+ derived prostate tumors. Such a correlation between loss of E-cadherin expression and Gleason grade has been shown in human prostate tumors (51). Cancer cell dedifferentiation via activation of EMT-associated pathways (52) is causal to the metastatic process. Our findings support the EMT effect in impaired TGF-β signaling, (reduction in E-cadherin and β-catenin and increased N-cadherin and Snail expression). Snail overexpression in epithelial cells causes their mesenchymal phenotype, via E-cadherin downregulation ((53)), by binding to the E-cadherin promoter to repress its transcription ((54). A mutant DNTGFβRII receptor can promote recruitment of monocytes/macrophages by the microenvironment, a significant observation considering that macrophages represent the major inflammatory component of tumor stroma (55). Lack of recruitment of macrophages at the tumor site leads to decreased tumorigenic growth (56); moreover clinical evidence suggests a correlation between a high tumor infiltration of macrophages and a poor prognosis of prostate cancer patients (55). TGF-β directly induces chemotaxis of monocytes/macrophages or indirectly causes immigration of inflammatory cells in combination with other modulatory cytokines or chemokines such as MCP-1 (57), towards regulating tumor-infiltrating monocytes/macrophages (48, 58).
In our model, targeted functional inactivation of TGF-β signaling in TRAMP mice provides a new insight into the impact of loss of TGF-β responsiveness on tumorigenesis: both via direct effects on cell proliferation and EMT, and indirectly via ligand up-regulation with associated paracrine effects on the tumor microenvironment. The dominant negative approach makes the dissection of individual contributions by each effector component to TGF-β signaling in cancer difficult, by rendering the microenvironment unpredictable. The interaction between TGF-β and AR can lead to potential new therapeutic options based on inhibition of vital exchanges between TGF-β, its receptors and AR, in the context of EMT during prostate tumor progression (59). Targeted restoration of TGF-β response in early carcinogenesis might constitute a potential therapeutic strategy for prostate cancer.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by an NIH R01 Grant DK5355-12 (NK) and a UPR Summer Research Grant in Toxicology (JC). Dustin Gayheart is a University of Kentucky College of Medicine Clinical Research Scholar. The authors thank Menglei Zhu for useful discussions and Lorie Howard for her assistance with the submission.
Abbreviations
- TGF-β
transforming growth factor-β
- DNTGFβRII
Dominant negative TGF-β receptor II
- TRAMP
transgenic adenocarcinoma of mouse prostate
- AR
androgen receptor
- TUNEL
Terminal deoxynucleotidyl tranafer5ase-mediated dUTP-biotin nick end labeling
- DAPI
4’-6’-Diamidino2-phenylindole
- EMT
epithelial-mesenchymal transition
- VEGF
endothelial growth factor
- MCP-1
monocyte chemoattractant protein 1
REFERENCES
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488–1490. doi: 10.1056/NEJMp048178. [DOI] [PubMed] [Google Scholar]
- 3.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 4.van der Poel HG. Androgen receptor and TGFbeta1/Smad signaling are mutually inhibitory in prostate cancer. European urology. 2005;48:1051–1058. doi: 10.1016/j.eururo.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Zeng L, Rowland RG, Lele SM, Kyprianou N. Apoptosis incidence and protein expression of p53, TGF-beta receptor II, p27Kip1, and Smad4 in benign, premalignant, and malignant human prostate. Human pathology. 2004;35:290–297. doi: 10.1016/j.humpath.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Guo Y, Jacobs SC, Kyprianou N. Down-regulation of protein and mRNA expression for transforming growth factor-beta (TGF-beta1) type I and type II receptors in human prostate cancer. International journal of cancer. 1997;71:573–579. doi: 10.1002/(sici)1097-0215(19970516)71:4<573::aid-ijc11>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 7.Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. The Journal of biological chemistry. 2003;278:47853–47861. doi: 10.1074/jbc.M305171200. [DOI] [PubMed] [Google Scholar]
- 8.Ayala GE, Dai H, Tahir SA, et al. Stromal antiapoptotic paracrine loop in perineural invasion of prostatic carcinoma. Cancer research. 2006;66:5159–5164. doi: 10.1158/0008-5472.CAN-05-1847. [DOI] [PubMed] [Google Scholar]
- 9.Chesire DR, Ewing CM, Gage WR, Isaacs WB. In vitro evidence for complex modes of nuclear beta-catenin signaling during prostate growth and tumorigenesis. Oncogene. 2002;21:2679–2694. doi: 10.1038/sj.onc.1205352. [DOI] [PubMed] [Google Scholar]
- 10.Tu WH, Thomas TZ, Masumori N, et al. The loss of TGF-beta signaling promotes prostate cancer metastasis. Neoplasia (New York, NY. 2003;5:267–277. doi: 10.1016/S1476-5586(03)80058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer research. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 12.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nature medicine. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 13.Hayes SA, Zarnegar M, Sharma M, et al. SMAD3 represses androgen receptor-mediated transcription. Cancer research. 2001;61:2112–2118. [PubMed] [Google Scholar]
- 14.Kang HY, Huang KE, Chang SY, Ma WL, Lin WJ, Chang C. Differential modulation of androgen receptor-mediated transactivation by Smad3 and tumor suppressor Smad4. The Journal of biological chemistry. 2002;277:43749–43756. doi: 10.1074/jbc.M205603200. [DOI] [PubMed] [Google Scholar]
- 15.Chipuk JE, Cornelius SC, Pultz NJ, et al. The androgen receptor represses transforming growth factor-beta signaling through interaction with Smad3. The Journal of biological chemistry. 2002;277:1240–1248. doi: 10.1074/jbc.M108855200. [DOI] [PubMed] [Google Scholar]
- 16.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 17.Tang B, de Castro K, Barnes HE, et al. Loss of responsiveness to transforming growth factor beta induces malignant transformation of nontumorigenic rat prostate epithelial cells. Cancer research. 1999;59:4834–4842. [PubMed] [Google Scholar]
- 18.Guo Y, Kyprianou N. Overexpression of transforming growth factor (TGF) beta1 type II receptor restores TGF-beta1 sensitivity and signaling in human prostate cancer cells. Cell Growth Differ. 1998;9(2):185–193. [PubMed] [Google Scholar]
- 19.Guo Y, Kyprianou N. Restoration of transforming growth factor beta signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer research. 1999;59:1366–1371. [PubMed] [Google Scholar]
- 20.Greenberg NM, DeMayo F, Finegold MJ, et al. Prostate cancer in a transgenic mouse. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate cancer and prostatic diseases. 1999;2:70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]
- 22.Gingrich JR, Barrios RJ, Morton RA, et al. Metastatic prostate cancer in a transgenic mouse. Cancer research. 1996;56:4096–4102. [PubMed] [Google Scholar]
- 23.Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer research. 1997;57:4687–4691. [PubMed] [Google Scholar]
- 24.Bottinger EP, Jakubczak JL, Roberts IS, et al. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. The EMBO journal. 1997;16:2621–2633. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmiter RD, Sandgren EP, Koeller DM, Brinster RL. Distal regulatory elements from the mouse metallothionein locus stimulate gene expression in transgenic mice. Molecular and cellular biology. 1993;13:5266–5275. doi: 10.1128/mcb.13.9.5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Cancer research. 2006;66:464–472. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suttie A, Nyska A, Haseman JK, Moser GJ, Hackett TR, Goldsworthy TL. A grading scheme for the assessment of proliferative lesions of the mouse prostate in the TRAMP model. Toxicologic pathology. 2003;31:31–38. doi: 10.1080/01926230390173842. [DOI] [PubMed] [Google Scholar]
- 28.Bello-DeOcampo D, Tindall DJ. TGF-betal/Smad signaling in prostate cancer. Current drug targets. 2003;4:197–207. doi: 10.2174/1389450033491118. [DOI] [PubMed] [Google Scholar]
- 29.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 30.Brattain MG, Ko Y, Banerji SS, Wu G, Willson JK. Defects of TGF-beta receptor signaling in mammary cell tumorigenesis. Journal of mammary gland biology and neoplasia. 1996;1:365–372. doi: 10.1007/BF02017392. [DOI] [PubMed] [Google Scholar]
- 31.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 32.Kim SJ, Im YH, Markowitz SD, Bang YJ. Molecular mechanisms of inactivation of TGF-beta receptors during carcinogenesis. Cytokine & growth factor reviews. 2000;11:159–168. doi: 10.1016/s1359-6101(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 33.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends in cell biology. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 34.Barcellos-Hoff MH, Medina D. New highlights on stroma-epithelial interactions in breast cancer. Breast Cancer Res. 2005;7:33–36. doi: 10.1186/bcr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett JM, Rovedo MA, Tajuddin AM, et al. Prostate cancer cells regulate growth anddifferentiation of bone marrow endothelial cells through TGFbeta and its receptor, TGFbetaRII. The Prostate. 2006;66:632–650. doi: 10.1002/pros.20370. [DOI] [PubMed] [Google Scholar]
- 36.Albertsen PC, Hanley JA, Fine J. 20-year outcomes following conservative management of clinically localized prostate cancer. Jama. 2005;293:2095–2101. doi: 10.1001/jama.293.17.2095. [DOI] [PubMed] [Google Scholar]
- 37.Potters L, Morgenstern C, Calugaru E, et al. 12-year outcomes following permanent prostate brachytherapy in patients with clinically localized prostate cancer. The Journal of urology. 2005;173:1562–1566. doi: 10.1097/01.ju.0000154633.73092.8e. [DOI] [PubMed] [Google Scholar]
- 38.Roth BJ. Prostate cancer chemotherapy: emerging from the shadows. J Clin Oncol. 2005;23:3302–3303. doi: 10.1200/JCO.2005.11.933. [DOI] [PubMed] [Google Scholar]
- 39.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer research. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 40.Mohler JL, Gregory CW, Ford OH, 3rd, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 41.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. Journal of cellular biochemistry. 2004;91:483–490. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 42.Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. Journal of cellular biochemistry. 2006;99:333–344. doi: 10.1002/jcb.20794. [DOI] [PubMed] [Google Scholar]
- 43.Wang G, Sadar MD. Amino-terminus domain of the androgen receptor as a molecular target to prevent the hormonal progression of prostate cancer. Journal of cellular biochemistry. 2006;98:36–53. doi: 10.1002/jcb.20802. [DOI] [PubMed] [Google Scholar]
- 44.Song K, Wang H, Krebs TL, Kim SJ, Danielpour D. Androgenic control of transforming growth factor-beta signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-beta receptor II. Cancer research. 2008;68:8173–8182. doi: 10.1158/0008-5472.CAN-08-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qi W, Gao S, Wang Z. Transcriptional regulation of the TGF-beta1 promoter by androgen receptor. The Biochemical journal. 2008;416:453–462. doi: 10.1042/BJ20080651. [DOI] [PubMed] [Google Scholar]
- 46.Zhu ML, Partin JV, Bruckheimer EM, Strup SE, Kyprianou N. TGF-beta signaling and androgen receptor status determine apoptotic cross-talk in human prostate cancer cells. The Prostate. 2008;68:287–295. doi: 10.1002/pros.20698. [DOI] [PubMed] [Google Scholar]
- 47.Buck MB, Fritz P, Dippon J, Zugmaier G, Knabbe C. Prognostic significance of transforming growth factor beta receptor II in estrogen receptor-negative breast cancer patients. Clin Cancer Res. 2004;10:491–498. doi: 10.1158/1078-0432.ccr-0320-03. [DOI] [PubMed] [Google Scholar]
- 48.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forrester E, Chytil A, Bierie B, et al. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer research. 2005;65:2296–2302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 50.Scopa CD, Tsamandas AC, Zolota V, Kalofonos HP, Batistatou A, Vagianos C. Potential role of bcl-2 and ki-67 expression and apoptosis in colorectal carcinoma: a clinicopathologic study. Digestive diseases and sciences. 2003;48:1990–1997. doi: 10.1023/a:1026178506348. [DOI] [PubMed] [Google Scholar]
- 51.Jaggi M, Johansson SL, Baker JJ, Smith LM, Galich A, Balaji KC. Aberrant expression of E-cadherin and beta-catenin in human prostate cancer. Urologic oncology. 2005;23:402–406. doi: 10.1016/j.urolonc.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 52.Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 53.Cano A, Perez-Moreno MA, Rodrigo I, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature cell biology. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 54.Arias AM. Epithelial mesenchymal interactions in cancer and development. Cell. 2001;105:425–431. doi: 10.1016/s0092-8674(01)00365-8. [DOI] [PubMed] [Google Scholar]
- 55.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 56.Lin EY, Li JF, Gnatovskiy L, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer research. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 57.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 58.Umemura N, Saio M, Suwa T, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. Journal of leukocyte biology. 2008;83:1136–1144. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- 59.Niu Y, Altuwaijri S, Lai KP, et al. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12182–12187. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.