

Abstract
A number of bicyclo[2.2.2]octenone-containing natural products have been isolated from the heartwood of Chamaecyparis obtusa var. formosana (Figure 1) including the Diels-Alder adducts[1] obtunone (1),[2] chamaecypanone C (2),[3] and the [4+2] dimer (+)-3.[2],[4] Compound (+)-2 was shown to exhibit potent cytotoxicity against several human cancer cells including human oral epidermoid carcinoma (KB) (IC50 = 190 nM).[3] The biosynthesis of 2 was proposed[3] to occur via endo [4+2] cycloaddition between 1-hydroxymentha-3,5-dien-2-one 4 (Figure 2) and 1,3-bis-arylcyclopenta-1,3-diene 5, followed by oxidation to an enone in accord with literature reports of cyclopentadienes as biosynthetic precursors to natural products.[1b] An alternative possibility involving the corresponding cyclopentadienone 6 as dienophile may also be considered in light of known biosyntheses involving reactive cyclopentadienones.[5] Herein, we report a concise synthesis of both enantiomers of chamaecypanone C involving a retro-DA/DA cascade of dimer 3, obtained utilizing copper-mediated asymmetric oxidative dearomatization,[6] as well as biological studies documenting that the cytotoxic action of (+)-2 involves mitotic arrest as a consequence of its binding in the colchicine site of tubulin.
Keywords: cycloaddition, total synthesis, natural product, cyclopentadienone, microtubule inhibitor
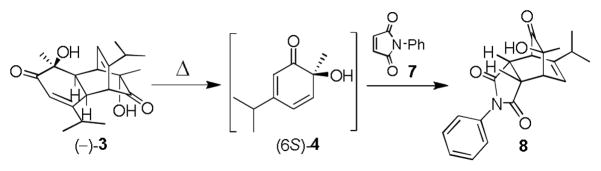
Inspired by literature reports of tandem retro-DA/DA reactions of dimers derived from o-quinols and masked o-benzoquinones (MOBs), [7], [8] we first evaluated reactions between the readily accessible dimer (−)-3[6] and N-phenylmaleimide (7) under thermolytic conditions in different solvents (Table 1). Although reactions in toluene (entry 1) and chlorobenzene (entry 2) generated the desired cycloadduct 8 in moderate to good conversion (12 h), reactions in mesitylene at 150 °C were found to give both excellent conversion and isolated yield of 8 in 1.5 h (entries 3 and 4).
Table 1.
Optimization of the Retro-DA/DA Cascade
 | |||||
|---|---|---|---|---|---|
| Entry | solvent | temp. (°C) | dienophile 7 (equiv.) | time (h) | conversiona (%) |
| 1 | toluene | 110 | 5 | 12 | 69 |
| 2 | chlorobenzene | 130 | 5 | 12 | 92 |
| 3 | mesitylene | 150 | 5 | 1.5 | >99 (98)b |
| 4 | mesitylene | 150 | 3 | 1.5 | >99 (97)b |
Conversion based on 1H-NMR analysis of 8 and starting material (−)-3;
Isolated yield of 8 in parenthesis.
Using these optimized conditions, a number of representative dienophiles were thermolyzed in the presence of dimer (−)-3 in mesitylene (Table 2). Reactions with methyl vinyl ketone (MVK, 9, entry 1), 2,3-dihydrofuran 10 (entry 2), and indene 11 (entry 3) successfully generated bicyclo[2.2.2]octenones 12 to 14 in good to excellent yields, which underscores the reactivity of o-quinols as both normal and inverse demand dienes. The observed regioselectivity for products of 12–14 is in agreement with that reported for related cyclohexadienones (MOBs).[7c,d] Reaction with β-myrcene (15, entry 4) smoothly generated an inseparable 1:1 mixture of ent-obtunone (1) and a decalin product, both of which were acetylated to afford 16 and 17.[9] Hydrolysis of 16 (aq. NaOH/MeOH) afforded optically pure product ent-1.[2], [10] Furthermore, cyclopentadiene dimer 18 (entry 5) was found to be very reactive, affording [4+2] adduct 19 as a single diastereomer in nearly quantitative yield.[7a] In contrast, reaction with cyclopentadienone dimer 20 (entry 6) produced 21 in moderate yield, probably due to side reactions of 20 at high temperature including decarbonylation.[11]
Table 2.
Tandem retro-DA/DA reactions using bicyclooctenone (−)-3a
| entry | Dienophile(eqiv) | cycloadduct | Time(h) | Yieldb (%) |
|---|---|---|---|---|
| 1 |
 (5 equiv) (5 equiv)9 |
 12c 12c
|
3 | 92 |
| 2 |
 (20 equiv) (20 equiv)10 |
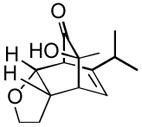 13 13
|
12 | 84 |
| 3 |
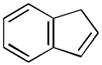 (10 equiv) (10 equiv)11 |
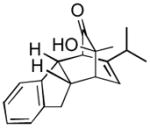 14 14
|
3 | 99 |
| 4d |
15 |
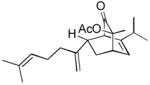 16 16
|
4 | 42 |
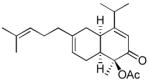 17 17
|
4 | 39 | ||
| 5 |
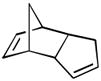 (5 equiv) (5 equiv)18 |
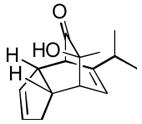 19 19
|
4 | 98 |
| 6 |
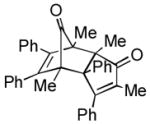 (2.5 equiv) (2.5 equiv)20 |
 21 21
|
5 | 57 |
Reaction conditions: dimer (−)-3, dienophile, mesitylene, 150 °C;
Isolated yield after column chromatography;
Approximately 6% of an inseparable minor product was observed by 1H-NMR;
Acetylation required for product separation.
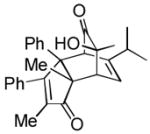
Based on our ability to trap (6S)-4 with a number of dienophiles, we proceeded to evaluate both cyclopentadienes and cyclopentadienones for the synthesis of 2. Accordingly, we targeted a single starting material for preparation of both precursors. Starting from the known bis-arylcyclopentene derivative 22,[12] allylic oxidation using selenium dioxide afforded alcohol 23 as major product (50%) along with small amount of enone 24 (Scheme 1). Although diaryl cyclopentadienes[13] were detected by GC-MS under acid-catalyzed dehydration conditions (cat. MP-TsOH, toluene, 110 °C, 1 h),[14] all attempts to isolate pure product 25, or trap it with reactive dienophiles (e.g. maleic anhydride, tetracyanoethylene (TCNE)) failed. Moreover, thermolysis of the crude mixture from either dehydration of 23 or base-promoted elimination of the derived mesylate derivative with dimer 3 also did not afford the desired cycloadduct 26.
Scheme 1.
a) 0.5 equiv SeO2, 2 equiv TBHP, DCE, 60 °C, 2 h, 50% (23), and 5% (24); b) cat. MP-TsOH, toluene, 110 °C, 1 h; or Martin sulfurane, CH2Cl2, r.t., 0.5 h; c) 0.2 equiv (−)-3, mesitylene, 150 °C. TBHP=tert-butyl hydroperoxide, DCE=1,2-dichloroethane.
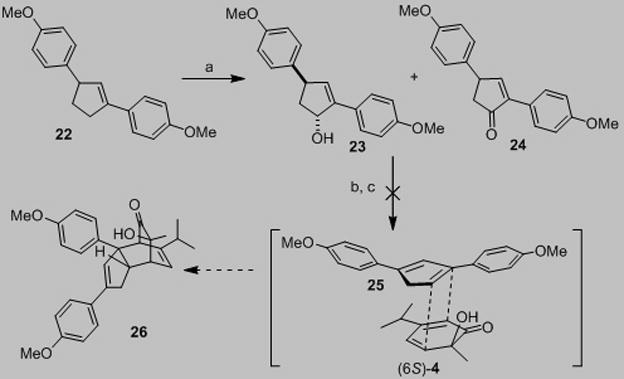
Alternatively, allylic alcohol 23 could be efficiently converted to cyclopentenone 24 using IBX as oxidant[15] (Scheme 2). After extensive experimentation, it was found that DDQ oxidation of 24[16] in the presence of dimer 3 afforded the desired cycloadduct 27 in good yield. The endo stereochemistry of cycloadduct 27 was unambiguously assigned by NOE experiments.[10] The transformation presumably proceeds via initial formation of the reactive cyclopentadienone 28 from cyclopentenone 24.[ 17 ] Unfortunately, all efforts to isolate either the cyclopentadienone monomer or derived dimers in control experiments have thus far failed. Finally, treatment of 27 with BBr3 effected smooth demethylation to afford (−)-chamaecypanone C (ent-2) (86%). To the best of our knowledge, this is the first example of generation of a 2,4-diarylcyclopentadienone and its usage in natural product synthesis.[18] The instability and high reactivity of the diarylcyclopentadienone intermediate[19] is likely due to the relief of antiaromaticity upon cycloadditionas suggested by Harmata and coworkers.[20]
Scheme 2.
a) 2.0 equiv IBX, toluene/DMSO (1 M, 2:1), 50 °C, 30 min, 90%; b) 1.5 equiv (−)-3, 2.0 equiv DDQ, o-dichlorobenzene, 150 °C, 1 h, 61%; c) 8.0 equiv BBr3, CH2Cl2, −78 °C to rt, 4 h, 86%. DMSO=dimethyl sulfoxide, DDQ=2,3-dichloro-5,6-dicyanobenzoquinone.
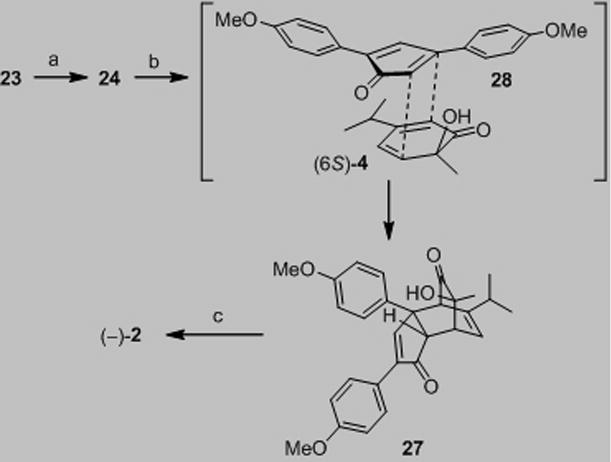
In a similar manner, we prepared (+)-chamaecypanone C (2, Scheme 3). Hydrogenation of 29 quantitatively generated 2,4-disubstituted phenol 30. An asymmetric hydroxylation-α – ketol rearrangement-dimerization sequence[6] afforded (+)-dimer 3 in moderate yield over two steps (>99% ee) which was further elaborated into (+)-chamaecypanone C (53%, two steps from enone 24). Synthetic 2 was confirmed to be identical with data reported for natural chamaecypanone C by comparison of 1H and 13C NMR spectra, mass spectrum, IR, and [α]D, thus confirming its absolute configuration.[10]
Scheme 3.
a) H-Cube® (Pd/C), H2 (40 bar), MeOH (0.03 M), 50 °C, 0.5 mL/min, quantitative; b) 1.0 equiv LiOH · H2O, EtOH/toluene, azeotrope; Cu(CH3CN)4PF6 (2.2 equiv.), (−)-sparteine (2.3 equiv), 3Ǻ molecular sieves, O2, THF, −78 °C, 16 h; c) benzene, reflux, 12 h, 47% (2 steps). THF=tetrahydrofuran.
Both enantiomers of 2 were tested in the NCI 60-cell single dose assay at 10−5 M. Confirming earlier studies on the natural product[3], (+)-2 inhibited tumor cell growth by an average of 71%, while (−)-2 had no effect. (+)-2 was then tested in a dose response format, where it displayed robust selectivity with a mean GI50 value of 0.21 μM. COMPARE analysis of the data[21] at the TGI level suggested that (+)-2 might act through interference with tubulin function, as high correlations were seen to the data for seven established tubulin inhibitors.[10] Examination of this hypothesis using an in vitro tubulin polymerization assay[22] found this to be the case, with an IC50 of 2.0±0.1 μM, while the (−)-enantiomer had no effect at 40 μM. (+)-2 was also tested for inhibition of colchicine binding[23] where it had moderate activity at 50 μM with 5 μM [3H]colchicine and 1 μM tubulin. Finally, we confirmed that (+)-2 had effects in cells consistent with its inhibitory effects on tubulin assembly. Cytotoxic concentration of (+)-2 arrested cells in mitosis concordant with inhibition of cell growth (Figure 3) and caused the disassembly of the intracellular microtubule network (Figure 4). [24]
Figure 3.
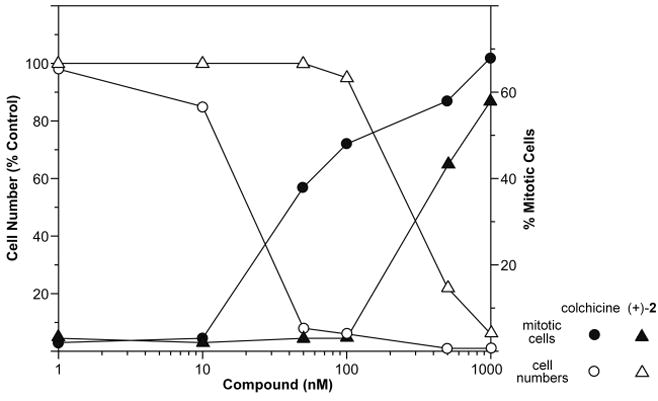
Human Burkitt lymphoma CA46 cells, obtained from the American Type Tissue Collection, were grown in suspension culture for 24 h at 37 °C in a humidified, 5% CO2 atmosphere. The medium was RPMI 1640 supplemented with 5% fetal bovine serum. Initially, the culture medium contained 20,000 cells/mL. For determination of cell growth, the increase in cell number was determined, with the cells counted in a Beckman Coulter model Z1 particle counter. For determination of mitotic cells, cells were harvested by centrifugation, briefly swollen in a hypotonic solution, fixed on a glass slide, and stained with Giemsa. The percentage of cells with condensed chromosomes was determined.
Figure 4.

Disruption of intracellular microtubule network by chamaecypanone C. Potorus tridactylis PtK2 kidney epithelial cells were obtained from the American Type Tissue Collection and were cultured in Minimal Essential Medium supplemented with 10% fetal bovine serum, 1 mM glutamine, and 1 mM sodium pyruvate. The cells were grown to confluence, disrupted by trypsinization, and seeded at about 35,000 cells into each compartment of a Chambered Coverglass System from Nunc with either no compound (A) or 0.5 μM chamaecypanone C (B) added to the culture medium. Following growth for 16 h at 37 °C in a humidified, 5% CO2 atmosphere, the cells on the coverglass were fixed with − 20 °C acetone, washed with phosphate-buffered saline, and stained with a monoclonal antibody to β-tubulin conjugated to the fluorescent dye Cy3 (Sigma product C4585), following instructions provided by the manufacturer. The coverglass was mounted on a slide with Antifade Mounting Solution and examined in a Nikon Eclipse E800 microscope with a 100x oil objective and using appropriate epifluorescence accessories. Images were captured with a Spot digital camera. The scale bar shown in panel A represents 20 μm.
In conclusion, we have accomplished total syntheses of both (+)- and (−)-chamaecypanone C. The key transformation involves Diels-Alder cycloaddition between an in situ-generated diarylcyclopentadienone and a chiral ortho-quinol derived from a retro-Diels-Alder reaction of its dimeric form. Initial biological studies indicate that (+)-chamaecypanone C is a potent tumor cell growth inhibitor[3] acting primarily through inhibition of tubulin polymerization. Further studies on preparation of (+)-chamaecypanone C analogues using a retro-DA/DA cascade, as well as biological evaluation of these compounds, are currently in progress and will be reported in due course.
Supplementary Material
{kind=link}
Figure 1.
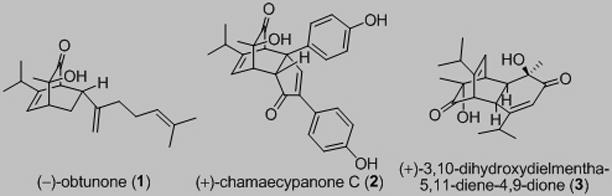
Representative Bicyclo[2.2.2]octenone-Containing Natural Products
Figure 2.
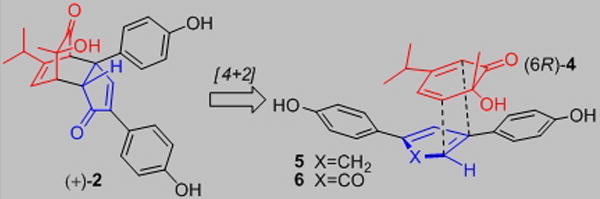
Plausible Biosyntheses of Chamaecypanone C
Footnotes
Financial support from the National Institutes of Health, (GM-073855 and P50 GM067041), Wyeth Pharmaceuticals, Merck Research Laboratories, and the NCI intramural, research program is gratefully acknowledged. We thank the, DTP, NCI for 60-cell assays, ThalesNano, Inc. (Budapest, Hungary) for assistance with the continuous-flow, hydrogenation reactor, and Prof. Dr. Corey Stephenson, (Boston University) for helpful discussions.
Supporting information for this article is available on the, WWW under http://www.angewandte.org.
References
- 1.For reviews on biosynthetic Diels-Alder reactions, see: Stocking EM, Williams RM. Angew Chem. 2003;115:3186–3223. doi: 10.1002/anie.200200534.Angew Chem Int Ed. 2003;42:3078–3115. doi: 10.1002/anie.200200534.Oikawa H, Tokiwano T. Nat Prod Rep. 2004;21:321–352. doi: 10.1039/b305068h.
- 2.Kuo YH, Chen CH, Huang SL. Chem Pharm Bull. 1998;46:181–183. [Google Scholar]
- 3.Chien SC, Chang JY, Kuo CC, Hsieh CC, Yang NS, Kuo YH. Tetrahedron Lett. 2007;48:1567–1569. [Google Scholar]
- 4.Carman RM, Lambert LK, Robinson WT, Van Dongen JMAM. Aust J Chem. 1986;39:1843–1850. [Google Scholar]
- 5.Al-Busafi S, Doncaster JR, Drew MGB, Regan AC, Whitehead RC. J Chem Soc, Perkin Trans. 2002;1:476–484. [Google Scholar]
- 6.Dong S, Zhu J, Porco JA., Jr J Am Chem Soc. 2008;130:2738–2739. doi: 10.1021/ja711018z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For use of ortho-quinol dimers in retro-DA/DA cascade, see: Singh VK, Deota PT, Bedekar AV. J Chem Soc Perkin Trans. 1992;1:903–912.Singh V, Samanta B. Tetrahedron Lett. 1999;40:1807–1810.For use of MOB-derived dimers, see: Liao CC, Peddinti RK. Acc Chem Res. 2002;35:856–866. doi: 10.1021/ar000194n.Chittimalla SK, Shiao HY, Liao CC. Org Biomol Chem. 2006;4:2267–2277. doi: 10.1039/b602928k.
- 8.For reviews on ortho-quinol and MOB-derived [4+2] cyclodimers, see: Magdziak D, Meek SJ, Pettus TRR. Chem Rev. 2004;104:1383–1430. doi: 10.1021/cr0306900.Quideau S, Pouysegu L, Deffieux D. Synlett. 2008:467–495.
- 9.Snider BB, Chen Z. Org Prep Proced Int. 1999;31:537–541. [Google Scholar]
- 10.See Supporting Information for complete experimental details.
- 11.Hafner D, Goliasch K. Angew Chem. 1960;72:781. [Google Scholar]
- 12.Prashad M, Tomesch JC, Wareing JR, Smith HC, Cheon SH. Tetrahedron Lett. 1989;30:2877–2880. [Google Scholar]
- 13.Datta S, Odedra A, Liu RS. J Am Chem Soc. 2005;127:11606–11607. doi: 10.1021/ja053674o. [DOI] [PubMed] [Google Scholar]
- 14.Szymoniak J, Besancon J, Dormond A, Moise C. J Org Chem. 1990;55:1429–1432. [Google Scholar]
- 15.Nicolaou KC, Zhong YL, Baran PS. J Am Chem Soc. 2000;122:7596–7597. [Google Scholar]
- 16.a) Buckle DR. In: Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. Vol. 3. John Wiley & Sons; Chichester, UK: 1995. p. 1699. [Google Scholar]; b) Walker D, Hiebert JD. Chem Rev. 1967;67:153–196. doi: 10.1021/cr60246a002. [DOI] [PubMed] [Google Scholar]
- 17.For syntheses of cyclopentadienones from cyclopentenones, see: Baraldi PG, Barco A, Benetti S, Pollini GP, Polo E, Sinoni D. Chem Commun. 1984:1049–1050.Harmata M, Barnes CL, Brackley J, Bohnert G, Kirchhoefer P, Kuerti L, Rashatasakhon P. J Org Chem. 2001;66:5232–5236. doi: 10.1021/jo015671+. and references therein.
- 18.For representative syntheses of cyclopentadienones, see: Wender PA, Paxton TJ, Williams TJ. J Am Chem Soc. 2006;128:14814–14815. doi: 10.1021/ja065868p. and references therein.For preparation and studies on 2,4-di-tert-butylcyclopentadienone, see: Garbisch EW, Jr, Sprecher RF. J Am Chem Soc. 1969;91:6785–6800.DeSelms RC, Schleigh WR. Synthesis. 1973:614–615.For a total synthesis of the 2,3-substituted cyclopentadienone-derived natural product manzamenone A, see: Al-Busafi S, Doncaster JR, Drew MGB, Regan AC, Whitehead RC. J Chem Soc, Perkin Trans. 2002;1:476–484.
- 19.The presumed cyclopentadienone 28 was also successfully trapped with dimethyl acetylenedicarboxylate (DMAD) to afford a tetrasubstituted aromatic product.[10] For examples of cyclopentadienones as 4π partners in cycloadditions, see: Ban T, Wakita Y, Kanematsu K. J Am Chem Soc. 1980;102:5415–5416.Jikyo T, Eto M, Harano K. J Chem Soc, Perkin Trans. 1998;1:3463–3470.
- 20.For deantiaromatization in electrocyclic reactions, see: Harmata M, Zheng P, Schreiner PR, Navarro-Vazquez A. Angew Chem. 2006;118:2000–2005. doi: 10.1002/anie.200503812.Angew Chem Int Ed. 2006;45:1966–1971.For use of reactive cyclopentadienones in cycloaddition, see: Harmata M, Gomes MG. Eur J Org Chem. 2006:2273–2277.
- 21.Paull KD, Lin CM, Malspeis L, Hamel E. Cancer Res. 1992;52:3892–3900. [PubMed] [Google Scholar]
- 22.Hamel E. Cell Biochem Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 23.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol Pharmacol. 1998;53:62–72. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 24.Chang and coworkers (National Health Research Institutes, Taipei, Taiwan) have also independently demonstrated that chamaecypanone C is a microtubule inhibitor, see: C. C. Hsieh, Y. H. Kuo, C. C. Kuo, C. Y. Chang, S. C. Chien, J. Y. Chang. The 13th Joint Congress of Oncology Societies of Taiwan (Taipei, Taiwan), May 3–4, 2008; Abstract #A–I-18.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.