

Summary
The heart is a force-generating organ that responds to self-generated electrical stimuli from specialized cardiomyocytes. This function is modulated by sympathetic and parasympathetic activity.
In order to contract and accommodate the repetitive morphological changes induced by the cardiac cycle, cardiomyocytes depend on their highly evolved and specialized cytoskeletal apparatus. Defects in components of the cytoskeleton, in the long term, affect the ability of the cell to compensate at both functional and structural levels. In addition to the structural remodeling, the myocardium becomes increasingly susceptible to altered electrical activity leading to arrhythmogenesis. The development of arrhythmias secondary to structural remodeling defects has been noted, although the detailed molecular mechanisms are still elusive. Here I will review the current knowledge of the molecular and functional relationships between the cytoskeleton and ion channels and, I will discuss the future impact of new data on molecular cardiology research and clinical practice.
Keywords: Arrhythmias, Ion Channel, SCN5A, Dystrophin, DGC, Caveolin-3, Cytoskeleton, Syntrophin, Potassium Channels
Electrocardiographic Changes in Heart Failure
Heart failure (HF) is amongst the major causes of death in the USA, with approximately 5 million cases nationwide, including 500,000 new cases each year and more than 800,000 hospitalizations per annum. 1
Myocardial dysfunction in the end-stage failing heart is very often associated with increasing susceptibility to ventricular tachycardia (VT) and ventricular fibrillation (VF), both of which are common causes of sudden cardiac death (SCD).
Among the various forms of HF, myocardial remodeling due to ischemic cardiomyopathy (ICM) or dilated cardiomyopathy (DCM) is characterized by alterations in baseline ECG, which includes the prolongation of the QT interval, as well as QT dispersion, ST-segment elevation, and T-wave abnormalities, especially during exercise. 1 In particular, subjects with severe left ventricular chamber dilation such as in DCM can have left bundle branch block (LBBB), while right bundle branch block (RBBB) is more characteristic of right ventricular failure. In either case, both LBBB and RBBB have been repeatedly associated with AV block in heart failure.1
The impact of volume overload on structural and electrocardiographic alterations has been noted in cardiomyopathy patients treated with left ventricular assist device (LVAD) therapy; this device therapy puts the heart at mechanical rest. In LVAD-treated subjects, QRS- and both QT- and QTc duration decreased, suggesting that QRS- and QT-duration are significantly influenced by mechanical load and that the shortening of the action potential duration contributes to the improved contractile performance after LVAD support. 2 Despite the increasing use of LVAD supporting either continuous or pulsatile blood flow in patients with severe HF, the benefit of this treatment in dealing with the risk of arrhythmias is still controversial.
In several studies on small patients group or single cases, a dramatic improvement in the management of ventricular arrhythmias was achieved employing a continuous blood flow DeBakey LVAD. 3–5
Large epidemiological studies, such as the REMATCH study, demonstrated that the employment of LVAD significantly improved survival rate and the quality of life, in comparison to optimal medical management. 6 An early postoperative period study after cardiac unloading therapy in 17 HF patients showed that in the first two weeks after LVAD implantation, HF was associated with a relatively high incidence of ventricular arrhythmias associated with QTc interval prolongation 7. In addition, a recent retrospective study of 100 adult patients with advanced HF, treated with an axial-flow HeartMate LVAD suggested that the rate of new-onset monomorphic ventricular tachycardia (MVT) was increased in LVAD-treated patients compared to patients given only medical treatment, while no effect was observed on the development of polymorphic ventricular tachycardia (PVT)/ventricular fibrillation (VF) 8.
Despite the unresolved controversy about the efficacy of mechanical unloading on the management of arrhythmias and its support of hemodynamic normalization, it is clear that altered cardiac contractility is associated with unbalanced ion homeostasis.
Contractile Apparatus and Arrhythmogenesis
The sarcomere
The myocardium is exposed to severe and continuous biomechanical stress during each contraction-relaxation cycle. When fiber tension remains uncompensated or simply unbalanced, it may represent a trigger for arrhythmogenesis caused by cytoskeletal stretching, which ultimately leads to altered ion channel localization, and subsequent action potential and conduction alterations. 9
Cytoskeletal proteins not only provide the backbone of the cellular structure, but they also maintain the shape and flexibility of the different sub-cellular compartments, including the plasma membrane, the double lipid layer, which defines the boundaries of the cell and where ion channels are mainly localized. The interaction between the sarcomere, which is the basic contractile unit of striated muscles, and the sarcolemma, the plasma membrane surrendering the muscle fibers in skeletal muscle and the muscle cell of the cardiomyocyte, determines the mechanical plasticity of the cell, enabling it to complete and re-initiate each contraction-relaxation cycle.
At the level of the sarcomere, actin (thin) and myosin (thick) filaments generate the contractile force, while other components such as titin, the largest protein known to date, are responsible for the passive force during diastole and for the restoring force during systole. Titin connects the Z-line to the M-line of the sarcomeric structure (Figure 1). 10 In addition to the strategic localization and mechanical spring function, titin also acts as a length-dependent sensor during stretch and promotes actin-myosin interaction. 11 Titin is stabilized by the cross-linking protein telethonin (T-Cap), which localizes at the Z-line and is also part of titin sensor machinery (Figure 1). 12 The complex protein interactions in the sarcomere entwine telethonin to other Z-line components through the family of the telethonin-binding proteins of the Z-disc, FATZ, also known as calsarcin and myozenin. FATZ binds to calcineurin, γ-filamin as well as the spectrin-like repeats (R3–R4) of α-actinin-2, the major component of the Z-line and a pivotal F-actin cross-linker (Figure 1).13
Figure 1. Sarcomere structure.
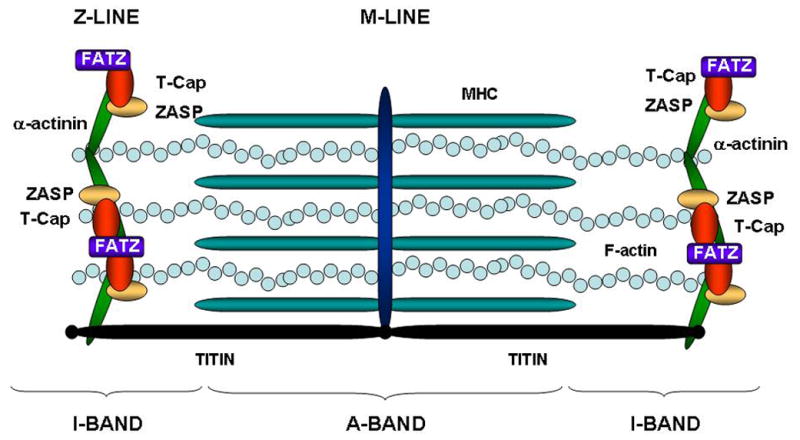
The diagram illustrates the sarcomeric structure. The Z-line determines the boundaries of the contractile unit, while Titin connects the Z-line to the M-line and acts as a functional spring during contraction/relaxation cycles.
Another cross-linker of α-actinin-2 in the complex Z-line scaffold is the Z-band alternatively spliced PDZ motif protein (ZASP), which has an important role in maintaining Z-disc stability in skeletal and cardiac muscle (Figure 1). 14 ZASP contains a PDZ motif at its N-terminus, which interacts with C-terminus of α-actinin-2, 12 and a conserved sequence called the ZASP-like motif (ZM) found in the alternatively spliced exons 4 and 6. 15 It has also been reported to bind to the FATZ (calsarcin) family of Z-disc proteins 16
Sarcomeric Proteins and Ion Channels
Safeguarding the structural integrity of the sarcomere is essential to maintain the mechanical properties of the contractile unit of the muscle, and failure to preserve sarcomeric organization has been associated with the development of cardiomyopathies in humans 17.
In addition to systolic dysfunction characteristic of dilated cardiomyopathy (DCM) and diastolic dysfunction featuring hypertrophic cardiomyopathy (HCM), the clinical phenotype of patients with severe cardiomyopathy is very often associated with a high incidence of cardiac arrhythmias. 1 Therefore, besides fiber stretch associated with mechanical and hemodynamic impairment, cytoskeletal alterations due to primary genetic defects or indirectly to alterations in response to cellular injury can potentially affect ion channel anchoring, and trafficking, as well as functional regulation by second messenger pathways, causing an imbalance in cardiac ionic homeostasis that will trigger arrhythmogenesis.
In the last few years, intense investigation of the sarcomeric actin network, the Z-line structure, and chaperone molecules docking in the plasma membrane, has shed new light on the molecular basis of cytoskeletal interactions in regulating ion channels. In 1991, Cantiello et al., demonstrated that although the epithelial sodium channel and F-actin are in close proximity, they do not co-localize. 18 Actin disruption using cytochalasin D, an agent that interferes with actin polymerization, increased Na+ channel activity in 90% of excised patches tested within 2 min, which indicated that the integrity of the filamentous actin (F-actin) network was essential for the maintenance of normal Na+ channel function. 18 Later, the group of Dr. Jonathan Makielski studied the action of cytochalasin D in rat and rabbit ventricular cardiomyocytes, and demonstrated that actin disruption induced a dramatic reduction in Na+ peak current and slowed current decay without affecting steady-state voltage-dependent availability or recovery from inactivation. These data were the first to support a role for the cytoskeleton in cardiac arrhythmias. 19
As previously described, F-actin is intertwined in a multi-protein complex that includes the composite Z-line structure. Interestingly, recent investigations show a direct binding between the major protein of the Z-line, α-actinin-2 and the voltage-gated K+ channel 1.5 (Kv1.5), 20 (Figure 2). The latter is expressed in human cardiomyocytes and localizes to the intercalated disk of the cardiomyocyte in association with connexin 43 and N-cadherin. 21 In their experiments, Maruoka et al. treated HEK293 cells stably expressing Kv1.5 with cytochalasin D, which led to a massive increase in ionic and gating IK+ currents. 20 The observed phenomenon was entirely prevented by pre-incubation with phalloidin, an F-actin stabilizing agent. 20 In addition, the Z-line protein telethonin binds to the cytoplasmic domain of minK, the beta subunit of the potassium channel KCNQ1 (Figure 2). 22 KCNQ1 is known to generate the slow rectifier current IKs involved in phase 3 of the cardiac action potential, and it has been implicated in long QT syndromes (LQTS). 23
Figure 2. Molecular interactions between the cytoskeleton and ion channels.
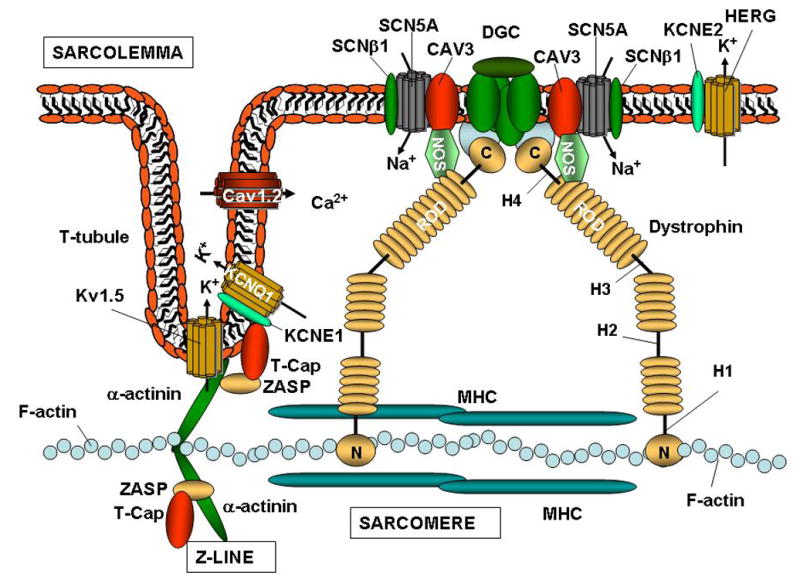
The figure illustrates the interactions between the ion channels on the sarcolemma, and the sarcomere in cardiac myocytes. Note that the Z-line is connected to the cardiac T-tubules. The diagram illustrates the complex protein-protein interactions that occur between structural components of the cytoskeleton and ion channels. The cytoskeleton is involved in regulating the metabolism of ion channels, modifying their expression, localization, and electrical properties. The cardiac sodium channel Nav1.5 associates with the DGC, while potassium channels such as Kv1.5, associate with the Z-line.
Interestingly, mutations in Z-line proteins T-Cap, MLP, 24 and ZASP 25 have been identified in patients with cardiomyopathy associated with some form of arrhythmias, suggesting that altered connection between the Z-line, T-tubules and ion channels may represent an arrhythmogenic mechanism in structurally damaged hearts (Figure 2). T-tubules are invaginations of the plasma membrane in striated muscle,
From the Cytoplasm to the Cell Membrane: Ion Trafficking
Ion Channel Subunits and Trafficking
Correct localization is essential for ion channel function and this is dependent upon the ability of auxiliary proteins to shuttle ion channels from the cytoplasm to their final destination such as the plasma membrane or other sub-cellular compartments. In this regard, Kvβ-subunits are cytoplasmic components known to assemble with the α-subunits of voltage-dependent K+ (Kv) channels at their N-terminus to form stable Kvα/β hetero-oligomeric channels. 26 When Kvβ is co-expressed with Kv1.4 or Kv1.5, it enhances Kv1.x channel trafficking to the cell membrane without changing the overall protein channel content. 27, 28 The regulatory Kvβ subunits, which are also expressed in cardiomyocytes, directly decrease K+ current by accelerating Kv1.x channel inactivation. 29,30 Therefore, altered expression or mutations in Kvβ subunits could cause abnormal ion channel transport to the cell surface, by reducing the number of channel units on the plasma membrane or by interfering with Kv1.x voltage dependence modulation and inactivation kinetics, thereby increasing the risk of cardiac arrhythmias.
Ion Channel Protein Motifs and Trafficking
Cell membrane trafficking in the Kv1.x family may occur in a Kvβ subunit-independent manner through specific motifs in their C-terminus. 31 Analysis of cell surface expression of truncated Kv1.4 channel proteins generated by deletion of sequential protein domains has shown the VXXSL motif to be sufficient for plasma membrane transport. 31 The LXXSL pattern confers the same ability to Kv1.5 channel proteins, although at lower efficiency. 31 Interestingly, Kv1.2, another component of the family contains the similar VXXSN motif, but requires Kvβ subunits for enhanced surface expression. 32 Mutagenesis of the final asparagine (N) in the Kv1.2 motif restores the leucine (L) of the Kv1.4 motif re-establishing high expression levels at the plasma membrane in a Kvβ-independent manner. 32
The existence of these specific Kv1.x protein motifs involved in ion channel cell surface transport suggests that genetic mutations changing any amino acidic residue in the VXXSN motif may alter channel trafficking, channel membrane density, and overall current, increasing abnormal ion homeostasis and susceptibility to arrhythmogenesis. Although in humans no such mutations have yet been found, they must be regarded as possible candidate genes for arrhythmias in cases with altered cardiac repolarization.
Cytoskeletal Proteins and Ion Channel Trafficking
Until recently, primary arrhythmias such as LQTS have been almost exclusively regarded as ion channelopathies. Mutations in genes encoding the cardiac Na+ channel Nav1.5 (SCN5A), 33 K+ channels KCNQ1, KCNH2 and KCNJ2 (Kir 2.1) and the ancillary subunits KCNE1, KCNE2, 34,35,36,37,38,39 as well as the alpha 1C subunit of the cardiac L-type calcium channel (CACNA1C) 40,41 have previously been linked to the prolongation of the QT-interval in humans.
However, in the last few years, the conviction that primary mutations in ion channels were solely responsible for the electrical defects associated with arrhythmias has been shaken by the identification of mutations in the ANK2 gene encoding the cytoskeletal protein ankyrin-B that is associated with LQTS in animal models and humans. 42,43
Ankyrin-B acts as a chaperone protein, which shuttles the cardiac sodium channel from the cytoplasm to the membrane. Immunohistochemical analysis has localized ankyrin-B to the Z-lines/T-tubules on the plasma membrane in the myocardium. Mutations in ankyrin-B associated with LQTS alter sodium channel trafficking due to loss of ankyrin-B localization at the Z-line/transverse (T)-tubules, while retaining expression at the M-line and intercalated disc level. 42 Reduced levels of ankyrin-B at cardiac Z-lines/T-tubules were associated with the deficiency of ankyrin-B-associated proteins such as Na/K-ATPase, Na/Ca exchanger (NCX) and inositol-1, 4, 5-trisphosphate receptors (InsP3R).
It remains to be understood whether the prolonged QT-interval and the development of arrhythmias is caused by reduced sodium current (INa+) due to Nav1.5 altered transport to the sarcolemma or alternatively by the dowregulation of Na/K-ATPase, NCX or IP3R. In either case, these data emphasize the importance of the sarcomere/sarcolemma connection through the Z-line/T-tubule as a physical anchorage, and a trafficking dock for ion channels in the plasma membrane. Therefore, disturbances of the sarcomere/sarcolemma link could result in action potential abnormalities, leading to a high risk of arrhythmogenesis.
An alteration in ion channel trafficking may not allow the ion channel to leave the endoplasmic reticulum (ER) where it is synthesized. An example of this is the R1432G mutation in Nav1.5 identified in a subject with Brugada syndrome (BrS), which resulted in the mutated Nav1.5 being sequestered by calnexin in the ER. 44 Given the observation that mutated ion channels retain normal kinetics but undergo abnormal metabolism and altered interaction with cytoskeletal components, it is not surprising that Nav1.5 mutations were recently identified in patients with severe autosomal dominant cardiac conduction disorders associated with sinus node dysfunction, arrhythmia, and associated DCM. 45, 46 These findings broaden the phenotypic outcome of sodium channel mutations, and emphasize the complex relationship between the cytoskeleton and ion channels, suggesting that disturbances in either component could account for arrhythmogenesis in the presence or absence of structural heart disease.
From the Sarcomere to the Sarcolemma: The Dystrophin Glycoprotein Complex (DGC)
Muscle contraction occurs at the sarcomeric level, but in order to be efficient the force generated in the contractile unit of the muscle must be transmitted to the entire muscle cell including the sarcolemma. In addition, synchronized contraction is essential for cardiomyocytes, which are connected to each other via the extracellular matrix (ECM) through the DGC.
Dystrophin is the major component of the DGC. The N-terminus domain of dystrophin binds F-actin, and connects it to the sarcomere, while the cysteine-rich (CR) C-terminus domain ensures its connection to the sarcolemma (Figure 2). The central portion of dystrophin, the rod domain, is composed of rigid spectrin-like repeats and four hinge portions (H1–H4) that determine the flexibility of the protein. 47 Recently, it has been demonstrated that, in addition to the N-terminus, dystrophin possesses another F-actin binding domain in the Rod domain region, between the basic repeats 11–17 (DysN-R17). 48 A truncated form of dystrophin containing the N-terminus along with the DysN-R17 could bind F-actin with the same efficiency of the full-length protein. 48 This suggests that, DysN-R17 not only contains two F-actin binding domains, but also provides a sufficient three-dimensional shape needed to effectively stabilize its connection to the actin network.
Dystrophin, the DGC and Ion Channels
Dystrophin, originally identified as the gene responsible for Duchenne and Becker muscular dystrophies (DMD/BMD), 49 and later for the X-linked form of dilated cardiomyopathy (XLCM), 50 exerts a major function in physical force transmission in striated muscle. 51
In addition to its structural significance, dystrophin and other DGC proteins such as syntrophins are required for the correct localization, clustering and regulation of ion channel function. Loss of dystrophin function in the mdx-mouse leads to altered spatial ion channel distribution in myocytes and a decreased density in the voltage-gated skeletal sodium channel (SkM1) in the extrajunctional sarcolemma that is associated with a reduction in the number of SkM1-rich fast-twitch IIb fibers in mdx muscle. 52 In addition, lowered sarcolemmal labeling for SkM1 was found in all mdx fibers independent of their metabolic subtype. 52 Although these studies were performed in skeletal muscle, the observations are intriguing, suggesting a possible role for dystrophin in altering cardiac ion channel metabolism..
In addition to dystrophin, the dystrophin-associated proteins, syntrophins, have also been implicated in ion channel regulation. 53, 54, 55 Syntrophins contain two pleckstrin homology (PH) domains, a PDZ domain, and a syntrophin-unique (SU) C-terminal region. The interaction between syntrophins and dystrophin occurs at the PH domain distal to the syntrophin N-terminus and through the highly conserved SU domain. 56, 57 Conversely, the PH domain proximal to the N-terminal portion of the protein and the PDZ domain interact with other membrane components such as phosphatidyl inositol-4, 5-bisphosphate, 58 neuronal NOS (nNOS), 59 aquaporin-4, 60 stress-activated protein kinase-3, 61 and Nav1.5, 62 thereby linking all these molecules to the dystrophin complex (Figure 2). 63
Among the five known isoforms of syntrophin, the 59 KDa α1-syntrophin isoform is the most highly represented in human heart, whereas in skeletal muscle it is only present on the sarcolemma of fast type II fibers. 64 In addition, the skeletal muscle γ2-syntrophin was found at high levels only at the postsynaptic membrane of the neuromuscular junctions. 65, 66 Alsoγ2-syntrophin has been detected in skeletal muscle, where it has been reported to localize to the sarcolemma 55 and to be able to bind the C-terminus of Nav1.5 through its PDZ domain, regulating the gating properties of the sodium channel. 67
In addition to syntrophin, other scaffolding proteins such as caveolin-3 (CAV3), which is present in the caveolae, flask-shaped plasma membrane microdomains, are involved in signal transduction and vesicle trafficking in myocytes, modulating cardiac remodeling during heart failure. 68 CAV3 and α1-syntrophin, localizes at the T-tubule and are part of the DGC. 69 In addition, α1-syntrophin binds Nav1.5, while caveolin-3 binds the Na+/Ca2+ exchanger, 70 Nav1.5 and the L-type Ca2+ channel 68 as well as nNOS and the DGC (Figure 2). 71, 72, 73 Therefore, α1-syntrophin, through its binding to F-actin, nNOS, and Nav1.5, appears to be involved in regulating structural and electrical functions as well as signal transduction in heart failure. 68
All these findings suggest that, although ankyrin-B is the only protein found mutated in patients with primary arrhythmias, other proteins such as caveolin-3 and the syntrophins if mutated may alter ion channel function. Due to the structural importance of the cytoskeleton, genetic variants or acquired abnormalities causing the loss of cytoskeletal integrity may lead to the progressive dysfunction observed in heart failure and the possible delocalization or abnormal regulation of ion channel expression, leading to ventricular arrhythmias.
Conclusions and Future Perspectives
The increasing biological and genetic complexity of the regulation of cardiac contractile and electrical function has been recognized by clinical investigators. It is important to be aware of the enormous variety of clinical presentations that derive from distinct variants in the same pool of genetic factors. Knowledge of these variants could facilitate tailoring the therapy of choice for each patient. In particular, the recent findings of structural and functional links between the cytoskeleton and ion channels could expand the therapeutic interventions in arrhythmia management in structurally abnormal myocardium, where aberrant binding between cytoskeletal proteins can directly or indirectly alter ion channel function.
It is likely that mutations or polymorphisms in structural proteins will soon be recognized to be modulators of arrhythmogenesis in the failing heart through their ability to directly condition ion channel function or to alter ion channel distribution and metabolism thus confering higher susceptibility to myocardial remodeling. In addition, functional polymorphisms in ion channels are likely to be genetic determinants of primary and secondary arrhythmogenesis due to altered ion channel anchorage to the cytoskeleton, which can be an arrhythmogenic trigger, even in a hemodynamically normal heart.
In this regard, genetic abnormalities in cytoskeletal proteins could also alter ion channel function and vice versa, suggesting a more complex genetic background in the pathogenesis of primary and secondary arrhythmias. Therefore, a broader spectrum of candidate genes, mutations, and molecular mechanisms leading to cardiac diseases could not only change the basic cardiovascular research perspective, but also the therapeutic approach to patient management, which ideally should be tailored to the subject’s specific genetic characteristics. Although further studies are needed to demonstrate the impact of cytoskeletal changes in the development of arrhythmias, the potential consequences of these phenomena could be of enormous importance for patients’ management. Finally, the increasing number of genetic targets for pathological mutations leading to rhythm disorders will help to explain the variable phenotypic penetrance as well as the diverse pharmacological response of subjects with heart failure and arrhythmias.
Executive Summary
Arrhythmogenesis and myocardial structure
Rhythm alterations can develop as a secondary consequence of myocardial structural abnormalities or as a result of a primary defect in the cardiac electric machinery.
Until recently, no molecular mechanism has been able to fully explain the occurrence of arrhythmogenesis in heart failure, however genetic defects that are found almost exclusively in ion channel genes account for the majority of primary arrhythmias such as long QT syndromes and Brugada syndrome.
The contractile apparatus is linked to ion channels
The sarcomere, which represents the contractile unit of the myocardium not only generates the mechanical force necessary to exert the pump function, but also provides localization and anchorage to ion channels.
Alpha-actinin-2, and telethonin, two members of the Z-line scaffolding protein complex in the striated muscle associate with the potassium voltage-gated channel alpha subunit Kv1.5 and the beta subunit KCNE1 respectively.
Mutations in KCNE1 have previously been associated with the development of arrhythmias in LQTS subjects.
Mutations in both alpha-actinin-2, and telethonin were identified in individuals with cardiomyopathy. The primary defect is structural leading to ventricular dysfunction, but the secondary consequence is arrhythmia.
Ion channel trafficking and sub-cellular compartments
Ion channel trafficking from the endoplasmic reticulum (ER) to the Golgi complex is an important check-point for regulating the functional channel molecules on the plasma membrane. Several molecules acting as chaperones bind to and shuttle the channel proteins to their final localization on the cell surface
Ion channel subunits such as Kvβ enhance Kv1.x ion channel presentation on the sarcolemma. The α subunits of the Kv1.x potassium channels can be shuttled in a Kvβ-independent manner through specific sequence motif at Kv1.x protein level.
In addition, cytoskeletal proteins such as ankyrin-G bind Nav1.5 and are involved in the sodium channel trafficking. Another member of the ankyrin family, ankyrin-B was found mutated in patients with LQTS but the pathological mechanism of ankyrin-B mutations is still obscure, although the sodium current intensity is dramatically reduced.
The sarcolemma and ion channels
The sarcolemma contains a wide range of ion channels, which are responsible for the electrical propagating force in the myocardium.
The DGC is a protein complex, which forms a scaffold for cytoskeletal components and ion channels.
Dystrophin is the major component of the DGC and mutations in dystrophin and DGC cause muscular dystrophies and X-linked cardiomyopathies (XLCM) in humans. Cardiomyopathies are associated with arrhythmias
Caveolin-3 and syntrophins associate with Nav1.5, and are part of the DGC. Syntrophins can directly modulate Nav1.5 channel function.
Conclusions
The role of the cytoskeleton in ion channel function has been hypothesized in the past, but only recently the mechanism underlying the development of arrhythmias in structurally impaired myocardium has become clearer.
The recently acknowledged role of the cytoskeleton in ion channel function suggests that genes encoding cytoskeletal proteins should be regarded as potential candidates for variants involved in the susceptibility to arrhythmias, as well as the primary target of genetic mutations in patients with arrhythmogenic syndromes such as LQTS and Brugada syndrome.
Studies of genotype-phenotype correlation and patient risk stratification for mutations in cytoskeletal proteins will help to tailor the therapy and management of patients with arrhythmias.
References
- 1*.Hombach V. Electrocardiogram of the failing heart. Card Electrophysiol Rev. 2002 Sep;6(3):209–214. doi: 10.1023/a:1016316706195. It is a comprehensive review of ECG abnormalities in the failing heart. [DOI] [PubMed] [Google Scholar]
- 2.Harding JD, Piacentino Vr, Gaughan JP, et al. Electrophysiological alterations after mechanical circulatory support in patients with advanced cardiac failure. Circulation. 2001 Sep 11;104(11):1241–1247. doi: 10.1161/hc3601.095718. [DOI] [PubMed] [Google Scholar]
- 3.Iqbal I, Ventura HO, Smart FW, et al. Difficult cases in heart failure: Left ventricular assist device implantation for the treatment of recurrent ventricular tachycardia in end stage heart failure. Congest Heart Fail. 1999 May;5(3):129–130. [PubMed] [Google Scholar]
- 4.Maile S, Kunz M, Oechslin E, et al. Intractable ventricular tachycardia and bridging to heart transplantation with a non-pulsatile flow assist device in a patient with isolated left-ventricular non-compaction. J Heart Lung Transplant. 2004 Jan;23(1):147–149. doi: 10.1016/s1053-2498(03)00101-3. [DOI] [PubMed] [Google Scholar]
- 5.Salzberg SP, Lachat ML, Zund G, et al. Left ventricular assist device (LVAD) enables survival during 7 h of sustained ventricular fibrillation. Eur J Cardiothorac Surg. 2004 Aug;26(2):444–446. doi: 10.1016/j.ejcts.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Rose EA, Gelijns AC, Moskowitz AJ, et al. Long-term mechanical left ventricular assistance for end-stage heart failure. N Engl J Med. 2001 Nov 15;345(20):1435–1443. doi: 10.1056/NEJMoa012175. [DOI] [PubMed] [Google Scholar]
- 7.Harding JD, Piacentino V, 3rd, Rothman S, et al. Prolonged repolarization after ventricular assist device support is associated with arrhythmias in humans with congestive heart failure. J Card Fail. 2005 Apr;11(3):227–232. doi: 10.1016/j.cardfail.2004.08.158. [DOI] [PubMed] [Google Scholar]
- 8.Ziv O, Dizon J, Thosani A, et al. Effects of left ventricular assist device therapy on ventricular arrhythmias. J Am Coll Cardiol. 2005 May 3;45(9):1428–1434. doi: 10.1016/j.jacc.2005.01.035. [DOI] [PubMed] [Google Scholar]
- 9*.Kamkin A, Kiseleva I, Isenberg G. Stretch-activated currents in ventricular myocytes: amplitude and arrhythmogenic effects increase with hypertrophy. Cardiovasc Res. 2000 Dec 2000;48(3):409–420. doi: 10.1016/s0008-6363(00)00208-x. This report provides evidence of the role of fiber stretch in the development of arrhythmias in cardiac hypertrophy. [DOI] [PubMed] [Google Scholar]
- 10.Agarkova I, Perriard JC. The M-band: an elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 2005 Sep;15(9):477–485. doi: 10.1016/j.tcb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Lange S, Himmel M, Auerbach D, et al. Dimerisation of myomesin: implications for the structure of the sarcomeric M-band. J Mol Biol. 2005 Jan 14;345(2):289–298. doi: 10.1016/j.jmb.2004.10.040. [DOI] [PubMed] [Google Scholar]
- 12**.Faulkner G, Pallavicini A, Formentin E, et al. ZASP: a new Z-band alternatively spliced PDZ-motif protein. J Cell Biol. 1999 Jul 26;146(2):465–475. doi: 10.1083/jcb.146.2.465. The ZASP gene and its alternative splicing isoforms, are an example of the complexity of the proteins found in the Z-line. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Faulkner G, Pallavicini A, Comelli A, et al. FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J Biol Chem. 2000 Dec 29;275(52):41234–41242. doi: 10.1074/jbc.M007493200. The discovery of the FATZ family of Z-line proteins. [DOI] [PubMed] [Google Scholar]
- 14*.Zhou Q, Ruiz-Lozano P, Martone ME, et al. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. J Biol Chem. 1999 Jul 9;274(28):19807–19813. doi: 10.1074/jbc.274.28.19807. The association of Cypher, the murine ZASP homolog, with alpha actinin. [DOI] [PubMed] [Google Scholar]
- 15.Klaavuniemi T, Ylanne J. Zasp/Cypher internal ZM-motif containing fragments are sufficient to co-localize with alpha-actinin-Analysis of patient mutations. Exp Cell Res. 2006 Feb 11; doi: 10.1016/j.yexcr.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 16.Frey N, Olson EN. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J Biol Chem. 2002 Apr 19;277(16):13998–14004. doi: 10.1074/jbc.M200712200. [DOI] [PubMed] [Google Scholar]
- 17**.Towbin JA, Bowles NE. The failing heart. Nature. 2002 Jan 10;415(6868):227–233. doi: 10.1038/415227a. The first investigation about the role of the cytoskeleton in ion channel function. [DOI] [PubMed] [Google Scholar]
- 18**.Cantiello HF, Stow JL, Prat AG, et al. Actin filaments regulate epithelial Na+ channel activity. Am J Physiol. 1991 Nov;261(5 Pt 1):C882–888. doi: 10.1152/ajpcell.1991.261.5.C882. The first report of sodium channel dysfunction following actin network disruption in cardiac cells. [DOI] [PubMed] [Google Scholar]
- 19*.Undrovinas AI, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. Am J Physiol. 1995 Jul;269(1 Pt 2):H203–214. doi: 10.1152/ajpheart.1995.269.1.H203. The first report demonstrating association between actinin-2 and Kv1.5. [DOI] [PubMed] [Google Scholar]
- 20.Maruoka ND, Steele DF, Au BP, et al. alpha-actinin-2 couples to cardiac Kv1.5 channels, regulating current density and channel localization in HEK cells. FEBS Lett. 2000 May 12;473(2):188–194. doi: 10.1016/s0014-5793(00)01521-0. [DOI] [PubMed] [Google Scholar]
- 21*.Mays DJ, Foose JM, Philipson LH, et al. Localization of the Kv1.5 K+ channel protein in explanted cardiac tissue. J Clin Invest. 1995 Jul;96(1):282–292. doi: 10.1172/JCI118032. The first report demonstrating association between telethonin and KCNE1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furukawa T, Ono Y, Tsuchiya H, et al. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol. 2001 Nov 2;313(4):775–784. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- 23**.Vatta M, Li H, Towbin JA. Molecular biology of arrhythmic syndromes. Curr Opin Cardiol. 2000 Jan;15(1):12–22. doi: 10.1097/00001573-200001000-00003. The first report showing that mutations in MLP and telethonin are associated with cardiomyopathy in humans and providing evidence of their role in mechanical sensing. [DOI] [PubMed] [Google Scholar]
- 24*.Knoll R, Hoshijima M, Hoffman HM, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002 Dec 27;111(7):943–955. doi: 10.1016/s0092-8674(02)01226-6. The first report identifying ZASP mutations in subjects with cardiomyopathy. [DOI] [PubMed] [Google Scholar]
- 25.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003 Dec 3;42(11):2014–2027. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 26*.Kuryshev YA, Wible BA, Gudz TI, et al. KChAP/Kvbeta1.2 interactions and their effects on cardiac Kv channel expression. Am J Physiol Cell Physiol. 2001 Jul;281(1):C290–299. doi: 10.1152/ajpcell.2001.281.1.C290. An elegant study demonstrating that Kvβ subunits are involved in ion channel trafficking. [DOI] [PubMed] [Google Scholar]
- 27.Shi G, Nakahira K, Hammond S, et al. Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron. 1996 Apr;16(4):843–852. doi: 10.1016/s0896-6273(00)80104-x. [DOI] [PubMed] [Google Scholar]
- 28.Peri R, Wible BA, Brown AM. Mutations in the Kv beta 2 binding site for NADPH and their effects on Kv1.4. J Biol Chem. 2001 Jan 5;276(1):738–741. doi: 10.1074/jbc.M008445200. [DOI] [PubMed] [Google Scholar]
- 29.Lombardi SJ, Truong A, Spence P, et al. Structure-activity relationships of the Kvbeta1 inactivation domain and its putative receptor probed using peptide analogs of voltage-gated potassium channel alpha- and beta-subunits. J Biol Chem. 1998 Nov 13;273(46):30092–30096. doi: 10.1074/jbc.273.46.30092. [DOI] [PubMed] [Google Scholar]
- 30*.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005 Oct;85(4):1205–1253. doi: 10.1152/physrev.00002.2005. The demonstration of a protein motif involved in ion channel plasma membrane transport in a Kvβ –independent manner. [DOI] [PubMed] [Google Scholar]
- 31.Li D, Takimoto K, Levitan ES. Surface expression of Kv1 channels is governed by a C-terminal motif. J Biol Chem. 2000 Apr 21;275(16):11597–1160232. doi: 10.1074/jbc.275.16.11597. [DOI] [PubMed] [Google Scholar]
- 32.Griffith LC. Potassium channels: the importance of transport signals. Curr Biol. 2001 Mar 20;11(6):R226–228. doi: 10.1016/s0960-9822(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 33.Jiang C, Atkinson D, Towbin JA, et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat Genet. 1994;8(2):141–147. doi: 10.1038/ng1094-141. [DOI] [PubMed] [Google Scholar]
- 34.Keating M, Dunn C, Atkinson D, et al. Consistent linkage of the long-QT syndrome to the Harvey ras-1 locus on chromosome 11. Am J Hum Genet. 1991;49(6):1335–1339. [PMC free article] [PubMed] [Google Scholar]
- 35.Barhanin J, Lesage F, Guillemare E, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 36.Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 37.Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97(2):175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 38.Derst C, Karschin C, Wischmeyer E, et al. Genetic and functional linkage of Kir5.1 and Kir2.1 channel subunits. FEBS Lett. 2001 Mar 2;491(3):305–311. doi: 10.1016/s0014-5793(01)02202-5. [DOI] [PubMed] [Google Scholar]
- 39.Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001 May 18;105(4):511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 40.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004 Oct 1;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 41**.Splawski I, Timothy KW, Decher N, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005 Jun 7;102(23):8089–8096. doi: 10.1073/pnas.0502506102. discussion 8086–8088. The first demonstration that mutations in the cytoskeletal protein ankyrin-B are found in patients with LQTS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003 Feb 6 2003;421(6923):634–639. doi: 10.1038/nature01335. An elegant study creating the first animal model for cytoskeletal protein ablation showing that it causes prolonged QT interval and suggesting that ankyrin-B is a candidate gene for LQTS. [DOI] [PubMed] [Google Scholar]
- 43*.Chauhan VS, Tuvia S, Buhusi M, et al. Abnormal cardiac Na(+) channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ Res. 2000 Mar 3;86(4):441–447. doi: 10.1161/01.res.86.4.441. The first demonstration of Nav1.5 trafficking abnormalities. [DOI] [PubMed] [Google Scholar]
- 44*.Baroudi G, Pouliot V, Denjoy I, et al. Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G) Circ Res. 2001 Jun 22 2001;88(2):E78–83. doi: 10.1161/hh1201.093270. The first demonstration of SCN5A mutations in DCM. [DOI] [PubMed] [Google Scholar]
- 45.McNair WP, Ku L, Taylor MR, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004 Oct 12;110(15):2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- 46.Olson TM, Michels VV, Ballew JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005 Jan 26;293(4):447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blake DJ, Weir A, Newey SE, et al. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002 Apr;82(2):291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 48.Rybakova IN, Humston JL, Sonnemann KJ, et al. Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem. 2006 Feb 13; doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- 49*.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. First demonstration that linked dystrophin to XLCM. [DOI] [PubMed] [Google Scholar]
- 50.Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87(6):1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 51*.Brown SC, Lucy JA. Dystrophin as a mechanochemical transducer in skeletal muscle. Bioessays. 1993;15:413–419. doi: 10.1002/bies.950150608. A report showing that the skeletal muscle of mdx mice has altered sodium channel localization and content. [DOI] [PubMed] [Google Scholar]
- 52.Ribaux P, Bleicher F, Couble ML, et al. Voltage-gated sodium channel (SkM1) content in dystrophin-deficient muscle. Pflugers Arch. 2001 Mar;441(6):746–755. doi: 10.1007/s004240000483. [DOI] [PubMed] [Google Scholar]
- 53.Ahn AH, Yoshida M, Anderson MS, et al. Cloning of human basic A1, a distinct 59-kDa dystrophin-associated protein encoded on chromosome 8q23–24. Proc Natl Acad Sci U S A. 1994 May 10;91(10):4446–4450. doi: 10.1073/pnas.91.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams ME, Dwyer TM, Dowler LL, et al. Mouse alpha 1- and beta 2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J Biol Chem. 1995 Oct 27;270(43):25859–25865. doi: 10.1074/jbc.270.43.25859. [DOI] [PubMed] [Google Scholar]
- 55.Piluso G, Mirabella M, Ricci E, et al. Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J Biol Chem. 2000 May 26;275(21):15851–15860. doi: 10.1074/jbc.M000439200. [DOI] [PubMed] [Google Scholar]
- 56.Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995 Feb. 1995;128(3):363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kachinsky AM, Froehner SC, Milgram SL. A PDZ-containing scaffold related to the dystrophin complex at the basolateral membrane of epithelial cells. J Cell Biol. 1999 Apr 19;145(2):391–402. doi: 10.1083/jcb.145.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chockalingam PS, Gee SH, Jarrett HW. Pleckstrin homology domain 1 of mouse alpha 1-syntrophin binds phosphatidylinositol 4,5-bisphosphate. Biochemistry. 1999 Apr 27;38(17):5596–5602. doi: 10.1021/bi982564+. [DOI] [PubMed] [Google Scholar]
- 59.Brenman JE, Chao DS, Gee SH, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996 Mar 8;84(5):757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 60.Frigeri A, Nicchia GP, Verbavatz JM, et al. Expression of aquaporin-4 in fast-twitch fibers of mammalian skeletal muscle. J Clin Invest. 1998 Aug 15. 1998;102(4):695–703. doi: 10.1172/JCI2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hasegawa M, Cuenda A, Spillantini MG, et al. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem. 1999 Apr 30 1999;274(18):12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- 62.Gee SH, Madhavan R, Levinson SR, et al. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998 Jan 1;18(1):128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams ME, Mueller HA, Froehner SC. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J Cell Biol. 2001 Oct 1;155(1):113–122. doi: 10.1083/jcb.200106158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peters MF, Adams ME, Froehner SC. Differential association of syntrophin pairs with the dystrophin complex. J Cell Biol. 1997 Jul 14;138(1):81–93. doi: 10.1083/jcb.138.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peters MF, Kramarcy NR, Sealock R, et al. beta 2-Syntrophin: localization at the neuromuscular junction in skeletal muscle. Neuroreport. 1994 Aug 15;5(13):1577–1580. [PubMed] [Google Scholar]
- 66*.Kramarcy NR, Sealock R. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci. 2000 Mar 2000;15(3):262–274. doi: 10.1006/mcne.1999.0823. The first demonstration of gamma 2 syntrophin regulating the gating properties of Nav1.5. [DOI] [PubMed] [Google Scholar]
- 67.Ou Y, Strege P, Miller SM, et al. Syntrophin gamma 2 regulates SCN5A gating by a PDZ domain-mediated interaction. J Biol Chem. 2003 Jan 17;278(3):1915–1923. doi: 10.1074/jbc.M209938200. [DOI] [PubMed] [Google Scholar]
- 68.Feron O, Kelly RA. Gaining respectability: membrane-delimited, caveolar-restricted activation of ion channels. Circ Res. 2002 Mar 8;90(4):369–370. doi: 10.1161/01.res.0000012911.90134.ef. [DOI] [PubMed] [Google Scholar]
- 69.Parton RG, Way M, Zorzi N, et al. Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol. 1997 Jan 13;136(1):137–154. doi: 10.1083/jcb.136.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bossuyt J, Taylor BE, James-Kracke M, et al. Evidence for cardiac sodium-calcium exchanger association with caveolin-3. FEBS Lett. 2002 Jan 30;511(1–3):113–117. doi: 10.1016/s0014-5793(01)03323-3. [DOI] [PubMed] [Google Scholar]
- 71.Venema VJ, Ju H, Zou R, et al. Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J Biol Chem. 1997 Nov 7;272(45):28187–28190. doi: 10.1074/jbc.272.45.28187. [DOI] [PubMed] [Google Scholar]
- 72.Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989 Mar 16;338(6212):259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 73.Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002 Jun;12(3):349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]