

Abstract
Staphylococcus aureus produces hospital- and community-acquired infections, with methicillin-resistant S. aureus posing a serious public health threat. The golden carotenoid pigment of S. aureus, staphyloxanthin, promotes resistance to reactive oxygen species and host neutrophil-based killing, and early enzymatic steps in staphyloxanthin production resemble those for cholesterol biosynthesis. We determined the crystal structures of S. aureus dehydrosqualene synthase (CrtM) at 1.58 angstrom resolution, finding structural similarity to human squalene synthase (SQS). We screened nine SQS inhibitors and determined the structures of three, bound to CrtM. One, previously tested for cholesterol-lowering activity in humans, blocked staphyloxanthin biosynthesis in vitro (median inhibitory concentration ~100 nM), resulting in colorless bacteria with increased susceptibility to killing by human blood and to innate immune clearance in a mouse infection model. This finding represents proof of principle for a virulence factor–based therapy against S. aureus.
Over the past 20 years, there has been an explosion in the prevalence of antibiotic-resistant bacterial infections, both in the hospital and in the general community; in the United States, more deaths are now attributable to methicillin-resistant Staphylococcus aureus (MRSA) infections than to HIV/AIDS (1, 2). Unfortunately, over the same time, there has been a decrease in the rate of discovery of new antibiotics, creating a pressing need for the development of novel infectious disease therapies. One approach, discussed at a recent National Research Council workshop (3), involves the specific neutralization of bacterial virulence factors to render pathogenic bacteria susceptible to innate immune system clearance. However, conventional screening campaigns are not well suited for selecting such inhibitors, because virulence factors typically do not affect bacterial cell growth but rather exert their activity in vivo. In S. aureus, an important virulence factor is the carotenoid pigment staphyloxanthin. This pigment acts as an antioxidant, with its numerous conjugated double bonds enabling the detoxification of host immune system–generated reactive oxygen species (ROS) such as O2−, H2O2, and HOCl (4, 5). Bacteria that lack the carotenoid pigment grow normally, but they are rapidly killed by ROS from host neutrophils and are deficient in skin abscess formation (4). Blocking staphyloxanthin biosynthesis is therefore a potentially attractive therapeutic target (3, 4), and the bright golden coloration of the virulence factor facilitates inhibitor screening.
The first committed step in staphyloxanthin biosynthesis, catalyzed by the S. aureus dehydrosqualene synthase (CrtM) enzyme, involves the 1,1′ or head-to-head condensation of two molecules of farnesyl diphosphate to produce the C30 species, presqualene diphosphate (5), which then undergoes skeletal rearrangement and further loss of diphosphate to produce dehydrosqualene (Fig. 1A). Successive dehydrogenations yield 4,4′-diaponeurosporene, which is then further oxidized, glycosylated, and esterified to give the carotenoid, staphyloxanthin (Fig. 1A). The structure of dehydrosqualene is very similar to that of the squalene used in cholesterol biosynthesis in humans, and both dehydrosqualene and squalene biosyntheses proceed through presqualene diphosphate. The pathways diverge at this intermediate, with a reduced nicotinamide adenine dinucleotide phosphate (NADPH)–catalyzed reductive step present in squalene synthesis but not in dehydrosqualene synthesis (Fig. 1B). Thus, S. aureus CrtM and human squalene synthase (SQS) might possess structural similarities, although only modest sequence homology (30% identity, 36% similarity) is found by Clustal (6) amino acid alignment (fig. S1). However, human SQS is known to have considerable structural similarity to other prenyl synthase enzymes, including farnesyl diphosphate synthase, pentalenene synthase, and 5-epi-aristolochene synthase, despite the lack of sequence homology (7).
Fig. 1.
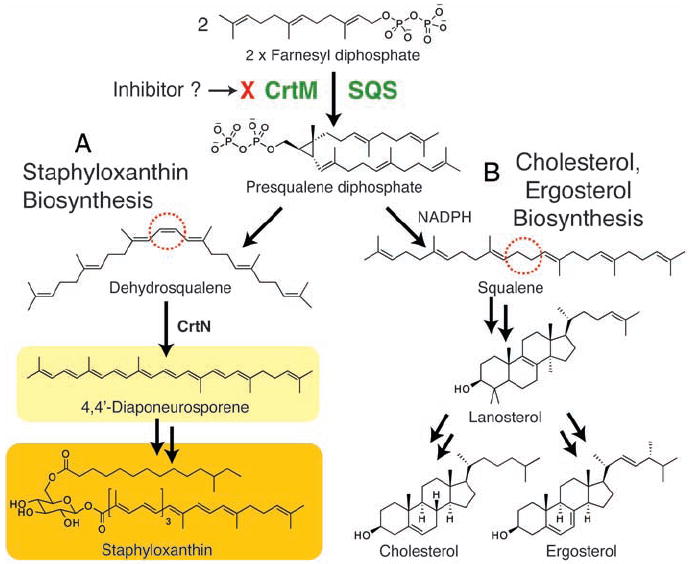
Biosynthetic pathways. (A) Staphyloxanthin biosynthesis (in S. aureus). (B) Cholesterol (in humans) and ergosterol (in, e.g., yeasts and some parasitic protozoa) biosynthesis. Each biosynthetic pathway involves initial formation of presqualene diphosphate, catalyzed by CrtM (S. aureus) or by squalene synthase (SQS). In S. aureus, the NADPH reduction step is absent, resulting in production of dehydrosqualene, not squalene.
To probe this structural question, we cloned, expressed, purified, and crystallized the CrtM protein from S. aureus and solved its structure by x-ray crystallography to 1.58 Å. Data collection and refinement statistics for the selenomethionine-substituted and wild-type proteins are presented in tables S1 and S2. CrtM crystallizes in the P3221 space group and there is one molecule per asymmetric unit. The overall fold (Fig. 2A) shows clear similarity to that seen in human SQS (PDB accession number 1EZF), as can be seen in the superposition (Fig. 2B), with a 5.5 Å root-mean-square (RMS) deviation between the Cα atoms in the two structures. CrtM is all helical and has a large central cavity (also seen in other isoprenoid synthases) capable of accommodating the C30 product, dehydrosqualene.
Fig. 2.
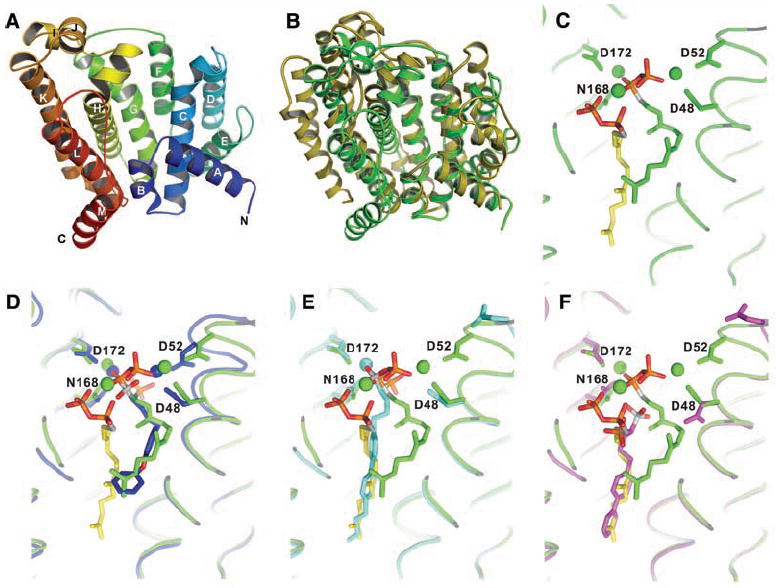
X-ray crystallographic structures. (A) X-ray structure of S. aureus CrtM. (B) Superposition of CrtM and human squalene synthase structures. There is a 5.5 Å Cα RMS deviation between the two structures. (C) Close-up view of FsPP bound to CrtM. (D) Close-up view of S. aureus CrtM with bound BPH-652. (E) S. aureus CrtM with bound BPH-698. (F) S. aureus CrtM with bound BPH-700. In (C) to (F), the FsPP ligands are in green or yellow; BPH-652, BPH-698, and BPH-700 (and associated Mg2+) are in blue, cyan, and magenta, respectively. Key contacts with Asp (D) and Asn (N) residues are indicated.
To see how inhibitors might bind to CrtM, we first crystallized the protein with farnesyl thiodiphosphate (FsPP, Fig. 3), a nonreactive analog of farnesyl diphosphate, the substrate for CrtM. FsPP has previously been found to be a substrate-analog inhibitor of other prenyl transferases, including geranylgeranyl diphosphate synthase and undecaprenyl diphosphate synthase (8). However, in each of those structures, only one FsPP binds per protein. In CrtM, however, two FPP molecules must come together to form dehydrosqualene, and indeed, two FsPP molecules were found in the large central cavity (Fig. 2C). Their diphosphate head groups interact with three Mg2+ ions, which in turn interact with Asp residues in the two conserved Asp-X-X-X-Asp repeats (fig. S1) seen in many prenyl synthases. The space group of FsPP·CrtM is P3121, and there are two molecules per asymmetric unit. Electron densities for the two ligands and key protein-ligand interactions are shown in figs. S2 and S3. This ligand binding pattern is likely to define the location of the presqualene diphosphate intermediate binding site.
Fig. 3.
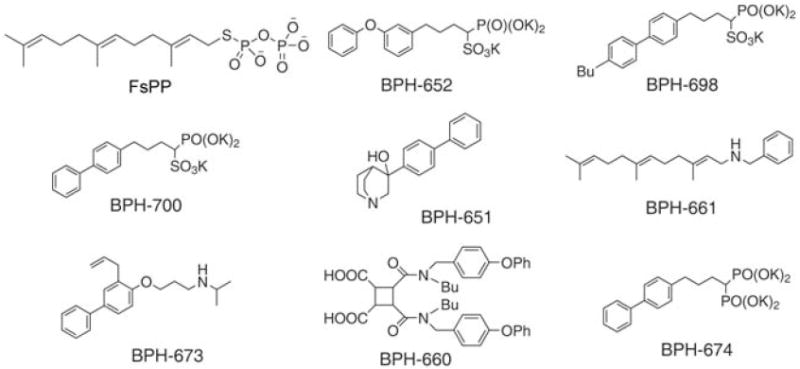
Chemical structures of farnesyl thiodiphosphate (FsPP) and eight squalene synthase inhibitors. There are many different types of SQS inhibitor, but only the phosphonosulfonates (and related bisphosphonates) inhibit CrtM. To date, only the phosphonosulfonates have been found to have activity in S. aureus.
Structural similarity raised the possibility that human SQS inhibitors developed as potential cholesterol-lowering drugs (9-12) might also be active against CrtM. We synthesized eight such compounds (Fig. 3) and tested them for activity in CrtM inhibition (13). There is considerable structural diversity among these compounds: Some mimic the FPP substrate (e.g., the phosphonosulfonates BPH-652, BPH-698, and BPH-700); others presumably act as analogs of carbocations or transition states (e.g., the amines, BPH-651, BPH-661, and BPH-673); and others, such as the dicarboxylic acid BPH-660, have a less certain mechanism of action.
Of the eight compounds tested, only the three phosphonosulfonates were found to be CrtM inhibitors (BPH-652, inhibitor constant Ki = 1.5 nM; BPH-698, Ki = 135 nM; BPH-700, Ki = 6 nM; fig. S4). Each phosphonosulfonate also had potent activity against S. aureus pigment formation in vitro, as shown for BPH-652 in Fig. 4A, with median inhibitory concentration (IC50) values in the range 100 to 300 nM (Fig. 4B). The bisphosphonate BPH-674 (Fig. 3) was a more potent CrtM inhibitor (Ki = 0.2 nM; fig. S4) than any of the phosphonosulfonates but had no activity at all in pigment inhibition in vitro, due perhaps to poor bacterial cell uptake or increased efflux. None of the four amine/carboxylate human SQS inhibitors had any activity in inhibiting CrtM or pigment formation, although all are known to be potent human SQS inhibitors. Taken together, these results pointed to phosphonosulfonates as being the most promising class of CrtM inhibitors for further investigation, beginning with structure determination.
Fig. 4.
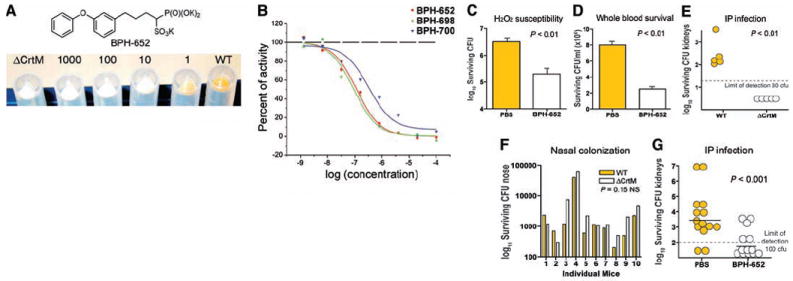
In vitro, in vivo, and CrtM inhibition results. (A) Inhibition of wild-type (WT) S. aureus pigmentation using 0 to 1000 μM BPH-652, with ΔCrtM control at left. The IC50 for pigment formation is ~110 nM. (B) Pigment inhibition by phosphonosulfonates and BPH-674 (no inhibition observed). Note that good CrtM inhibition does not guarantee activity in S. aureus. (C) Effect of BPH-652 on S. aureus susceptibility to H2O2 (1.5%, 1 hour). (D) Effect of BPH-652 on S. aureus susceptibility to killing in human whole blood. (E) CFU isolated from kidneys 3 days after intraperitoneal inoculation of mice with either WT or ΔCrtM S. aureus. (F) CFU isolated from noses 5 days after intranasal inoculation of mice with 1:1 mixture of WT and ΔCrtM S. aureus. (G) Effect of BPH-652 on S. aureus survival in the kidney after intraperitoneal infection. In vitro data shown are representative of at least three identical experiments. In vivo data in (G) are compiled from two sets of experiments, using different numbers of mice, performed under the same conditions (13).
All three phosphonosulfonates, BPH-652, BPH-698, and BPH-700, yielded well-resolved 2Fobs − Fcalc densities (fig. S5); the refined structures obtained are shown in Fig. 2, D to F, superimposed on the FsPP structure. Full crystallographic details are given in table S2, and ligand interaction results are shown in figs. S6 to S8. Interestingly, in all three structures, we found evidence for only one phosphonosulfonate bound per CrtM, not the two ligands that might have been anticipated from the FsPP structure. Moreover, all three inhibitors have different binding modes. BPH-652 (Fig. 2D) binds into the FsPP-1 site with two Mg2+, BPH-698 binds into the FsPP-2 site (Fig. 2E) with only one Mg2+, and BPH-700 binds into the FsPP-2 site with no Mg2+ (Fig. 2F). The phosphonosulfonate side chains do, however, closely track the locations of the two FsPP inhibitor side chains. FsPP-1 is highly bent (green structure in Fig. 2C), and BPH-652 is able to track this bent geometry (Fig. 2D), due at least in part to the presence of its central ether O atom. On the other hand, the (linear) biphenyl side chains in the BPH-698 and BPH-700 structures can be closely superimposed on the fully extended farnesyl side chain in the FsPP-2 site (yellow in Fig. 2, C, E, and F). Additional views of all superimposed ligands are shown in fig. S9.
Although the aryl side chains in the two biphenyl phosphonosulfonates are closely aligned (Fig. 2, E and F), the head groups bind very differently. With BPH-698, the phosphonosulfonate head group is involved in electrostatic interactions with one Mg2+, but in BPH-700 (which lacks the 4′-butyl side chain), no Mg2+ is found in the structure; instead, the phosphonosulfonate is involved in electrostatic or hydrogen bond interactions with His18 and Tyr41. With all three phosphonosulfonate inhibitors, electrostatic interactions between ligand and protein are important, with eight or nine highly hydrophobic amino acid side chains contacting the inhibitor side chains.
To probe the possible use of a phosphonosulfonate CrtM inhibitor as an antimicrobial, we performed a series of experiments to determine whether such a compound was (i) nontoxic to mammalian cells, (ii) able to restore S. aureus killing by oxidants and phagocytes, and (iii) able to limit S. aureus disease progression in vivo. BPH-652 was selected for these experiments because it has a good IC50 in pigment inhibition (110 nM; Fig. 4B) and because S-BPH-652 (and its analogs) have been advanced through preclinical testing in rats and in two human clinical trials (14, 15) as cholesterol-lowering agents.
As expected, BPH-652 had no effect on the growth of three human cell lines (MCF-7, NCI-H460, and SF-268), because only cholesterol biosynthesis is targeted and cholesterol is generally abundant in serum (or diet). This indicates low toxicity, consistent with the results of the clinical trials (on S-BPH-652). Also, incubation with up to 2 mM BPH-652 affected neither S. aureus growth characteristics nor survival through 48 hours in culture. However, after incubation with 100 μM BPH-652, the resulting nonpigmented (white) S. aureus were more susceptible to killing by 1.5% hydrogen peroxide by a factor of ~15, and, relative to normally pigmented S. aureus treated with phosphate-buffered saline (PBS) control, were less able to survive in freshly isolated human whole blood by a factor of ~4 (Fig. 4, C and D), as expected because they contained no carotenoid pigment to act as an antioxidant.
Finally, we sought to establish whether inhibition of staphyloxanthin biosynthesis by BPH-652 could represent a purely virulence factor–based therapy against S. aureus infection. We first extended our previous observations on the contribution of staphyloxanthin to abscess formation (4) to a systemic S. aureus infection model. Staphyloxanthin was found to promote invasive disease potential, because mice inoculated with 108 colony-forming units (CFU) of wild-type S. aureus (by intraperitoneal injection) developed a sustained infection, with bacteria recovered from the kidneys 72 hours later; an isogenic S. aureus mutant lacking the CrtM enzyme was cleared by the host after similar challenge (Fig. 4E). Conversely, the contribution of S. aureus pigmentation to mucosal colonization after intranasal inoculation was negligible (Fig. 4F). These data are consistent with a primary function of staphyloxanthin in resisting the host oxidant-based phagocytic defenses that are present in disease contexts but are absent on mucosal surfaces without inflammation.
Under the same intraperitoneal challenge used in Fig. 4E, we then treated one group of mice (n = 14) with 0.5 mg of BPH-652 twice per day (days −1, 0, 1, and 2), and a second group (n = 13) with equivalent volume injections of PBS control. Upon killing at 72 hours, S. aureus bacterial counts in the kidneys of the mice treated with BPH-652 were significantly lower than those of the control group (P < 0.001), with 8 of 13 below the detection threshold, versus only 2 of 14 in the control group (Fig. 4G); on average, this result corresponds to a 98% decrease in surviving bacteria in the treatment group.
Thus, the CrtM (dehydrosqualene synthase) enzyme from S. aureus is a target for anti-infective therapy based on virulence factor neutralization, and a drug candidate already tested in humans in the context of cholesterol-lowering therapy provides a good lead. These results therefore provide a basis for rational drug design against this major human pathogen, and provide proof of principle of the utility of an anti-infective drug without direct bactericidal properties that instead renders the pathogen susceptible to normal host innate immune clearance. Our approach, and other virulence factor-based concepts (16, 17) for highly specific antistaphylococcal therapy, also offer theoretical advantages for reducing selection pressure toward the emergence of resistance, both in the pathogen and in our normal commensal microflora (3, 18).
Supplementary Material
Footnotes
www.sciencemag.org/cgi/content/full/1153018/DC1
Materials and Methods
Figs. S1 to S9
Tables S1 and S2
References
References and Notes
- 1.Bancroft EA. JAMA. 2007;298:1803. doi: 10.1001/jama.298.15.1803. [DOI] [PubMed] [Google Scholar]
- 2.Klevens RM, et al. JAMA. 2007;298:1763. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.National Research Council. Treating Infectious Diseases in a Microbial World: Report of Two Workshops on Novel Antimicrobial Therapeutics. National Academies Press; Washington, DC: 2006. pp. 21–22. [PubMed] [Google Scholar]
- 4.Liu GY, et al. J Exp Med. 2005;202:209. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clauditz A, Resch A, Wieland KP, Peschel A, Gotz F. Infect Immun. 2006;74:4950. doi: 10.1128/IAI.00204-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandit J, et al. J Biol Chem. 2000;275:30610. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 8.Guo RT, et al. Proc Natl Acad Sci U S A. 2007;104:10022. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiyoshi H, et al. J Lipid Res. 2000;41:1136. [PubMed] [Google Scholar]
- 10.Nishimoto T, et al. Br J Pharmacol. 2003;139:911. doi: 10.1038/sj.bjp.0705332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishihara T, et al. Bioorg Med Chem. 2003;11:2403. doi: 10.1016/s0968-0896(03)00143-3. [DOI] [PubMed] [Google Scholar]
- 12.Magnin DR, et al. J Med Chem. 1996;39:657. doi: 10.1021/jm9507340. [DOI] [PubMed] [Google Scholar]
- 13.See supporting material on Science Online.
- 14.Sharma A, Slugg PH, Hammett JL, Jusko WJ. Pharm Res. 1998;15:1782. doi: 10.1023/a:1011977116543. [DOI] [PubMed] [Google Scholar]
- 15.Sharma A, Slugg PH, Hammett JL, Jusko WJ. J Clin Pharmacol. 1998;38:1116. [PubMed] [Google Scholar]
- 16.Ton-That H, Schneewind O. J Biol Chem. 1999;274:24316. doi: 10.1074/jbc.274.34.24316. [DOI] [PubMed] [Google Scholar]
- 17.Lyon GJ, Mayville P, Muir TW, Novick RP. Proc Natl Acad Sci U S A. 2000;97:13330. doi: 10.1073/pnas.97.24.13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alksne LE, Projan SJ. Curr Opin Biotechnol. 2000;11:625. doi: 10.1016/s0958-1669(00)00155-5. [DOI] [PubMed] [Google Scholar]
- 19.Supported by U.S. Public Health Service grants AI07482 (G.Y.L), HD051796 (V.N.), and GM073216 and GM65307 (E.O.); Academia Sinica and National Core Facility of High-Throughput Protein Crystallography grant NSC95-3112-B-001-015-Y (A.H.-J.W.); a Burroughs-Wellcome Career Award (G.Y.L.); and a Leukemia and Lymphoma Society Special Fellowship (Y.S.). We thank K. Krysiak for carrying out the mammalian cell growth inhibition assays. Diffraction data were obtained at the National Synchrotron Radiation Research Center of Taiwan, SPring-8 and the Photon Factory in Japan, and at the Advanced Light Source, Berkeley, CA. The coordinates for the CrtM, FsPP, BPH-652, BPH-698, and BPH-700 structures have been deposited in the Protein Data Bank under accession numbers 2ZCO, 2ZCP, 2ZCQ, 2ZCR, and 2ZCS, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.