

Abstract
In the heart, oxidative stress caused by exogenous H2O2 has been shown to induce early afterdepolarizations (EADs) and triggered activity by impairing Na current (INa) inactivation. Since H2O2 activates Ca2+/calmodulin kinase II (CaMKII), which also impairs INa inactivation and promotes EADs, we hypothesized that CaMKII activation may be an important factor in EADs caused by oxidative stress. Using the patch clamp and intracellular Ca (Cai) imaging in Fluo-4 AM-loaded rabbit ventricular myocytes, we found that exposure to H2O2 (0.2–1mM) for 5-15 min consistently induced EADs that were suppressed by the INa blocker tetrodoxtin (TTX, 10 μM), as well as the ICa,L blocker nifedipine. H2O2 enhanced both peak and late ICa,L, consistent with CaMKII-mediated facilitation. By prolonging the AP plateau and increasing Ca influx via ICa,L, H2O2-induced EADs were also frequently followed by DADs in response to spontaneous (i.e. non ICa,L-gated) SR Ca release after repolarization. The CaMKII inhibitor KN-93 (1 μM, n=4), but not its inactive analog KN-92 (1 μM, n=5), prevented H2O2-induced EADs and DADs, and the selective CaMKII peptide inhibitor AIP (2 μM) significantly delayed their onset. In conclusion, H2O2-induced afterdepolarizations depend on both impaired INa inactivation to reduce repolarization reserve, and enhancement of ICa,L to reverse repolarization, both facilitated by CaMKII activation. Our observations support a link between increased oxidative stress, CaMKII activation and afterdepolarizations as triggers of lethal ventricular arrhythmias in diseased hearts.
Keywords: Reactive oxidative species, early afterdepolarization, triggered activity, arrhythmia, CaM Kinase
Introduction
Reactive oxygen species (ROS) are generated as natural byproducts of normal oxygen metabolism and play important roles in cell signaling. However, under pathological conditions, such as heart failure and ischemia-reperfusion, ROS levels can become elevated and predispose the heart to arrhythmias1 2. Oxidative stress induced by exposure to hydrogen peroxide (H2O2) and other agents has been shown to induce early afterdepolarizations (EADs), delayed afterdepolarizations (DADs) and triggered activity (TA), 3-6, primarily attributed in previous studies to impaired inactivation of Na current (late INa) 4, 5. However, H2O2 also affects other ion channel and transporters, including the L-type Ca current (ICa,L) 7-9, K currents 10, ryanodine receptors (RyR) 11, 12, sarcoplasmic reticulum (SR) Ca pump SERCA2a 13, and the Na-Ca exchanger (INCX) 14, 15, which also may influence EADs and TA.
Recently, H2O2 has been shown to activate Ca/calmodulin (CaM) kinase II (CaMKII) in cardiac myocytes and other cells 16-18, by direct oxidation of paired Met residues (Met281/282) in the regulatory domain. It is notable that binding of Ca2+/CaM is required to expose the redox sites in the regulatory domain in order for oxidation to persistently activate CaMKII 18. In addition, ventricular myocytes isolated from transgenic mice overexpressing CaMKIV have been shown to exhibit EADs and TA. Increased endogenous CaMKII activity accompanied EADs and VT/VF observed during telemetry in this mouse model19. Alterations in ICaL properties have been implicated as a major factor by which CaMKII activation promotes EADs 19, 20, but a recent study demonstrated that CaMKII activation also increases the late INa 21, similar to H2O2. CaMKII inhibition has been shown to suppress EADs and triggered activity. In a genetic heart failure model, for example, CaMKII inhibition prevented EADs, triggered activity and improved mortality19. The ability of pharmacologic agents such as clofilium to induce EADs in isolated rabbit ventricular myocytes 22 was also suppressed by CaMKII inhibition.
Taken together, this evidence led us to hypothesize that activation of CaMKII by oxidative stress may be an important factor in arrhythmogenic effects of H2O2. Accordingly, we used isolated patch-clamped rabbit ventricular myocytes and Cai imaging to analyze the ionic mechanism(s) underlying H2O2-induced afterdepolarizations in the absence and presence of CaMKII inhibition. Our findings support a direct link between oxidative stress, CaMKII activation and the genesis of TA due to afterdepolarizations.
Methods
Cell isolation
Ventricular myocytes were enzymatically isolated from adult rabbit hearts. Briefly, hearts were removed from adult New Zealand white rabbits (2-3 kg) anesthetized with intravenous pentobarbital, and perfused retrogradely in Langendorff fashion at 37°C with nominally Ca-free Tyrode's solution containing ∼1.4 mg/ml collagenase (Type II; Worthington) and 0.1 mg/ml protease (type XIV, Sigma) for 25-30 minutes. After washing out the enzyme solution, the hearts were removed from the perfusion apparatus, and swirled in a culture dish. The Ca concentration was slowly increased to 1.8 mM and the cells were stored at room temperature and used within 8 hours.
Intracellular Ca measuremen
Myocytes were loaded with the Ca indicator Fluo-4 by incubating them for ∼30 minutes in bath solution containing 4 μM Fluo-4 AM (Molecular Probes) and 0.016% (wt/wt) pluronic (Molecular Probes), after which the cells were washed and placed in a heated chamber on an inverted microscope. Cai fluorescence was recorded using an Andor Ixon Charge Coupled Device (CCD) camera (Andor Technology) operating at ∼100 frames per second with a spatial resolution of 512 × 180 pixels. The fluorescence intensity was recorded in arbitrary units.
Patch Clamp Methods
Myocytes were patch-clamped using the whole-cell configuration of the patch clamp technique. For AP recordings, patch pipettes (resistance 2–4 MΩ) were filled with pipette solution containing (in mM): 110 K-Aspartate, 30 KCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 MgATP, 5 creatine phosphate, 0.05 cAMP, pH 7.2 with KOH. In some experiments, the CaMKII inhibitor peptide AIP (2-10 μM) (Biomol, or Sigma) was added directly to the pipette solution. The cells were superfused with standard Tyrode's solution containing (in mM): 136 NaCl, 5.4 KCl, 0.33 Na2PO4, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES, pH 7.4 adjusted with NaOH. To isolate ICa,L, patch pipettes (resistance 2–4 MΩ) were filled with pipette solution containing (in mM): 110 Cs-Aspartate, 30 CsCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 MgATP, 5 creatine phosphate, 0.05 cAMP, pH 7.2 with KOH, and the cells were superperfused with a modified Tyrode's solution in which KCl was replaced by CsCl. H2O2 (0.2-1 mM) was added directly to the bath superfusate. Nifedipine (Sigma), KN-93 and KN-92 (Biomol) were dissolved in dimethyl sulfoxide (DMSO) as stock solution before diluting into the superfusate solution at the final concentration. The maximum DMSO concentration was <1/500 (v/v). Chemicals and reagents were purchased from Sigma unless indicated. Voltage or current signals were measured with an Axopatch 200B patch-clamp amplifier controlled by a personal computer using a Digidata 1200 acquisition board driven by pCLAMP 8.0 software (Axon Instruments, Foster City, CA). Action potentials were elicited with 2 ms, 2-4 nA square pulses at pacing cycle lengths (PCL) of 6 sec.
All experiments were performed at 34-36°C. Data are presented as mean ± SEM. Statistical significance was assessed using unpaired Student's t-tests, with p < 0.05 considered significant.
Results
H2O2-induced EADs and DADs in adult rabbit ventricular myocytes
APs were recorded from isolated rabbit ventricular myocytes using whole-cell current clamp mode. After AP duration (APD) and morphology reached steady state, cells were superfused with 0.2-1 mM H2O2. The generation of EADs by H2O2 was highly dependent on the pacing cycle length (PCL). Typically, at long PCLs (e.g. 6 sec), EADs occurred with every AP, and at short PCL (e.g. 1 s), no EADs occurred. In the intermediate range, EADs occurred irregularly. Therefore, to elicit EADs most reliably, we used a PCL of 6 sec throughout. EADs appeared after an average exposure time of 6.9 ± 1.1 min with 0.2 mM H2O2 (n=10), and 6.6 ± 0.8 min with 1 mM H2O2 (n=12). As shown in Fig. 1A, EADs could be irregular, single or multiple with an oscillating membrane potential before repolarization (Fig. 1 A & B). DADs were also observed after prolonged perfusion of H2O2 and occasionally triggered APs (Fig 1B, right panel). DADs often occurred following a previous AP with an EAD (Fig. 1B middle panel and 1C), presumably because the prolonged APD allowed maintained ICa,L to overload the SR with Ca. Prolonged perfusion (15-20 min) of H2O2 eventually caused gradual depolarization of the resting membrane potential to less than -40 mV and the cells became inexcitable.
Fig. 1. Afterdepolarizations and triggered activity during H2O2 exposure in isolated rabbit ventricular myocytes.

A. Action potentials (APs) were elicited with 2 ms-2nA pulses under current clamp conditions at pacing cycle length (PCL) of 6 sec. Values of consecutive APD90 are plotted over time. H2O2 (200 μM) was perfused continuously as indicated by the horizontal bar above the plot. EADs are indicated by the sudden and dramatic increases of APD90 (at ∼ 6 min after H2O2 application). Insets show APs under control condition, and after H2O2 with an EAD. B. Examples of afterdepolarizations and TA during exposure to 1 mM H2O2, including multiple oscillatory EADs (left panel), an EAD followed by a DAD (middle panel), and an EAD followed by a DAD causing TA (right panel). C. Successive APs recorded during H2O2 exposure, illustrating the irregular occurrence of EADs (asterisks) and associated DADs (arrows).
Ionic mechanisms of H2O2-induced EADs and DADs
We next used pharmacologic interventions to analyze the ionic and cellular mechanism(s) underlying H2O2-induced aftedepolarizations. As shown in Fig. 2A, H2O2-induced EADs were reversibly suppressed by the selective Na current blocker TTX (10 μM), which also shortened APD. Ranolazine (20 μM), a more selective blocker of late INa, also eliminated H2O2-incuded EADs and shortened APD (data not shown), consistent with previous reports by Song et al 4, 5 implicating late INa as playing a key role. Unlike H2O2, anemone toxin II (ATX), an agent that selectively delays the late phase inactivation of INa 23, failed to induce frank EADs, producing instead only small (1-2 mV) irregular oscillations during phase 2 of the AP. Note that APD was prolonged by ATX to a greater extent than after H2O2 (Fig. 2B), yet no EADs were observed. This finding indicates that activation of late INa is not, by itself, sufficient to produce EADs under these experimental conditions, implying that other actions of H2O2 are also required.
Fig. 2. The involvement of both INa and ICa,L in H2O2-induced EAD.

A. The specific INa blocker TTX (10 μM) reversibly suppressed EADs and shortened APD. B. ATX (30 nM), a selective activator of persistent INa, prolonged APD but did not generate cause frank EADs with a distinct upstroke. C. The amplitude of H2O2-induced EADs depended on their take-off potentials (arrows). D. The ICa,L blocker nifedipine (Nif, 10 μM) suppressed the EAD upstroke, although AP plateau remained prolonged, unless TTX (10 μM) was also added.
Fig. 2C shows that EAD amplitude after H2O2 depended on its take-off potential during repolarization, such that the more repolarized the take-off potential, the larger the EAD amplitude. This relationship parallels the voltage dependence of ICa,L reactivation and the ICa,L window current 24, 25. Consistent with this interpretation, the selective ICa,L blocker nifedipine (10 μM) eliminated EADs (Fig. 2D), even though APD remained markedly prolonged due to the effect of H2O2 on late INa (as indicated by shortening of APD with subsequent application of 10 μM TTX or 20 μM ranolazine). Taken together, these results suggest that at least two actions of H2O2 are required for EAD formation: activation of late INa to reduce repolarization reserve (i.e. prolong APD) and modification of ICa,L to enhance its reactivation properties so as to generate the EAD upstroke.
Whereas the effects of H2O2 on late INa have been well documented in previous studies 4, 5, the reported effects of H2O2 on ICa,L have been variable. Therefore, we examine how H2O2 affected ICa,L in rabbit ventricular myocytes under our experimental conditions. Fig. 3A and B show the time course of both peak ICa,L and the residual pedestal ICa,L at the end of a 300ms voltage clamp to 0 mV in a representative myocyte during exposure to 1 mM H2O2. Both peak and pedestal ICa,L increased, from 7.3 ± 0.8 to 12.1 ± 1.8 pA/pF (n=5) and from 0.5 ± 0.1 to 3.2 ± 0.3 pA/pF (n=5), respectively. To examine how these changes affect ICa,L during the repolarization phase of the AP, we performed AP clamp experiments before and after exposure to 1 mM H2O2. Fig. 3C shows that H2O2 induced a prominent ICa,L hump during the late phase of the AP plateau, consistent with the timing of the EAD (compare to Fig. 1 &2). The recordings shown in Fig. 3 B & C represent the nifedipine-sensitive (subtracted) current, indicating that the increase of the inward current (including the hump in the late phase) is mostly due to ICa,L, although minor contamination by the Na-Ca exchange current (INCX) cannot be excluded.
Fig. 3. Enhancement of ICa,L by H2O2.

A. Time course of the peak and residual pedestal (ped) ICa,L during a 300 ms voltage clamp pulse to 0 mV (voltage protocol shown in B). B. Voltage clamp pulse (above) and superimposed current traces showing ICa,L before (black) and ∼5 min after perfusion of 1mM H2O2 (red). The difference current is shown in the bottom trace. C. Same as B, but with an AP clamp waveform replacing the square voltage clamp pulse.
The role of Cai cycling
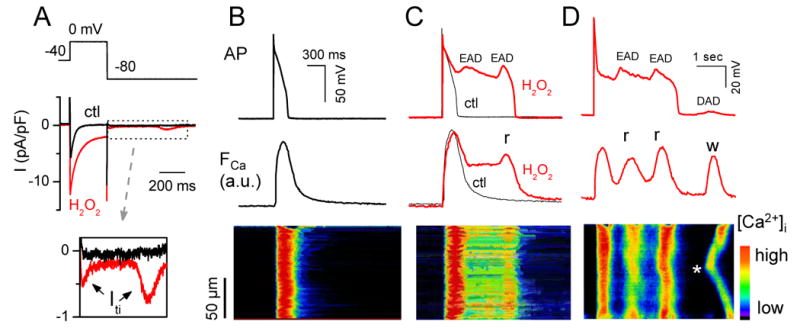
After H2O2 exposure, we often observed a transient inward current (Iti) following repolarization to -80 mV (Fig. 4A and inset), consistent with spontaneous SR Ca release as a result of the enhanced Ca influx via ICa,L causing SR Ca overload. To investigate the relationship to the EAD-induced DADs observed in Fig. 1, we simultaneously imaged Cai in Fluo-4-AM loaded myocytes (Fig. 4B-D). During H2O2-induced EADs, Cai remained elevated (Fig. 4C) or increased (Fig. 4D), consistent with additional SR Ca release due to reactivation of ICa,L. The line scan in Fig. 4D shows that the rise in Cai during the second EAD was not spatially homogeneous. This may indicate that Ca-induced Ca release from the SR, in addition to ICa,L enhancement, contributed to EAD formation by enhancing Iti (since H2O2 has been reported to enhance Na-Ca exchange 14). Fig. 4D documents that after repolarization, a spontaneous Cai wave originating from the center of the cell and propagating towards both ends was associated with a DAD. This can be attributed to increased SR Ca loading during the preceding AP, which was markedly prolonged by EADs.
Fig. 4. Transient inward current (Iti) and Cai transients during H2O2–induced EADs and DADs.

A. Similar to Fig. 3B, but with an Iti following repolarization to -80 mV in the presence of H2O2 (expanded in bottom trace). B. AP (top), simultaneous whole-cell Cai transient (middle), and a line-scan image along the long axis of the myocyte (bottom) before H2O2. C & D. Same following H2O2 exposure. In C, two EADs occur, leading to persistent elevation in Cai and additional Ca2+ release (r) during the second EAD. In D (different cell), two EADs (associated with additional ICa,L-gated SR Ca2+-release) are followed by a DAD due to a non-voltage gated spontaneous Cai wave (w) starting from the middle of the cell (*).
To further investigate the role of Cai cycling in H2O2-induced afterdepolarizations, we examined the effects of pre-loading myocytes with BAPTA-AM to buffer changes in Cai (Fig. 5A). We also pre-treated myocytes with thapsigargin (0.2 μM) and ryanodine (10 μM) to suppress SR Ca cycling (Fig. 5B). Both interventions completely prevented EADs and DADs during exposure to 1 mM H2O2 (Fig. 5), indicating the intact Cai cycling plays a critical role. However, APD remained prolonged under these conditions, probably due to reduced Ca-induced inactivation of ICa,L when the intracellular Cai transient was suppressed 26.
Fig. 5. Dependence of H2O2-induced EADs on intact Cai cycling.

A. Time course of APD90 in a myocyte preloaded with BAPTA-AM (4 μM) for 30 min exposure to 1 mM H2O2. Representative APs at points a (control), b (∼10 min in H2O2), and c (H2O2 + TTX) are shown below. B. Same, but in a myocyte pre-treated with 10 μM Ryanodine (Ry) + 200 nM Thapsigargin (Th) prior to H2O2.
The role of CaMKII signaling
Similar to the effects of H2O2, Ca2+-dependent CaMKII activation has been shown to modify INa inactivation, enhance ICa,L, and promote EADs 19-21. CaMKII could either be activated by Cai, due to the effects of H2O2 on Cai cycling proteins, or directly by oxidative stress, as shown recently in cardiac myocytes 16, 18. The Cai-dependence is still consistent H2O2-mediated activation of CaMKII, since persistent activation of CaMKII by oxidation requires Ca-CaM binding to CaMKII to expose its redox sites 18. We examined the effects of CaMKII inhibition to test these possibilities. Pretreatment with the CaMK inhibitor KN-93 (1 μM) prevented the emergence of EADs during exposure to H2O2 (1 mM for up to 20-30 min) in 4 myocytes (Fig. 6A), compared to non-pretreated myocytes in which H2O2 (1 mM) consistently induced EADs within 5-10 min of H2O2 exposure (Fig. 1A, 6B). In addition, KN-93 also suppressed EADs after they were induced by H2O2 perfusion (Fig. 6B).
Fig. 6. Suppression of H2O2–induced EADs by the CaMKII inhibitor KN-93 and AIP.
A. Time course of APD90 in a myocyte treated with the CaMKII inhibitor KN-93 prior to exposure to 1 mM H2O2. APs under control (a), in the presence of KN-93 and after perfusion with KN93 + H2O2 for 9 min (c) and 19 min (d) are shown in the lower panels. B. Same, but with KN-93 applied after EADs were induced by H2O2. In both protocols, KN-93 suppressed EADs. C. Bar graph showing the average H2O2 (200 μM) perfusion time to cause EAD appearance in the absence and presence of 2 μM AIP.
In contrast, 1μM KN-92, an inactive analog of KN-93, was ineffective (Fig. 7A and B), with EADs appearing after an average of 7.5 ± 1.2 min (n = 5) after exposure to 1 mM H2O2 (n=5). BAPTA-AM, however, remained effective at suppressing H2O2-induced EADs in the presence of KN-92 (Fig. 7B).
Fig. 7. Failure of KN-92, an inactive analog of KN-93, to prevent H2O2–induced EADs.

A. Same protocol as in Fig. 6A, but with KN-92 initially in place of KN-93. KN-92 failed to prevent EADs during exposure to 1 mM H2O2, which were subsequently partially suppressed by KN-93. APs at points a (control), b (∼ 2 min after KN-92), c (KN-92 + H2O2) and d (KN-93 + H2O2) are shown below. B. Same as A, but applying BAPTA-AM to suppress EADs after KN-92 + H2O2.
Since both KN-93 and KN-92 have nonspecific effects, such as blockade of Ca and K currents 22, 27, 28, we also examined the effects of the selective CaMKII inhibitor peptide AIP (2 μM). In 8 myocytes, AIP added to patch pipette dialyzing the cytoplasm delayed the appearance of EADs during exposure to 200 μM H2O2, from 6.6 ± 0.8 (n=10) to 13.9 ± 2.7 mins (Fig 6C) (p<0.05). Moreover, AIP dialysis also prolonged APD in a concentration-dependent manner, presumably due to nonspecific peptide effects, such that higher AIP concentrations were ineffective at preventing H2O2-induced EADs.
Discussion
In the present study, we investigated the mechanisms by which oxidative stress caused by exposure to H2O2 induces afterdepolarization and triggered activity in rabbit ventricular myocytes. The novel findings are: 1) ICa,L modification, in addition to INa modification, plays a crucial role in EAD generation by H2O2; 2) CaMKII activation, either directly via oxidative stress, or indirectly via elevated Cai, is critical for these effects; 3) By enhancing SR Ca loading and promoting spontaneous Cai waves, H2O2–induced EADs also cause DADs, providing a direct link between these two types of afterdepolarizations which compounds the arrhythmogenic potential of oxidative stress.
Ionic mechanisms of EADs and DADs induced by H2O2
Two conditions are required to generate an EAD. First, repolarization reserve must be reduced during phases 2 & 3 of the AP, by an increase in inward current, a decrease in outward current, or both. Second, once repolarization reserve has been compromised, reactivation of ICa,L window current, and/or additional SR Ca release augmenting Ca-sensitive inward currents such as INCX, must be sufficiently powerful to reverse repolarization and generate the EAD upstroke. In ventricular muscle, inward currents influencing repolarization reserve include INa, ICaL, and INCX, while outward currents include the rapid and slow delayed rectifier K+ currents (IKr, and IKs), the transient outward current (Ito) and the inward rectifier potassium current (IK1). Previous studies analyzing the mechanisms of H2O2-induced EADs have implicated impaired INa inactivation as the primary mechanism reducing repolarization reserve 4, 5. However, H2O2 has also been reported to affect other currents and transporters. In the present study, we show that in addition to modification of INa, modification of ICa,L by H2O2 plays a key role in EAD genesis. Not only did H2O2 substantially increase the peak amplitude of ICa,L, but it also impaired ICa,L inactivation (increase of pedestal current as shown in Fig. 3A & B). The consequence was a large increase in the late phase of ICa,L, as seen during the AP clamp in Fig. 3C, which was not successfully overcome by outward currents, accounting for the genesis of the EAD. The experiments using multiple ion channel blockers (Fig, 2) showed that despite prolonging APD, INa modification was not by itself sufficient to induce EADs when ICa,L was blocked. Both INa and ICa,L modification could be attributed to CaMKII activation, since, as shown in Fig. 6A, H2O2 failed to prolong APD significantly or cause EADs when CaMKII was blocked by KN-93.
Oxidative stress also modulates the properties of other Cai-cycling proteins, including ryanodine receptors 11, 12 and SERCA2a 13, which could potentially promote EAD genesis by modulating SR Ca release during repolarization. Indeed, some recent studies have shown that EADs and DADs can share a common mechanism under Cai overload conditions 29-31. Non-ICa,L-gated SR Ca2+-release during repolarization has been proposed to contribute to EAD genesis by activating Ca-sensitive inward currents 29, but it is difficult to unequivocally distinguish from SR Ca release triggered by reactivated L-type Ca channels.
It has been reported that voltage can directly activate SR Ca2+-release in cardiac myocytes, which is enhanced by cAMP 32, 33. However, EADs were readily induced by H2O2 using cAMP-free pipette solution (data not shown, also see Song et al5), excluding an essential role of a cAMP-sensitive voltage-activated Ca release in EAD generation.
Contribution of SR Ca handling to the EADs and DADs induced by H2O2
CaMKII can phosphorylate RyR2, which may enhance SR Ca release dependent triggered activity 34. However, while it is well accepted that CaMKII phosphorylates RyR2, the functional consequences are controversial. Bers' group found increased SR Ca leak from CaMKII-mediated RyR2 phosphorylation 34, 35, but Yang et al 36 reported a suppression of Ca sparks and Ca waves. The cause of this discrepancy remains to be determined. In addition, oxidative stress has direct effects on RyR and SR function. The interdependencies between SR Ca cycling, ICa,L, INCX, and repolarization reserve are complex and nonlinear, making it very challenging, if not impossible, to assign a simple mechanistic role of SR Ca cycling to EAD generation. For example, SR depletion could suppress EADs by any of the following interactive mechanisms: 1) preventing CaMKII activation by oxidative stress (since the Ca-CaM interaction is still required); 2) increasing repolarization reserve by suppressing INCX; 3) preventing Ca waves during the repolarization phase; 4) altering activation/inactivation properties of other ionic currents.
In the presence of BAPTA, the AP has a higher rectangular plateau (Fig. 5), which may also hinder the reactivation of ICa,L and EAD formation, consistent with a recent study showing that triangulation of the AP favors ICa,L reactivation 37.
The Role of CaMKII Signaling in H2O2-induced EADs
The enhancement of ICa,L by H2O2 in the present study is consistent with activation of CaMKII by H2O2, although direct redox modifications of L-type Ca channel subunits may also contribute. CaMKII is known to mediate ICa,L facilitation 38, and CaMKII activation has been shown to promote EADs in a variety of settings 19, 22. Moreover, CaMKII activation has recently been reported to impair INa inactivation 21, consistent with a common underlying pathogenesis of H2O2-induced and CaMKII-induced EADs. Our finding that the CaMKII inhibitor KN-93, but not its inactive analog KN-92, suppressed EAD formation by H2O2, generally supports a common mechanism. On the other hand, both KN-93 and KN-92 have nonspecific effects, including substantial block of ICa,L27 which is critical in EAD formation. However, the selective CaMKII peptide inhibitor AIP also significantly delayed the onset of EADs during exposure to 0.2 mM H2O2. Theoretically, AIP should be equally effective at preventing CaMKII activation by oxidative stress as by elevated Cai, since both activation modes require initial interaction of CaMKII's regulatory domain with Ca-CaM complexes, to expose the redox and autophosphorylation sites on CaMKII's catalytic domain 18. This also explains why BAPTA-AM prevents EADs during H2O2 exposure, by preventing Cai from rising sufficiently for Ca-CaM to interact with CaMKII and trigger the subsequent persistent activation by redox modification.
Based on these observations, the role of CaMKII activation in H2O2-induced EADs can be summarized as follows: 1) CaMKII activation by H2O2 contributes to both impaired INa inactivation and ICa,L facilitation; 2) Both factors enhance cellular Ca loading by prolonging APD and increasing Ca influx via ICa,L, promoting further CaMKII activation via Ca-CaM; 3) KN-93 synergistically suppresses EADs by preventing CaMKII activation by H2O2 and reducing Ca influx by directly blocking ICa,L. Without concomitantly inhibiting CaMKII activation, partial block of ICa,L by KN-92 is not sufficient to suppress EAD formation; 4) Direct redox modification of ion channel proteins may also contribute, since selective CaMKII inhibition with AIP delayed the onset of, but did not entirely suppress, H2O2-induced EADs. We do not have a ready explanation for why AIP showed less effectiveness than KN-93 in our present study. KN-93 also blocks the L-type Ca current upon which EAD generation depends, which may account for its more complete potency. Alternatively, AIP may have had nonspecific effects which reduced repolarization reserve, since it modestly prolonged APD.
EAD-associated DADs
Associations between EADs and DADs have been noted previously 6, 31, 39, 40, but the underlying mechanisms have not been analyzed in detail. For example, in atrial myocytes, Song et al 40 found that ATX-induced EADs were also associated with DADs. They postulated that DADs were due to excessive Ca-loading as a result of APD prolongation by EADs, leading to spontaneous SR Ca release following repolarization. Here we directly documented this postulated mechanism for H2O2-induced EADs, by using optical Cai imaging to detect the spontaneous Cai wave inducing a transient inward current causing the DAD (Fig. 4). It is also conceivable that oxidative stress directly sensitizes the SR to non ICa,L-gated SR Ca2+-release through direct effects on Cai cycling proteins such as RyR 11, 12, but this remains to be established. In either case, our findings suggest that both EADs and EAD-induced DADs may both contribute to TA during oxidative stress, amplifying the arrhythmogenic consequences.
Clinical Implications
Increased oxidative stress is believed to be an important factor predisposing the diseased heart to lethal arrhythmias. Here we have shown how oxidative stress caused by exogenous H2O2 predisposes the heart to both EADs and DADs, and demonstrated a link to CaMKII activation, which is also involved in other aspects of heart failure 41, 42. Other free radical sources have also been reported to produce similar results, i.e. inducing EADs, DADs and triggered activities 6. Although it is still under debate, H2O2 levels up to ∼35 μM have been reported in human blood plasma 43. It is also well known that ROS levels can increase during ischemia and reperfusion by as much as 100-fold 44, and also with age by 7.5 fold 45. Since ROS are short-lived radicals, it is conceivable that the local concentrations near sites of production are considerably higher than circulating concentrations. Therefore, the concentrations (100-200 μM) used in most of our experiments are reasonable approximations of the pathophysiologically relevant range. Targeting CaMKII signaling may have therapeutic potential to prevent arrhythmias in the failing heart, although negative inotropic actions 46 may limit the use of CaMKII inhibitors.
Acknowledgments
Sources of Funding: The study was supported by NIH/NHLBI grants P50 HL53219 and P01 HL078931, and Laubisch and Kawata Endowments.
Footnotes
Disclosures: None.
References
- 1.Lakatta EG, Sollott SJ. The “heartbreak” of older age. Mol Interv. 2002;2:431–446. doi: 10.1124/mi.2.7.431. [DOI] [PubMed] [Google Scholar]
- 2.Slezak J, Tribulova N, Pristacova J, Uhrik B, Thomas T, Khaper N, Kaul N, Singal PK. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X-ray microanalysis. Am J Pathol. 1995;147:772–781. [PMC free article] [PubMed] [Google Scholar]
- 3.Corretti MC, Koretsune Y, Kusuoka H, Chacko VP, Zweier JL, Marban E. Glycolytic inhibition and calcium overload as consequences of exogenously generated free radicals in rabbit hearts. J Clin Invest. 1991;88:1014–1025. doi: 10.1172/JCI115361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500:631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 6.Barrington PL, Meier CF, Jr, Weglicki WB. Abnormal electrical activity induced by H2O2 in isolated canine myocytes. Basic Life Sci. 1988;49:927–932. doi: 10.1007/978-1-4684-5568-7_151. [DOI] [PubMed] [Google Scholar]
- 7.Guo J, Giles WR, Ward CA. Effect of hydrogen peroxide on the membrane currents of sinoatrial node cells from rabbit heart. Am J Physiol Heart Circ Physiol. 2000;279:H992–999. doi: 10.1152/ajpheart.2000.279.3.H992. [DOI] [PubMed] [Google Scholar]
- 8.Hudasek K, Brown ST, Fearon IM. H2O2 regulates recombinant Ca2+ channel alpha1C subunits but does not mediate their sensitivity to acute hypoxia. Biochem Biophys Res Commun. 2004;318:135–141. doi: 10.1016/j.bbrc.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Goldhaber JI, Liu E. Excitation-contraction coupling in single guinea-pig ventricular myocytes exposed to hydrogen peroxide. J Physiol. 1994;477:135–147. doi: 10.1113/jphysiol.1994.sp020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berube J, Caouette D, Daleau P. Hydrogen peroxide modifies the kinetics of HERG channel expressed in a mammalian cell line. J Pharmacol Exp Ther. 2001;297:96–102. [PubMed] [Google Scholar]
- 11.Anzai K, Ogawa K, Ozawa T, Yamamoto H. Oxidative modification of ion channel activity of ryanodine receptor. Antioxid Redox Signal. 2000;2:35–40. doi: 10.1089/ars.2000.2.1-35. [DOI] [PubMed] [Google Scholar]
- 12.Zissimopoulos S, Docrat N, Lai FA. Redox sensitivity of the ryanodine receptor interaction with FK506-binding protein. J Biol Chem. 2007;282:6976–6983. doi: 10.1074/jbc.M607590200. [DOI] [PubMed] [Google Scholar]
- 13.Morris TE, Sulakhe PV. Sarcoplasmic reticulum Ca(2+)-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic Biol Med. 1997;22:37–47. doi: 10.1016/s0891-5849(96)00238-9. [DOI] [PubMed] [Google Scholar]
- 14.Hinata M, Matsuoka I, Iwamoto T, Watanabe Y, Kimura J. Mechanism of Na+/Ca2+ exchanger activation by hydrogen peroxide in guinea-pig ventricular myocytes. J Pharmacol Sci. 2007;103:283–292. doi: 10.1254/jphs.fp0060015. [DOI] [PubMed] [Google Scholar]
- 15.Goldhaber JI. Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am J Physiol. 1996;271:H823–833. doi: 10.1152/ajpheart.1996.271.3.H823. [DOI] [PubMed] [Google Scholar]
- 16.Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282:10833–10839. doi: 10.1074/jbc.M611507200. [DOI] [PubMed] [Google Scholar]
- 17.Howe CJ, Lahair MM, McCubrey JA, Franklin RA. Redox regulation of the calcium/calmodulin-dependent protein kinases. J Biol Chem. 2004;279:44573–44581. doi: 10.1074/jbc.M404175200. [DOI] [PubMed] [Google Scholar]
- 18.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac L-type Ca2+ current: a cellular mechanism for long Q-T arrhythmias. Am J Physiol. 1999;276:H2168–2178. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- 21.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson ME, Braun AP, Wu Y, Lu T, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 23.Mantegazza M, Franceschetti S, Avanzini G. Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firing properties of rat neocortical pyramidal neurones. J Physiol. 1998;507:105–116. doi: 10.1111/j.1469-7793.1998.105bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: induction with the Ca2+ channel agonist Bay K 8644. Circ Res. 1988;62:563–571. doi: 10.1161/01.res.62.3.563. [DOI] [PubMed] [Google Scholar]
- 25.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 26.Pott C, Yip M, Goldhaber JI, Philipson KD. Regulation of cardiac L-type Ca2+ current in Na+-Ca2+ exchanger knockout mice: functional coupling of the Ca2+ channel and the Na+-Ca2+ exchanger. Biophys J. 2007;92:1431–1437. doi: 10.1529/biophysj.106.091538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- 28.Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinn amyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- 29.Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salama G. Arrhythmia genesis: aberrations of voltage or Ca2+ cycling? Heart Rhythm. 2006;3:67–70. doi: 10.1016/j.hrthm.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 31.Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R, Szabo B. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997;34:348–359. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- 32.Ferrier GR, Howlett SE. Cardiac excitation-contraction coupling: role of membrane potential in regulation of contraction. Am J Physiol Heart Circ Physiol. 2001;280:H1928–1944. doi: 10.1152/ajpheart.2001.280.5.H1928. [DOI] [PubMed] [Google Scholar]
- 33.Hobai IA, Howarth FC, Pabbathi VK, Dalton GR, Hancox JC, Zhu JQ, Howlett SE, Ferrier GR, Levi AJ. “Voltage-activated Ca release” in rabbit, rat and guinea-pig cardiac myocytes, and modulation by internal cAMP. Pflugers Arch. 1997;435:164–173. doi: 10.1007/s004240050496. [DOI] [PubMed] [Google Scholar]
- 34.Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 35.Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–398. doi: 10.1161/01.RES.0000258172.74570.e6. [DOI] [PubMed] [Google Scholar]
- 36.Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG, Xiao RP, Cheng H. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 37.Guo D, Zhao X, Wu Y, Liu T, Kowey PR, Yan GX. L-type calcium current reactivation contributes to arrhythmogenesis associated with action potential triangulation. J Cardiovasc Electrophysiol. 2007;18:196–203. doi: 10.1111/j.1540-8167.2006.00698.x. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y, Kimbrough JT, Colbran RJ, Anderson ME. Calmodulin kinase is functionally targeted to the action potential plateau for regulation of L-type Ca2+ current in rabbit cardiomyocytes. J Physiol. 2004;554:145–155. doi: 10.1113/jphysiol.2003.053314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol. 1990;258:H1796–1805. doi: 10.1152/ajpheart.1990.258.6.H1796. [DOI] [PubMed] [Google Scholar]
- 40.Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–2039. doi: 10.1152/ajpheart.01357.2007. [DOI] [PubMed] [Google Scholar]
- 41.Bers DM, Guo T. Calcium signaling in cardiac ventricular myocytes. Ann N Y Acad Sci. 2005;1047:86–98. doi: 10.1196/annals.1341.008. [DOI] [PubMed] [Google Scholar]
- 42.Couchonnal LF, Anderson ME. The role of calmodulin kinase II in myocardial physiology and disease. Physiology (Bethesda) 2008;23:151–159. doi: 10.1152/physiol.00043.2007. [DOI] [PubMed] [Google Scholar]
- 43.Halliwell B, Clement MV, Long LH. Hydrogen peroxide in the human body. FEBS Lett. 2000;486:10–13. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 44.Dhalla NS, Duhamel TA. The paradoxes of reperfusion in the ischemic heart. Heart Metab. 2007;37:31–34. [Google Scholar]
- 45.Maurel A, Hernandez C, Kunduzova O, Bompart G, Cambon C, Parini A, Frances B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol Heart Circ Physiol. 2003;284:H1460–1467. doi: 10.1152/ajpheart.00700.2002. [DOI] [PubMed] [Google Scholar]
- 46.Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]