

Abstract
Methanogenic oxidation of butyrate to acetate requires a tight cooperation between the syntrophically fermenting Syntrophomonas wolfei and the methanogen Methanospirillum hungatei, and a reversed electron transport system in S. wolfei was postulated to shift electrons from butyryl coenzyme A (butyryl-CoA) oxidation to the redox potential of NADH for H2 generation. The metabolic activity of butyrate-oxidizing S. wolfei cells was measured via production of formazan and acetate from butyrate, with 2,3,5-triphenyltetrazolium chloride as electron acceptor. This activity was inhibited by trifluoperazine (TPZ), an antitubercular agent known to inhibit NADH:menaquinone oxidoreductase. In cell extracts of S. wolfei, the oxidation of NADH could be measured with quinones, viologens, and tetrazolium dyes as electron acceptors, and also this activity was inhibited by TPZ. The TPZ-sensitive NADH:acceptor oxidoreductase activity appeared to be membrane associated but could be dissociated from the membrane as a soluble protein and was semipurified by anion-exchange chromatography. Recovered proteins were identified by peptide mass fingerprinting, which indicated the presence of an NADH:acceptor oxidoreductase as part of a three-component [FeFe] hydrogenase complex and a selenocysteine-containing formate dehydrogenase. Furthermore, purification of butyryl-CoA dehydrogenase (Bcd) activity and peptide mass fingerprinting revealed two Bcd proteins different from the Bcd subunit of the Bcd/electron-transfer flavoprotein complex (Bcd/EtfAB) predicted from the genome sequence of S. wolfei. The results suggest that syntrophic oxidation of butyrate in S. wolfei involves a membrane-associated TPZ-sensitive NADH:acceptor oxidoreductase as part of a hydrogenase complex similar to the recently discovered “bifurcating” hydrogenase in Thermotoga maritima and butyryl-CoA dehydrogenases that are different from Bcd of the Bcd/EtfAB complex.
Butyrate is fermented to methane and CO2 by syntrophic communities in which a methanogenic partner organism maintains a low hydrogen partial pressure to allow the oxidation of butyrate to acetate (19, 20, 29). Only under such conditions can butyrate-oxidizing bacteria such as Syntrophomonas wolfei gain energy from the latter reaction in a range of approximately −20 kJ per mol of butyrate, which is just sufficient to support microbial growth (29). It was postulated that S. wolfei has to invest some of the ATP that is formed in the acetate kinase reaction during the β-oxidation of butyrate into an ATP-driven reversed electron transport in order to shift electrons from butyryl coenzyme A (butyryl-CoA) oxidation to the redox potential of NADH (34).
Experimental evidence for the involvement of a proton gradient and of ATPase activity in this process was obtained with intact cell suspensions (36), and it was hypothesized that menaquinone-7 could play an essential role in this reaction (36). This would imply that membrane-bound enzymes similar to complex I of the aerobic respiratory chain, i.e., NADH dehydrogenase (NDH), operate in reverse to reduce NAD+ with butyrate electrons.
Another option for a reversed electron transport during butyrate oxidation and hydrogen formation in S. wolfei could be a reversal of the so-called Buckel-Thauer reaction. In this reaction that was described for ethanol-acetate fermentation by Clostridium kluyveri, electrons from NADH are disproportionated to reduce both crotonyl-CoA and ferredoxin simultaneously. The reaction is catalyzed by the cytoplasmic butyryl-CoA dehydrogenase/electron-transfer flavoprotein (Bcd/EtfAB) complex (13, 18). Very recently, another “bifurcating” electron pathway has been described for an NADH- and ferredoxin-coaccepting di-iron hydrogenase complex in Thermotoga maritima (30). Here, electrons from NADH and from ferredoxin are combined to produce hydrogen, and the genome sequence of S. wolfei has been shown to contain candidate genes for such a three-component hydrogenase complex (30). Nonetheless, the energetic situation of syntrophic butyrate oxidation is basically different from that of ethanol or glucose degradation: electrons arise at comparably positive redox potentials, i.e., at −125 mV/−10 mV (12, 28) and −250 mV, and there is no oxidation step involved that could be coupled directly with ferredoxin reduction.
In the present study, we report that butyrate oxidation by S. wolfei cell suspensions can be inhibited by trifluoperazine (TPZ), an antitubercular agent that has been shown to inhibit type II NADH:menaquinone oxidoreductase NDH-2 in Mycobacterium tuberculosis (40), and that a TPZ-sensitive NADH:acceptor oxidoreductase activity can be measured in cell extracts of S. wolfei cells. This enzyme system and a butyryl-CoA dehydrogenase were enriched by anion-exchange chromatography, and the obtained proteins were identified by peptide mass fingerprinting.
MATERIALS AND METHODS
Organisms and cultivation.
S. wolfei subsp. wolfei (21, 22) was purchased from the DSMZ, Braunschweig, Germany, as an actively growing coculture with Methanospirillum hungatei JF1 (DSM 2245B). For further growth experiments with cocultures, M. hungatei M1h isolated in our lab was used.
Cocultures were grown in anoxic, bicarbonate-buffered, and sulfide-reduced freshwater medium (38, 39) containing 0.05% yeast extract and 20 mM sodium butyrate. Axenic cultures of S. wolfei were grown with 20 mM sodium crotonate (36). In addition, the medium contained resazurine (0.4 mg liter−1) as a redox indicator, EDTA, and a decreased amount of iron to minimize the precipitation of iron sulfide (25). The seven-vitamin solution of the original freshwater medium was supplemented with lipoic acid (200 μg liter−1) and thiamine (400 μg liter−1) to improve the growth of S. wolfei as described earlier (2). The medium was prepared in 4-liter jars and distributed to 1-liter or 120-ml infusion bottles after autoclaving for 40 min as described earlier (25). Larger volumes of medium for protein purification were prepared directly in 10-liter culture vessels. Cultures were incubated at 28 to 30°C in the dark under an N2/CO2 (80:20) atmosphere. Growth was monitored by determining the optical density (OD) against sterile medium. Prior to measurement, a few grains of sodium dithionite were added to the cuvettes to keep resazurine in its reduced state.
Anoxic buffers for cell harvest or cell suspension experiments were prepared at the concentration and pH value as stated below, transferred to infusion bottles, sealed with rubber stoppers, and evacuated via needles threaded through the stopper using a vacuum pump while stirring the buffer vigorously with a magnetic stirrer. After 30 min, an atmosphere of 100% N2 with a slight overpressure (∼0.3 bar) was applied to the bottle for 10 min to saturate the buffer. This process was repeated three times.
Preparation of cell suspensions.
Cocultures of S. wolfei and M. hungatei were harvested at the end of the exponential growth phase (OD578 = 0.1 to 0.18 after about 10 days) in an anoxic chamber (Coy, Ann Arbor, MI) by centrifugation in polypropylene beakers at 16,300 × g for 10 min at 4°C in a Sorvall RC-5B centrifuge (Du Pont de Nemours, Bad Homburg, Germany) as described previously (25). Large culture volumes of 10 or 20 liters were concentrated under air with a tangential filter device (MiniTan; Millipore, Bedford, MA). All subsequent manipulations were done in an anoxic chamber. Cells were washed twice by repeated centrifugation under the conditions described above in anoxic 0.05 M potassium phosphate buffer (pH 7.5) and resuspended in 4 to 6 ml of the same buffer. Cells of S. wolfei and M. hungatei were separated in two or four centrifuge tubes containing 20 ml of a Percoll gradient from 55 to 70% (3). After centrifugation at 2,200 × g for 1 h in SS-34 centrifuge tubes, the two cell types were separated, and the upper, S. wolfei-containing layer was transferred to two 120-ml infusion bottles and washed twice by centrifugation at 2,600 × g for 20 min in 100 ml of buffer. The pellet was suspended in 5 ml of 0.02 M Tris-HCl (pH 8.0) for cell lysis or 0.05 mM potassium phosphate buffer (pH 7.5) for cell suspension experiments. The separated S. wolfei cell fractions contained almost no cells of M. hungatei, as confirmed by microscopy.
Cell suspension experiments.
Metabolic activity of butyrate-grown cells was tested with 2,3,5-triphenyltetrazolium chloride (TTC) as an electron acceptor. Dense cell suspensions (OD578 = 2) of S. wolfei after separation on a Percoll density-gradient were prepared in 0.05 M potassium phosphate buffer (pH 7.5) containing 5 mM TTC and degassed to remove traces of hydrogen by repeatedly evacuating and regassing with 100% N2. From this suspension, 2-ml aliquots were transferred to rubber-stoppered tubes containing an atmosphere of 100% N2 gas. Experiments were started by adding 20 mM sodium butyrate to each tube. The inhibited treatments received, in addition, 0.1 mM TPZ, whereas butyrate was omitted in the negative controls. Each treatment was started in triplicate, and the tubes were incubated at 30°C on a shaker at 150 rpm. Samples of 250 μl were taken with syringes at the start of the experiment and at intervals thereafter. For high-pressure liquid chromatography (HPLC) analysis, 200-μl samples were stopped with 20 μl of 2 M H2SO4, and centrifuged at 15,700 × g for 5 min. The supernatant was transferred to HPLC vials and analyzed as described below. The remaining 50 μl of sample was mixed with 950 μl of absolute ethanol to give a final concentration of 95%, incubated for 5 min at room temperature to extract formazan from the cells, and then centrifuged at 15,700 × g for 5 min. Finally, the absorbance of the supernatant was recorded at a 483-nm wavelength against ethanol, and the formazan concentration was calculated (ɛ483 = 21 mM−1 cm−1 [1]).
Cell lysis and subcellular fractionation.
Cells were opened by three passages through an anoxic, cold French pressure cell operated at 137 MPa. The cell lysate was collected in an 8-ml serum vial, and the cell debris and unbroken cells were removed by centrifugation in an SS-34 rotor at 3,000 × g for 20 min using rubber adaptors. The supernatant thus obtained (cell extract) was further fractionated in an Optima TL-ultracentrifuge using the TLA-100.4-rotor (Beckman, Munich, Germany) at 236,000 × g for 30 min, which yielded the soluble fraction (supernatant) and the membrane fraction (pellet). The fractions were stored on ice under N2 gas.
Solubilization of membrane proteins.
Cells were disrupted as described above, but with only one passage through the French pressure cell (see Results). Unbroken cells and debris were spun down at 3,000 × g for 20 min, and the supernatant crude extract was removed and stored on ice. The pellet was resuspended in 4 ml of potassium phosphate buffer, subjected to a second passage through the French pressure cell, and centrifuged. The crude extracts were pooled and fractionated by ultracentrifugation as described above. Membrane particles were washed once in anoxic potassium phosphate buffer, centrifuged again at 236,000 × g for 30 min and finally suspended in 4 ml of 0.02 M Tris-HCl (pH 8.0) containing 0.5% dodecyl β-d-maltoside (DDM). After incubation for 30 min on ice, this mixture was ultracentrifuged again, and the supernatant contained the solubilized membrane protein (modified as described previously [31]).
Enrichment of NADH:quinone oxidoreductase.
Both the soluble and the membrane fraction of S. wolfei cells from 10- or 20-liter cultures contained high activities of NADH:quinone oxidoreductase and were used for enzyme purification. Stability tests showed that the activity was stable for several hours at room temperature and under air. Therefore, all purification steps were done at room temperature under air, but all manipulations were kept as short as possible, and active fractions were stored on ice under 100% N2 gas in between the purification steps to minimize activity losses. Purification was done on a fast protein liquid chromatography (FPLC) gradient system (Pharmacia, Uppsala, Sweden). Protein preparations were applied to a packed DEAE-Sepharose CL-6B column (bed volume of ∼10 ml) equilibrated with 0.02 M Tris-HCl (pH 8.0). For purification of solubilized membrane proteins, all column buffers contained 0.01% DDM. Separation was performed at a flow rate of 1 ml per min with a shallow gradient of 0.300 M to 0.360 M NaCl in 0.02 M Tris-HCl (pH 8.0) over 160 min. Fractions were tested for activity by mixing 50 μl of sample with 10 μl of 7 mg of iodonitrotetrazolium chloride (INT)/ml and 10 μl of 10 mM NADH in microtiter plates. Rapid development of red coloration (within 5 min) indicated the presence of NADH:INT oxidoreductase. Further red-colored fractions appeared after longer incubation (>15 min) but were neglected since they were considered to be due to unspecific INT reduction. These fractions were also tested for butyryl-CoA dehydrogenase activity, as described below. Active fractions were pooled and concentrated with centrifugal devices (Amicon Ultra-15, 10K nominal molecular weight limit; Millipore), and the presence of NADH:menadione oxidoreductase was verified as outlined in the enzyme assay section. Pooled fractions were concentrated and adjusted to a 4-ml volume with 0.02 M Tris-HCl (pH 8.0) and applied to a MonoQ column (Pharmacia). Proteins were eluted in the same buffer with a gradient of 0.260 to 0.360 M NaCl over 160 min at 1 ml min−1. The combined active fractions were again concentrated and rebuffered in 0.02 M Tris-HCl (pH 7.5) using PD-10 columns (Amersham Biosciences, Freiburg, Germany). Subsequently, a second run on MonoQ was performed, but this time in 0.02 M Tris-HCl (pH 7.5), and eluted by a shallow gradient of 0.250 to 0.310 M NaCl over 160 min. The fractions that eluted at 0.28 M NaCl showed NADH:INT oxidoreductase activity and were concentrated to a final volume of 1 ml and defined as the partially purified NADH:menadione oxidoreductase.
Enrichment of Bcd.
The activity of butyryl-CoA dehydrogenase (Bcd) was present in a late protein peak during the first NDH-purification step on DEAE-Sepharose eluting at 0.7 M NaCl at pH 8.0. Fractions of this peak were combined, concentrated, and desalted as described above and were almost pure after this step (see Results).
Electrophoresis and peptide mass fingerprinting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done according to the method of Laemmli (17). Gels containing 12% polyacrylamide in the resolving gel and 4% polyacrylamide in the stacking gel were cast either as minigels (Protean II; Bio-Rad) or as large gels (Protean XI; Bio-Rad) for the excision of bands to be analyzed by peptide mass fingerprinting. Protein samples were mixed 1:2 with loading buffer (0.125 M Tris-HCl [pH 6.8], 2% [wt/vol] SDS, 25% glycerol, 0.01% [wt/vol] bromophenol blue, and 5% β-mercaptoethanol or 0.1 M dithiothreitol [DTT]) and heated at 100°C for 5 min prior to loading. Where indicated, samples were prepared in the same buffer at pH 8.0, boiled for 5 min, and subsequently alkylated by adding 2% iodacetamide, followed by incubation for 30 min at room temperature (37). Gels were run at 15 mA for the stacking gel, and when the marker front reached the resolving gel the current was increased to 30 mA. Gels were stained with colloidal Coomassie blue (26). Stained protein bands to be identified were excised and sent to TopLab GmbH (Martinsried, Germany) for tryptic digest and peptide mass fingerprinting; the fingerprints were matched (Mascot search engine) against the amino acid sequences derived from the primary genome sequence of Syntrophomonas wolfei subsp. wolfei strain Goettingen (A. Copeland et al., complete sequence of Syntrophomonas wolfei subsp. wolfei strain Goettingen, GenBank accession no. NC_008346).
Nondenaturing (native) gels were prepared as described above, except that 8% polyacrylamide in the resolving gel and no SDS or reducing agent was used in the loading and electrode buffers. Native gels were stained either with colloidal Coomassie blue or by activity stain. Activity staining was done by immersing the gels in 0.05 M potassium phosphate buffer (pH 7.5) containing 0.1% INT and 0.1 mM NADH (16). Active enzymes appeared as red bands after 15 min of incubation at room temperature.
Enzyme assays.
All enzymes were assayed in anoxic rubber-stoppered cuvettes at 30°C in a spectrophotometer 100-40 (Hitachi, Tokyo, Japan) connected to an analogous recorder (SE 120 Metrawatt; BBC Goerz, Vienna, Austria). Substrates and inhibitors were added by using a syringe. All measurements were done in triplicates.
NADH:quinone oxidoreductase was measured in 950 μl of anoxic 0.05 M potassium phosphate buffer (pH 7.5) containing 0.2 mM NADH unless indicated otherwise. Then, 5- to 20-μl samples were added, and NADH oxidation was monitored at 340 nm (ɛ340 = 6.3 mM−1 cm−1 [42]). After 2 to 3 min, 0.1 mM menadione or other quinones were added, resulting in an increase of the NADH oxidation rate. Where indicated, 0.1 or 0.2 mM TPZ was added at least 3 min before the addition of quinone (modified as described in references 16 and 40). The electron acceptors used were menadione (2-methyl-1,4-naphthoquinone), 1,4-naphthoquinone, duroquinone, phenazine methosulfate, menaquinone-4, and ubiquinone. One unit was defined as 1 μmol of NADH oxidized per min and mg of protein.
NADH:acceptor oxidoreductase was measured at the wavelength specific for each acceptor tested. In all cases, a 5- to 20-μl samples were incubated in 950 μl of anoxic 0.05 M potassium phosphate buffer (pH 7.5), with the respective electron acceptor. Reactions were started by addition of 0.2 mM NADH. Electron acceptors tested were TTC, INT, nitroblue tetrazolium, benzyl viologen, methyl viologen, or ferredoxin from Clostridium pasteurianum. Concentrations, wavelengths, and extinction coefficients are indicated for the respective test. One unit was defined as 1 μmol of acceptor reduced per min and mg of protein (modified as described in reference 18). The activity of butyryl-CoA dehydrogenase was measured by using ferricenium hexafluorophosphate as an electron acceptor as described before (18).
A possible electron bifurcation reaction (Bcd/EtfAB complex) was tested as the oxidation of crotonyl-CoA with NADH and TTC, as described before (18). The reverse reaction was tested in a reaction mixture containing 0.02 M Tris-HCl (pH 7.5), 0.375 mM NAD+, and 10 μM ferredoxin from C. pasteurianum and 1 mM titanium citrate to keep the ferredoxin in its reduced state. The cell extract or soluble fraction was then added, and the increase in NADH concentration was measured at 340 nm (ɛ340 = 6.3 mM−1 cm−1 [42]). Then, 50 μM butyryl-CoA was added, and the absorption increase was monitored further at 340 nm (as described by W. Buckel, unpublished data).
Analytical methods.
Acetate was analyzed by HPLC (15) using an Aminex HPX-87H ion-exchange column (Bio-Rad) and an LC-10AT vp pump (Shimadzu). Sodium acetate was used as a standard. Protein concentrations were determined by using a microprotein assay (5) against bovine serum albumin as a standard.
Chemicals.
All chemicals were of analytical- or higher-grade quality and were obtained from Boehringer (Mannheim, Germany), Eastman Kodak (Rochester, NY), Fluka (Neu-Ulm, Germany), Merck (Darmstadt, Germany), Pharmacia (Freiburg, Germany), Serva (Heidelberg, Germany), and Sigma (Deisenhofen, Germany). Gases were purchased from Messer-Griesheim (Darmstadt, Germany) and Sauerstoffwerke Friedrichshafen (Friedrichshafen, Germany). Trifluoperazine dihydrochloride was purchased from Sigma and used as freshly prepared aqueous stock solutions.
Sequence analysis.
Basic sequence analysis was done using a Lasergene package version 5.5 from DNAstar (Madison, WI). Database searches were done using BLAST at the National Center for Biotechnology Information (NCBI) website, and the general domains and motifs in protein sequences were scanned in the NCBI Conserved Domain Search database. Transmembrane helices were scanned in the programs TMHMM 2.0 and SignalP 3.0.
RESULTS
Experiments with intact cells.
In order to assign activities specifically to the syntrophically fermenting bacterium, cells of cocultures were separated by density gradient centrifugation on a Percoll gradient as described previously (3). Butyrate oxidation by S. wolfei cells was measured as the production of formazan and acetate from butyrate, using TTC as an electron acceptor (Fig. 1). The cell suspensions produced 0.84 mM acetate from butyrate and 0.4 mM formazan from TTC after 180 min. Thereafter, the concentrations did not increase further (not shown). Control reactions in the absence of butyrate produced 27 μM formazan and no detectable acetate during 180 min (Fig. 1). When TPZ (0.1 mM) was tested as a potential inhibitor of a presumed type II NADH:menaquinone oxidoreductase in S. wolfei, the cell suspensions produced average concentrations of 0.087 mM formazan and 0.12 mM acetate during 180 min (Fig. 1). Thus, TPZ inhibited the metabolic activity of S. wolfei cells by 86 and 78%, as measured via acetate and formazan production, respectively. In mixed cell suspensions of S. wolfei and M. hungatei, TPZ inhibited butyrate oxidation by 63 to 68% (data not shown). Thus, inhibition of butyrate oxidation by TPZ was clearly associated with S. wolfei cells, presumably through inhibition of an NADH:menaquinone oxidoreductase-like enzyme that is involved in butyrate oxidation.
FIG. 1.
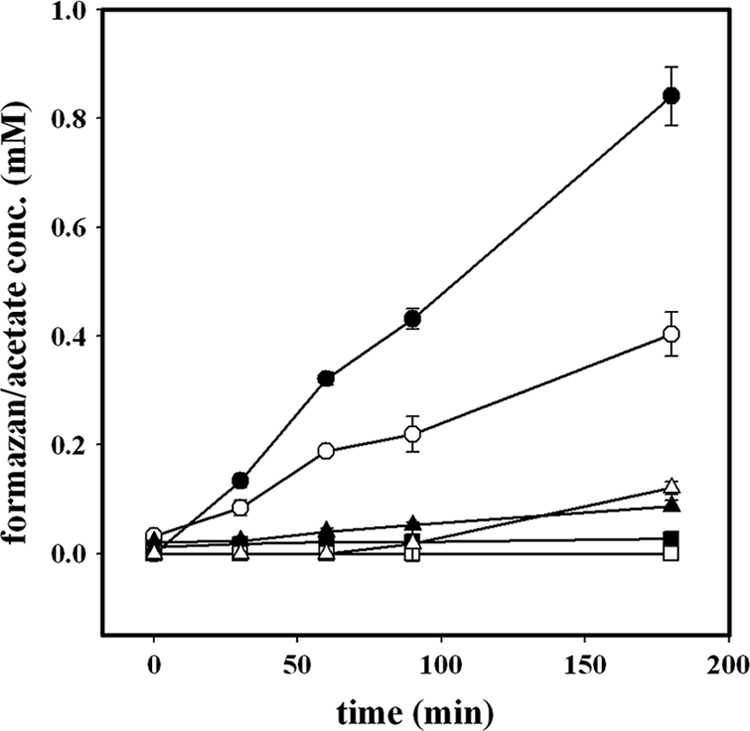
Oxidation of butyrate with TTC in cell suspensions of butyrate-grown, Percoll-separated S. wolfei cells. Shown are the concentrations of acetate and formazan. Open symbols, formazan concentration; filled symbols, acetate concentration. Circles, no inhibitor added; triangles, cell suspensions containing 0.1 mM TPZ; squares, negative control without butyrate (n = 3, mean values ± the standard deviation are indicated).
Enrichment and characterization of NADH:quinone oxidoreductase.
S. wolfei cell suspensions separated by density gradient centrifugation were opened by French press treatment, and cell extracts were assayed for NADH:quinone oxidoreductase activity. Activity was found predominantly in the soluble fraction of cell extracts (Table 1), but the activity was present also in the membrane fraction after one to three passages through the French press cell. After up to eight passages, almost all activity was in the soluble fraction. This suggested that the activity was membrane associated but could be separated from the membranes by this treatment. Enrichment of the activity was possible only by anion-exchange chromatography (DEAE and MonoQ [see Materials and Methods]), whereas after alternative treatments (hydroxyapatite, POROS HQ/H, and gel filtration on Superdex 200) the activity was permanently lost. NADH:menadione oxidoreductase activity was enriched 17-fold from 41 mg of soluble protein of S. wolfei, with a final specific activity of 59.3 U mg−1 (Table 1). In another preparation run, NADH:acceptor oxidoreductase was enriched from the membrane fraction to confirm whether the enzyme is membrane associated. S. wolfei cells were opened by only one passage through the French pressure cell (see above), and the washed membranes were solubilized by using DDM, followed by purification with the same methods as described for the soluble fraction. This preparation yielded 0.49 mg of partially purified enzyme with a specific activity of 26.5 U mg−1 (Table 1).
TABLE 1.
Partial purification of NADH:menadione oxidoreductase from S. wolfei
| Purification step | Protein amt (mg) | Sp act (U/mg)a
|
Yield (%) | Purification factor | |
|---|---|---|---|---|---|
| NADH oxidationb | NADH:menadione oxidoreductasec | ||||
| First preparation run | |||||
| Cell extract | 63.9 | 0.3 | 1.7 | NAd | NA |
| Soluble fraction | 41.4 | 0.3 | 3.4 | 100 | 1 |
| DEAE-Sepharose CL-4B (pH 8.0) | 10.2 | 0.56 | 9.9 | 72 | 2.9 |
| MonoQ (pH 8.0) | 1.07 | 2.49 | 19.9 | 15 | 5.8 |
| POROS HQ/H (pH 8.0) | 0.6 | 1.65 | 16.5 | 7 | 4.8 |
| MonoQ (pH 7.5) | 0.04 | 2.34 | 59.3 | 1.6 | 17.4 |
| Second preparation run | |||||
| Cell extract | 72 | 1.7 | 5.9 | NA | NA |
| Soluble fraction | 45.4 | 3.0 | 4.4 | NA | NA |
| Membrane fraction | 28.8 | 0.3 | 1.55 | 100 | 1 |
| Solubilized membrane protein | 14.16 | 0.37 | 5.14 | 163 | 3.3 |
| DEAE-Sepharose CL-6B (pH 8.0) | 7.92 | 0.2 | 4.75 | 84.3 | 3.06 |
| MonoQ (pH 8.0) | 0.88 | 1.46 | 20.2 | 39.8 | 13.03 |
| MonoQ (pH 7.5) | 0.48 | 0.66 | 26.46 | 12.7 | 17.07 |
Mean activities of three independent measurements are shown.
Unspecific oxidation of NADH before the addition of menadione.
Values corrected for unspecific NADH-oxidizing activity.
NA, not applicable.
The partially purified enzyme from the soluble fraction showed high NADH:menadione oxidoreductase activity but also reacted with other water-soluble quinones, as shown in Table 2. Activities were maximal with 0.2 mM NADH and 0.1 mM menadione. With 0.1 mM NADH and 0.1 mM menadione, the activity was ca. 50% of that observed with 0.2 mM NADH. All quinone-reducing activities were inhibited by TPZ. The extent of inhibition depended on the respective concentrations of NADH and quinone: with 0.2 mM TPZ, 0.2 mM NADH, and 0.1 mM menadione, the inhibition was maximal, resulting in a remaining activity of 23% of the noninhibited enzyme, whereas at equimolar concentrations of NADH and menadione almost no inhibition was observed (Table 3). A minimum concentration of 0.1 mM TPZ was needed to cause a significant inhibition at all.
TABLE 2.
Reactivity of partially purified NADH:quinone oxidoreductase with various electron acceptors
| Electron acceptor (concn [mM]) | Redox potential (mV) | Extinction coefficient (mM−1 cm−1); wavelength (nm) | TPZ concn (mM) | Sp act of NADH:acceptor oxidoreductase (U/mg) |
|---|---|---|---|---|
| Duroquinone (0.2) | +35d | NAg | 0 | 52.3 |
| 0.1 | 20.1 | |||
| Duroquinone (0.1) | +35d | NA | 0 | 15.7 |
| 0.1 | 4.02 | |||
| 1,4-Naphthoquinone (0.1) | +64d | NA | 0 | 80.4 |
| 0.1 | 37.4 | |||
| Phenazine methosulfate (0.1) | +80c | NA | 0 | 189.3 |
| 0.1 | 23.7 | |||
| INT (0.4) | -90a | 17.4; 465f | 0 | 68.9 |
| NBT (0.4) | -50a | 40.2; 605f | 0 | 0.47 |
| TTC (0.4) | -83,-240,-415b | 9.1; 546e | 0 | 41.2 |
| Benzyl viologen (1) | -360d | 8.65; 578d | 0 | 456.6 |
| Methyl viologen (1) | -440d | 9.7; 578d | 0 | 23.2 |
TABLE 3.
Inhibition of partially purified NADH:quinone oxidoreductase by TPZ
| Concn (mM)
|
Sp act (U/mg)
|
Remaining activity (%) | |||
|---|---|---|---|---|---|
| TPZ | NADH | Menadione | NADH oxidation | NADH:menadione oxidoreductase | |
| 0 | 0.2 | 0.1 | 2.34 | 59.3 | 100 |
| 0.1 | 0.2 | 0.1 | 1.61 | 26.2 | 44 |
| 0.2 | 0.2 | 0.1 | 0 | 14.1 | 23 |
| 0 | 0.1 | 0.1 | 1.0 | 19.8 | 100 |
| 0.1 | 0.1 | 0.1 | 1.31 | 19.1 | 96 |
| 0 | 0.1 | 0.04 | 1.91 | 32.3 | 100 |
| 0.1 | 0.1 | 0.04 | 1.0 | 18.8 | 58 |
No activity was observed with the commercially available menaquinone-4 or ubiquinone, and the enzyme did not react with ferredoxin of C. pasteurianum (results not shown). The activity of NADH:acceptor oxidoreductase with TTC or ferredoxin could not be stimulated by the addition of crotonyl-CoA, either in the soluble fraction or in the partially purified enzyme preparation. When testing the reverse reaction with butyryl-CoA and NAD+ and with ferredoxin kept in its reduced state by titanium citrate (see Materials and Methods), the enzyme reacted with NAD+ already before addition of butyryl-CoA, thus exhibiting ferredoxin:NAD+ oxidoreductase activity, in the range of 0.017 to 0.081 U mg−1. This activity did not increase in the presence of butyryl-CoA.
The partially purified NADH:menadione oxidoreductase activity from solubilized membranes was separated on a nondenaturing gel (Fig. 2) and appeared in a Coomassie blue stain two prominent bands of ∼130 kDa apparent size (Fig. 2A), and both prominent bands were stained positive by NADH:iodonitrotetrazolium oxidoreductase activity staining (Fig. 2B). The same picture was obtained with NADH:menadione oxidoreductase activity purified from the soluble fraction on a nondenaturing gel (data not shown). The two active bands in the nondenaturing gel (Fig. 2B) were excised, heated in SDS buffer containing DTT, and loaded onto a denaturing SDS gel (see Materials and Methods). Both excised bands (N1 and N2 in Fig. 2B) were resolved by SDS-PAGE into a set of four major bands (Fig. 2C), visible each at about 97, 60, 46, and 15 kDa, respectively; the additional protein apparent at ∼90 kDa (from band N2) was assumed to represent partially refolded or nondenatured 97-kDa protein (see below).
FIG. 2.

Partially purified NADH:quinone oxidoreductase from solubilized membranes separated on an nondenaturing gel stained with Coomassie or by activity staining, and the two active bands were further resolved on a denaturing gel. (A) Separated proteins stained with Coomassie G-250. From left to right: marker, solubilized membrane protein, after DEAE CL-6B, first MonoQ, second MonoQ. (B) Activity stain with INT and NADH as outlined in Materials and Methods. From left to right: marker, solubilized membrane protein, after DEAE CL-6B, first MonoQ, second MonoQ. (C) Further separation of the two active bands from the nondenaturing gel (bands N1 and N2 in panel B) after excision and transfer onto a denaturing gel.
These four prominent bands were consistently observed when the different preparations of NADH:menadione oxidoreductase obtained from the FPLC purification steps were separated on denaturing gels (Fig. 3), whether purified from the membrane or from the soluble fraction (cf. Fig. 3A and B), appearing as major bands at about 97, 60, 46, and 15 kDa (Fig. 3C, bands A1, A2, A3, and A7, respectively). These bands were excised and subjected to peptide mass fingerprinting (see below). Other bands which were not correlated with NADH:menadione oxidoreductase activity (bands A4, A5, and A6 in Fig. 3C) but which appeared to be enriched during the activity purifications from either the membrane fraction (cf. Fig. 3A, bands A5 and A6), or from the soluble protein fraction (cf. Fig. 3B, band A4), were also excised and submitted to peptide mass fingerprinting (see below).
FIG. 3.

SDS-PAGE of the partially purified NADH:quinone oxidoreductase of S. wolfei. (A) Purification from solubilized membrane protein. From left to right: marker, solubilized protein, after DEAE-Sepharose CL-6B, after first run on MonoQ, after second run on MonoQ. (B) Purification from the soluble fraction. From left to right: marker, soluble fraction, after DEAE-Sepharose CL-6B, after first run on MonoQ, after second run on MonoQ. (C) Comparison of membrane and soluble protein. From left to right: marker, partially purified protein from membranes, partially purified protein from soluble fraction. A1 to A7 indicate excised and identified bands. (D) Partially purified enzyme under different denaturing conditions. From left to right: marker, protein reduced with mercaptoethanol, protein sample with DTT, alkylated protein, no reducing agent added, sample not reduced and not boiled, marker.
Treatment of the preparations under various denaturing conditions did not produce differences in the SDS-PAGE banding pattern (Fig. 3D), which confirmed that the apparent protein complex was fully denatured when separated by SDS-PAGE (for the presumably selenocysteine-linked 97-kDa protein, see below).
Peptide mass fingerprinting of proteins associated with NADH:quinone oxidoreductase activity.
In all, seven different bands were excised from SDS-PAGE (see Fig. 3C), the proteins were digested, and the derived peptides were analyzed by mass spectrometry. The obtained peptide mass fingerprints were aligned against the S. wolfei genome sequence. A total of nine predicted genes could be attributed by peptide mass fingerprinting (Table 4): one band (band A7, at 15 kDa, see below) obviously represented a mixtures of two proteins with the same apparent molecular mass on SDS-PAGE, and one band (band A1, at 97 kDa) represented apparently one protein which appeared to be encoded by two predicted genes (presumably selenocysteine linked [see below]). Furthermore, several of the identified proteins appeared to be encoded by genes that are located in clusters in the genome sequence, thus in presumed operons, as detailed below.
TABLE 4.
Protein identification by peptide mass fingerprinting
| Protein band | Mass (Da) estimated by SDS-PAGE | Protein identified (by PMF) | Mass (Da) predicted by PMF | Gene locus tag | Sequence coverage (%) |
|---|---|---|---|---|---|
| A1 | 97,000 | NADH dehydrogenase I chain G | 39,714 | Swol_0785 | 84 |
| Formate dehydrogenase alpha subunit | 58,564 | Swol_0786 | 60 | ||
| A2 | 60,000 | Iron hydrogenase, small subunit | 64,259 | Swol_1017 | 70 |
| A3 | 46,000 | NADH dehydrogenase I (quinone) | 44,406 | Swol_1018 | 85 |
| A4 | 40,000 | Acetate/butyrate kinase | 43,734 | Swol_0768 | 93 |
| A5 | 39,000 | ABC transporter, periplasmic substrate binding protein | 37,964 | Swol_2479 | 72 |
| A6 | 30,000 | Extracellular solute-binding protein | 28,360 | Swol_0316 | 78 |
| A7 | 15,000 | NADH dehydrogenase I chain E | 16,563 | Swol_0783 | 62 |
| Fe-dependent hydrogenase gamma subunit | 16,181 | Swol_1019 | 67 | ||
| B1 | 65,000 | Acyl-CoA dehydrogenase | 68,811 | Swol_1933 | 73 |
| B2 | 64,000 | Acyl-CoA dehydrogenase | 68,798 | Swol_2052 | 80 |
| B3 | 40,000 | Putative iron-sulfur binding reductase (only C-terminal half) | 82,333 | Swol_0698 | 46 |
The prominent band at 46 kDa (band A3 in Fig. 3C) was attributed to predicted gene Swol_1018 (derived 44.4 kDa; see Table 4), which was annotated as the NADH-binding subunit gene of a [FeFe] hydrogenase/NADH:ubiquinone oxidoreductase complex (NuoF-like; COG1894). No other identified protein (see below) could be attributed to the same enzyme function (NADH:acceptor oxidoreductase). Hence, this gene was most likely responsible for the apparent NADH:menadione reductase activity of our semipurified enzyme. The deduced amino acid sequence showed the highest level of identity to three sequences derived from paralog genes in S. wolfei (Swol_1024, 1828, and 0784, with 76, 73, and 64% amino acid identity [id-aa], respectively), and to a sequence derived from the genome of Pelotomaculum thermopropionicum (PTH_2011; 56% id-aa). Furthermore, the Swol_1018 protein sequence was closely related to those of characterized [FeFe] hydrogenase components, e.g., HydB in Thermoanaerobacter tengcongensis (33) (TTE0893; 55%), HndC in Desulfovibrio fructosovorans (7) (U07229; 50%), and HydB in Thermotoga maritima (30) (TM1425; 46%). Notably, the Swol_1018 sequence lacked the N-terminal part (141 amino acids) of the HydB sequences of T. tengcongensis and T. maritima, which contain an additional [2Fe2S] binding site (33), thus showing more similarity in its domain structure to HndC of D. fructosovorans (above) and to NuoF of NADH:ubiquinone oxidoreductase complex I in Escherichia coli (locus tag b2284; 40% id-aa).
In the genome sequence, this attributed gene (Swol_1018) was part of an apparent three-gene operon (not shown). Peptide-mass fingerprints corresponding to the two other genes, Swol_1019 and Swol_1017, were obtained for the prominent protein bands that were excised from SDS-PAGE at ∼15 kDa (band A7 in Fig. 3) and 60 kDa (band A2), respectively (Table 4). The attributed gene Swol_1019 (derived mass of 15.9 kDa) was predicted to encode a [2Fe2S] ferredoxin component of a [FeFe] hydrogenase complex (NuoE-like; COG1905). The deduced amino acid sequence showed the highest level of identity to sequences derived from paralog genes in S. wolfei (four candidates, each with 66% id-aa), and with <41% homology to ferredoxin subunits in other sequenced organisms, e.g., HydC of the characterized [FeFe] hydrogenase complex in T. tengcongensis (TTE0890; 33%) and T. maritima (TM1424; 31%). The other gene, Swol_1017 (derived mass of 62.9 kDa), was annotated to encode a [FeFe] hydrogenase catalytic subunit (COG4624). The best BLAST hit was obtained for a paralog gene (Swol_2436; 68% id-aa), the second best BLAST hit was obtained for a gene predicted in P. thermopropionicum (PTH_2010; 60%); furthermore, the protein sequence appeared to be closely related to the characterized hydrogenase component HydA of T. tengcongensis (TTE0894; 55%), HndD of D. fructosovorans (7) (U07229; 51%), and HydA of T. maritima (TM1426; 40%). Notably, the Swol_1017 protein sequence lacked the C-terminal part (89 amino acids) of the HydA sequences in T. tengcongensis and T. maritima, which contain an additional [2Fe2S] binding site (33), thus showing in its domain structure more similarity to HndD of D. fructosovorans (above) and monomeric hydrogenase of C. pasteurianum (P29166; 40% id-aa). Thus, the peptide mass fingerprinting and sequence analysis suggested that these three proteins are coexpressed from a presumed operon in S. wolfei (Swol_1017-1019) during syntrophic growth with butyrate and constitute most likely a multimeric [FeFe] hydrogenase complex (HydABC-like). The subunit structure of the apparent NADH:quinone oxidoreductase activity (see above) confirmed that these three proteins (bands A2, A3, and A7 in Fig. 3C) form a protein complex in vivo which appears to be associated with the membrane.
The band which was excised at ∼15 kDa (band A7) yielded also sufficiently distinctive peptide mass fingerprint signals to attribute a second NuoE-like protein to be present in our protein fraction, which was encoded by gene Swol_0783 (see Table 4). This gene was suggested to be part of an apparent four-gene operon in S. wolfei (data not shown). Corresponding peptide mass fingerprints were obtained for two of the three other genes, Swol_0785 and Swol_0786 (Table 4). Interestingly, the fingerprint derived from the 97-kDa band (band A1 in Fig. 3C) matched the individual peptide masses of the genes Swol_0785 and Swol_0786, which are predicted to encode 39.7- and 58.6-kDa proteins, respectively (Table 4). The open reading frames of Swol_0785 and Swol_0786 appeared to be in-frame, with an apparent noncoding spacing of 48 bp (not shown). However, when the two derived protein sequences were conjoined (predicted size of 98.1 kDa), including the noncoding spacing (16 amino acids), and aligned against the database, BLAST hits were obtained for contiguous protein sequences with either selenocysteine or cysteine at the position of the stop codon of Swol_0785 (not shown). The best BLAST hit (55% id-aa) was obtained for the selenocysteine-containing formate dehydrogenase protein of Eubacterium acidaminophilum (FdhA-II, CAC39239); a predicted formate dehydrogenase sequence with cysteine instead of selenocysteine was found in Pelobacter propionicus (YP_903170; 52% id-aa). Peptide mass fingerprinting thus suggested that a selenocysteine-containing formate dehydrogenase was expressed in S. wolfei cells during syntrophic growth with butyrate, which appeared to copurify with our NADH:quinone oxidoreductase activity.
The bands submitted to peptide mass fingerprinting that were not correlated to NADH:quinone oxidoreductase activity (see above, bands A4, A5, and A6 in Fig. 3C), were attributed to the predicted gene Swol_0768 (band A4) annotated to code for acetate kinase (EC:2.7.2.1), and to predicted genes Swol_2479 (band A5) and Swol_0316 (band A6), which were annotated each to code for periplasmic components of ABC-type transport systems (Table 5).
TABLE 5.
Partial purification of butyryl-CoA dehydrogenase
| Purification step | Protein amt (mg) | Sp act (U/mg) | Yield (%) | Purification factor |
|---|---|---|---|---|
| Cell extract | 72 | NDa | NAb | NA |
| Soluble fraction | 45.4 | 12.19 | 100 | 1 |
| DEAE-Sepharose CL-6B (pH 8.0) | 5.14 | 40.72 | 37.8 | 3.34 |
| Membrane fraction | 28.8 | 0.11 | 100 | 1 |
| Solubilized membrane protein | 14.16 | 0.53 | 236 | 4.81 |
| DEAE-Sepharose CL-6B (pH 8.0) | 0.765 | 14.19 | 342 | 129 |
ND, not determined.
NA, not applicable.
Identification of butyryl-CoA dehydrogenase activity and of a DUF224 protein in S. wolfei.
The genome sequence of S. wolfei harbors at least nine candidate genes for butyryl-CoA dehydrogenases (bcd), one of which (Swol_0268) was colocated in a putative bcd-etfBA operon in synteny to that in C. kluyveri, and a second one (Swol_2126) was located several genes apart from a paralog set of etfBA genes in S. wolfei. Other bcd candidate genes were found in presumed operons with other butyryl-metabolic genes (e.g., Swol_0788, Swol_1483, and Swol_1933 [not shown]). The soluble protein fraction of butyrate-grown S. wolfei cells exhibited high activity of Bcd (Table 5), and the activity eluted as one peak during the first FPLC purification step (DEAE), in a fraction well separated from the NADH:menadione oxidoreductase activity (data not shown). The fraction exhibited a green-yellow color, suggestive of a high content of flavins (not shown). SDS-PAGE (Fig. 4) showed two proteins in the 66-kDa size range that were analyzed by peptide mass fingerprinting (bands B1 and B2; see also Table 5). The proteins were attributed to predicted genes Swol_1933 and Swol_2052, respectively, each annotated as FAD-containing acyl-CoA dehydrogenase genes (COG1960) with high sequence identity (75% id-aa). Interestingly, the derived Bcd sequences showed much lower identity (<20% id-aa) to the sequence encoded by the bcd genes which are associated etfBA genes in the C. kluyveri genome (bcd-etfBA operon; YP_001393857) or in the S. wolfei genome (see above). Thus, the butyryl CoA dehydrogenase activity in S. wolfei cells during syntrophic growth with butyrate was represented by two similar enzymes that copurified during FPLC, and these enzymes appeared to be different from the Bcd(s) associated with the described Buckel-Thauer reaction complex, the Bcd/EtfAB complex.
FIG. 4.

SDS-PAGE of partially purified butyryl-CoA dehydrogenase. From left to right: solubilized membrane protein, DEAE CL-6B, soluble fraction, soluble fraction after DEAE CL-6B, marker. B1 to B3 indicate excised and identified bands.
When the butyryl-CoA dehydrogenase activity was purified from the membrane fraction, an additional protein appeared on SDS-PAGE (band B3, at ∼40 kDa in Fig. 4). This band was identified by peptide mass fingerprinting to represent the predicted gene Swol_0698, annotated to encode a domain-of-unknown function 224 protein (DUF224; COG0247), which comprises a predicted transmembrane domain (N terminal) and a predicted 4Fe4S-cluster binding domain (C terminal). The predicted size of the derived protein sequence (81.4 kDa) did not match the size of the protein observed on SDS-PAGE (∼40 kDa), and the peptide signals obtained from peptide mass fingerprinting solely matched to, and fully comprised, the C-terminal half of the predicted sequence (∼40 kDa) (data not shown). We conclude that S. wolfei expressed a DUF224-like protein during growth with butyrate, the N-terminal transmembrane domain of which was either lost during the purification procedure or absent in the protein in vivo. Interestingly, this protein appeared to be encoded in a presumed operon with a third paralog pair of etfBA genes in the S. wolfei genome.
DISCUSSION
Release of electrons from butyryl-CoA oxidation to crotonyl-CoA in the form of molecular hydrogen requires a reversed electron transport system to overcome the redox potential difference between the donor (E°′ = −125/−10 mV [12, 28]) and the acceptor system (E°′ = −414 mV; −300 mV at 10−4 atm H2 [29]). An early hypothesis how such a reversed electron transport could operate in S. wolfei assumed the involvement of a menaquinone (29, 36). This concept seemed plausible also for glycolate-oxidizing syntrophs (8, 29). After investigation of the genome sequence of Syntrophus aciditrophicus, this hypothesis was supported by a more refined model involving an NADH:quinone oxidoreductase enzyme complex. It was stated that NADH produced from menaquinone can be oxidized by proton- or Na+-translocating Rnf-proteins reducing ferredoxin which in turn could be reoxidized by a classical ferredoxin-dependent hydrogenase (23). However, in the genome sequence of S. wolfei, we found no gene cluster for Rnf proteins as in Syntrophus acidotrophicus (encoded by SYN_01664-01659 [23]), especially no significant homologies for predicted integral membrane proteins RnfA, RnfD, RnfE, and RnfG (23).
After the discovery of a dismutation reaction between NADH, ferredoxin, and crotonyl-CoA in C. kluyveri, Herrmann et al. (13) regarded a reversed electron transport via quinones as unlikely because the genome of S. aciditrophicus lacks the complete set of enzymes for complex I. Instead, participation of a Bcd/EtfAB complex in butyrate oxidation by syntrophic fatty acid oxidizers was postulated (13, 18; W. Buckel, unpublished data); this was supported by the fact that a gene cluster of bcd-etfBA genes was found in the genome of S. wolfei. This assumption was tested in the present study by repeating the enzyme assays of Li et al. (18) with cell extracts of S. wolfei. However, rapid oxidation of NADH with several electron acceptors, including the artificial electron acceptor TTC, never required or showed an increased oxidation rate by crotonyl-CoA, as would be typical of NADH oxidation by the Bcd/EtfAB complex of C. kluyveri (18). Reduction of crotonyl-CoA with ferredoxin plus NADH could not be tested since this reaction would require hydrogenase in the assay, which is not commercially available, to keep ferredoxin in the oxidized state.
We found high activity of butyryl-CoA dehydrogenase with ferricenium as an electron acceptor in cell extracts, a reaction catalyzed by the Bcd/EtfAB complex as a side activity (18). This activity could be enriched from both the soluble fraction and the membrane fraction of S. wolfei, and two acyl-CoA dehydrogenases distinct from the Bcd of the Bcd/EtfAB complex were identified. This is consistent with earlier observations that up to 18% of the butyryl-CoA dehydrogenase activity in S. wolfei is located in the membrane fraction (36) and implies that the butyryl-CoA dehydrogenases of S. wolfei might react with components of the membrane. Therefore, it appears unlikely that the Bcd/EtfAB complex is involved in reversed electron transport during butyrate oxidation by S. wolfei. Nonetheless, this complex may be expressed under different growth conditions, e.g., during growth by dismutation of crotonate to acetate and butyrate. Even if one would assume an operation of a Bcd/EtfAB complex working in reverse during butyryl-CoA oxidation with NAD+, driven by reduced ferredoxin, there remains the problem of how the reduced ferredoxin could be regenerated, since no Rnf proteins are encoded in the genome of S. wolfei and there is no oxidation step involved in butyrate oxidation that could be coupled directly to ferredoxin reduction.
Several genes for butyrate metabolism are predicted to be clustered around the two identified Bcd genes (Swol_1933 and Swol_2052) in presumed operons (data not shown), i.e., enoyl-CoA hydratase (Swol_1936), 3-hydroxyacyl-CoA dehydrogenase (Swol_1935), and acetyl-CoA C-acyltransferases (Swol_1934 and Swol_2051). We therefore suggest that these genes are likely to be coexpressed in S. wolfei during growth with butyrate. Furthermore, the copurification of a DUF224 predicted transmembrane FeS-binding reductase (Swol_0698) with the Bcd activity from the membrane fraction (Fig. 4 and Table 4) and the fact that this gene is encoded in a presumed operon together with a (third) paralog set of predicted etfBA genes (Swol_0696 and Swol_0697; no bcd candidate) makes it tempting to speculate that these genes might be coexpressed during growth with butyrate and constitute a membrane-associated Bcd/EtfAB/DUF224 complex that funnels electrons from the butyryl-CoA/crotonyl-CoA redox couple into the membrane.
The high NADH-oxidizing activity observed in cell extracts promised to be an alternative starting point to approach the proposed reversed electron transport in S. wolfei. For purification of the NADH-oxidizing enzyme of S. wolfei, the methanogenic partner had to be removed because M. hungatei has been reported to exhibit NADH diaphorase activity (24). Menadione was found to be an electron acceptor of this NADH-oxidizing enzyme in S. wolfei, and NADH:menadione oxidoreductase activity was shown earlier to be an attribute of NADH dehydrogenases of the NDH-2-type (40) and of complex I-like enzymes (NDH-1) (16), although menadione is an artificial, water-soluble quinone lacking the alkyl side chain of natural quinones. The activity appeared to be associated with the membrane but could be resolved into the soluble protein fraction, thus showing another attribute typical of NDH-2-type and complex I-like NADH dehydrogenases, both of which are membrane-associated, but nontransmembrane proteins (16, 40). In experiments with cell suspensions, TPZ, an antitubercular agent acting against NADH:menaquinone oxidoreductase (NDH-2) in M. tuberculosis (40), was found to inhibit butyrate oxidation in S. wolfei, probably by blocking an NADH oxidoreductase enzyme. Accordingly, the partially purified NADH:menadione oxidoreductase activity from butyrate-grown cells of S. wolfei was also inhibited by TPZ. The enzyme appeared to be essential for butyrate oxidation because TPZ also inhibited butyrate oxidation and acetate production by intact S. wolfei cells with TTC as an electron acceptor; TTC has been reported to accept electrons over a wide range of redox potentials from −83 mV to −415 mV (27, 32) and was proven to be an indicator for the activity of complex I in mitochondria (27). Hence, we anticipated that the NADH:menaquinone oxidoreductase activity of S. wolfei is represented by an enzyme similar to the membrane-associated NDH-2 or complex-I-type NADH dehydrogenases, i.e., linked to the quinone pool in the membrane, and that this enzyme is involved in the proposed reversed electron transport in S. wolfei.
The semipurified NADH:menaquinone oxidoreductase activity separated by nondenaturing gels showed two prominent, active bands (Fig. 2A and B) that each could be resolved into the same set of four subunits by denaturing SDS-PAGE (Fig. 2C). We conclude that the semipurified activity was represented by a protein complex that appeared as a double band on nondenaturing gels, perhaps due to differences in charge, folding, or subunit assembly. The cumulative mass of the complex calculated from the masses of its subunits (97, 60, 46, and 15 kDa; Fig. 2C) was >210 kDa, whereas the bands of the complex on native gels were found close to the 140-kDa marker (Fig. 2A and B). Such a discrepancy is not uncommon because native gels do not allow reliable protein mass estimation. The four attributed subunits of the complex were consistently enriched both from the membrane and the soluble fraction, thus confirming that the NADH:menaquinone oxidoreductase activity was purified as a four-subunit protein complex.
This NADH-oxidizing protein complex contains a 46-kDa subunit (band A3 in Fig. 3), as indicated by peptide mass fingerprinting (Table 4), which was attributed as the NADH-accepting subunit of a [FeFe] hydrogenase/NADH:ubiquinone oxidoreductase complex (HydB/NuoF-like). The corresponding gene was part of an apparent three-gene operon (Swol_1017 to Swol_1019), and the 60- and 15-kDa proteins of the complex (bands A2 and A7 in Fig. 3) matched the other two predicted genes: the gene for the 60-kDa subunit (Swol_1019) was indicated to encode a catalytic [FeFe] hydrogenase subunit (HydA-like), and the gene for the 15-kDa subunit was indicated to encode a ferredoxin (HydC-like). Thus, surprisingly, the protein complex that we purified as NADH:menaquinone oxidoreductase activity was ascribed to contain the subunit homologues of a three-component hydrogenase complex, for example, of Thermotoga maritima, which was recently described as an electron-bifurcating hydrogenase (30). The fourth protein that consistently was copurified apparently as part of this protein complex, the 97-kDa protein (band A1 in Fig. 3), was attributed as a selenocysteine-containing catalytic subunit of a formate dehydrogenase complex encoded by two genes (Swol_0785 and Swol_0786) that are translated as one protein through selenocysteine insertion (41; P. Worm and C. Plugge, unpublished data). The homologous enzyme in Eubacterium acidaminophilum (FdhA-II) has been implicated to play a role also in interspecies formate transfer (10, 43).
The association of these attributed proteins as subunits of a membrane-associated protein complex during syntrophic growth of S. wolfei with butyrate appears likely based on their purification and separation properties discussed above, and the TPZ inhibition experiments indicated a crucial role for the NADH oxidoreductase component of the protein complex in butyrate oxidation. Other catalytic features attributed to the complex by sequence analysis, i.e., the predicted hydrogenase and formate dehydrogenase activities, must be confirmed in future biochemical studies. The observation that this complex could contain an electron-bifurcating hydrogenase leads to speculation as to whether NADH oxidation and hydrogen formation could be catalyzed in S. wolfei also via electron bifurcation, i.e., proton reduction with NADH plus reduced ferredoxin, as described for the Thermotoga enzyme system (30). However, for S. wolfei the same problem would arise as with the proposed electron-bifurcating Bcd/EtfAB complex, i.e., how ferredoxin could be reduced first to drive the bifurcation reaction (see above). Since the protein complex appears to be membrane associated and resembles NADH:quinone oxidoreductases, we assume that it could indeed be supplied with electrons from the butyryl-CoA/crotonyl-CoA redox couple, perhaps via a proton-translocating menaquinone cycle. Menaquinone-7 was previously found to be present in the membranes of S. wolfei (36) and might likely be the physiological reaction partner of this enzyme. The necessary proton motive force could be provided by ATP hydrolysis, thus sacrificing part of the ATP produced via substrate-level phosphorylation in butyrate oxidation as proposed previously (34, 36). Shifting electrons from the level of reduced menaquinol (E0′ = −74 mV [35]) to the level of NADH (E0′ = −320 mV [35]) would require an energy expenditure of +46 kJ/mol, which can be provided by two protons pumped across the membrane. Experimental evidence for the involvement of a proton gradient in butyrate oxidation by S. wolfei was provided thus far only with intact cells (36). To show this linkage in a cell-free system will be a matter of future investigations.
Acknowledgments
We thank Diliana Simeonova and Karin Denger for numerous helpful suggestions concerning protein purification and identification. We also thank Petra Worm and Caroline Plugge, University of Wageningen, for help with the interpretation of sequence data. We also thank Antje Wiese for technical assistance, especially for preparation of the growth medium and of the ferredoxin from C. pasteurianum. The publicly available genome sequence established by DoE-JGI is greatly appreciated.
This project was funded by the Deutsche Forschungsgemeinschaft, Bonn-Bad Godesberg, and by research funds of the University of Constance.
Footnotes
Published ahead of print on 31 July 2009.
Dedicated to Rudolph K. Thauer on the occasion of his 70th birthday.
REFERENCES
- 1.Altman, F. P. 1976. Tetrazolium salts and formazan. Prog. Histochem. Cytochem. 9:1-51. [DOI] [PubMed] [Google Scholar]
- 2.Beaty, P. S., and M. J. McInerney. 1990. Nutritional features of Syntrophomonas wolfei. Appl. Environ. Microbiol. 56:3223-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaty, P. S., N. Q. Wofford, and M. J. McInerney. 1987. Separation of Syntrophomonas wolfei from Methanospirillum hungatei in syntrophic cocultures by using Percoll gradients. Appl. Environ. Microbiol. 53:1183-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmeyer, H. U. 1974. Methoden der enzymatischen Analyse, 3rd ed. Verlag Chemie, Weinheim, Germany.
- 5.Bradford, M. M. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 6.Denke, E., T. Merbitz-Zahradnik, O. M. Hatzfeld, C. H. Snyder, T. A. Link, and B. L. Trumpower. 1998. Alteration of the midpoint redox potential and catalytic activity of the Rieske iron-sulfur protein by changes of amino acids forming hydrogen bonds to the iron-sulfur cluster. J. Biol. Chem. 273:9085-9093. [DOI] [PubMed] [Google Scholar]
- 7.Dermoun, Z., G. De Luca, M. Asso, P. Bertrand, F. Guerlesquin, and B. Guigliarelli. 2002. The NADP-reducing hydrogenase from Desulfovibrio fructosovorans: functional interaction between the C-terminal region of HndA and the N-terminal region of HndD subunits. Biochim. Biophys. Acta 1556:217-225. [DOI] [PubMed] [Google Scholar]
- 8.Friedrich, M. W., and B. Schink. 1993. Hydrogen formation from glycolate driven by reversed electron transport in membrane vesicles of a syntrophic glycolate-oxidizing bacterium. Eur. J. Biochem. 217:233-240. [DOI] [PubMed] [Google Scholar]
- 9.Reference deleted.
- 10.Graentzdoerffer, A., D. Rauh, A. Pich, and J. R. Andreesen. 2003. Molecular and biochemical characterization of two tungsten- and selenium-containing formate dehydrogenases from Eubacterium acidaminophilum that are associated with components of an iron-only hydrogenase. Arch. Microbiol. 179:116-130. [DOI] [PubMed] [Google Scholar]
- 11.Reference deleted.
- 12.Gustafson, W. G., B. A. Feinberg, and J. T. McFarland. 1986. Energetics of β-oxidation. Reduction potentials of general fatty acyl-CoA dehydrogenase, electron transfer flavoprotein and fatty acyl-CoA substrates. J. Biol. Chem. 261:7733-7741. [PubMed] [Google Scholar]
- 13.Herrmann, G., E. Jayamani, G. Mai, and W. Buckel. 2008. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190:784-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karmakar, S. S., A. G. E. Pearse, and A. M. Seligman. 1960. Preparation of nitrotetrazolium salts containing benzothiazole. J. Org. Chem. 25:575-578. [Google Scholar]
- 15.Klebensberger, J., O. Rui, E. Fritz, B. Schink, and B. Philipp. 2006. Cell aggregation of Pseudomonas aeruginosa strain PAO1 as an energy-dependent stress response during growth with sodium dodecyl sulfate. Arch. Microbiol. 185:417-427. [DOI] [PubMed] [Google Scholar]
- 16.Krebs, W., J. Steuber, A. C. Gemperli, and P. Dimroth. 1999. Na+ translocation by the NADH:ubiquinone oxidoreductase (complex I) from Klebsiella pneumoniae. Mol. Microbiol. 33:590-598. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 18.Li, F., J. Hinderberger, H. Seedorf, J. Zhang, W. Buckel, and R. K. Thauer. 2008. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri J. Bacteriol. 190:843-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McInerney, M. J. 1986. Transient and persistent associations among prokaryotes, p. 293-338. In J. S. Poindexter and E. R. Leadbetter (ed.), Bacteria in nature, vol. 2. Plenum Press, Inc., New York, NY.
- 20.McInerney, M. J. 1988. Anaerobic degradation of proteins and lipids, p. 373-415. In A. J. B. Zehnder (ed.), Biology of anaerobic microorganisms. John Wiley & Sons, Inc., New York, NY.
- 21.McInerney, M. J., M. P. Bryant, and N. Pfennig. 1979. Anaerobic bacterium that degrades fatty acids in syntrophic association with methanogens. Arch. Microbiol. 122:129-135. [DOI] [PubMed] [Google Scholar]
- 22.McInerney, M. J., M. P. Bryant, R. B. Hespell, and J. W. Costerton. 1981. Syntrophomonas wolfei gen. nov. sp. nov., an anaerobic, syntrophic, fatty acid-oxidizing bacterium. Appl. Environ. Microbiol. 41:1029-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McInerney, M. J., L. Rohlin, H. Mouttaki, U. Kim, R. S. Krupp, L. Rios-Hernandez, J. Sieber, C. G. Struchtemeyer, A. Bhattacharyya, J. W. Campbell, and R. P. Gunsalus. 2007. The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. USA 104:7600-7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKellar, R. C., K. M. Shaw, and G. D. Sprott. 1981. Isolation and characterization of a FAD-dependent NADH diaphorase from Methanospirillum hungatei strain GP1. Can. J. Biochem. 59:83-91. [DOI] [PubMed] [Google Scholar]
- 25.Müller, N., B. M. Griffin, U. Stingl, and B. Schink. 2008. Dominant sugar utilizers in sediment of Lake Constance depend on syntrophic cooperation with methanogenic partner organisms. Environ. Microbiol. 10:1501-1511. [DOI] [PubMed] [Google Scholar]
- 26.Neuhoff, V., N. Arold, D. Taube, and W. Erhardt. 1988. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9:255-262. [DOI] [PubMed] [Google Scholar]
- 27.Rich, P. R., L. A. Mischis, S. Purton, and J. T. Wiskich. 2001. The sites of interaction of triphenyltetrazolium chloride with mitochondrial respiratory chains. FEMS Microbiol. Lett. 202:181-187. [DOI] [PubMed] [Google Scholar]
- 28.Sato, K., Y. Nishina, C. Setoyama, R. Miura, and K. Shiga. 1999. Unusually high standard redox potential of acrylyl-CoA/propionyl-CoA couple among enoyl-CoA/acyl-CoA couples: a reason for the distinct metabolic pathway of propionyl-CoA from longer acyl-CoAs. J. Biochem. 126:668-675. [DOI] [PubMed] [Google Scholar]
- 29.Schink, B. 1997. Energetics of syntrophic cooperation in methanogenic cooperation. Microbiol. Mol. Biol. Rev. 61:262-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schut, G. J., and M. W. Adams. 2009. The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production. J. Bacteriol. 191:4451-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinegina, L., M. Wikstrm, M. I. Verkhovsky, and M. L. Verkhovskaya. 2005. Activation of isolated NADH:ubiquinone reductase I (complex I) from Escherichia coli by detergent and phospholipids: recovery of ubiquinone reductase activity and changes in EPR signals of iron-sulfur clusters. Biochemistry 44:8500-8506. [DOI] [PubMed] [Google Scholar]
- 32.Slater, T. F., B. Sawyer, and U. Sträuli. 1963. Studies on succinate-tetrazolium reductase systems. III. Points of coupling of four different tetrazolium salts. Biochim. Biophys. Acta 77:383-393. [DOI] [PubMed] [Google Scholar]
- 33.Soboh, B., D. Linder, and R. Hedderich. 2004. A multisubunit membrane-bound [NiFe] hydrogenase and an NADH-dependent Fe-only hydrogenase in the fermenting bacterium Thermoanaerobacter tencongensis. Microbiology 150:2451-2463. [DOI] [PubMed] [Google Scholar]
- 34.Thauer, R. K., and J. G. Morris. 1984. Metabolism of chemotrophic anaerobes: old views and new aspects, p. 123-168. In D. P. Kelly and N. G. Carr (ed.), The microbe 1984. II. Prokaryotes and eukaryotes. Cambridge University Press, Cambridge, United Kingdom.
- 35.Thauer, R. K., K. Jungermann, and K. Decker. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41:100-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wallrabenstein, C., and B. Schink. 1994. Evidence of reversed electron transport in syntrophic butyrate or benzoate oxidation by Syntrophomonas wolfei and Syntrophus buswellii. Arch. Microbiol. 162:136-142. [Google Scholar]
- 37.Westermeier, R. 1990. Elektrophorese Praktikum. Verlag Chemie, Weinheim, Germany.
- 38.Widdel, F., and F. Bak. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p. 3352-3378. In A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K. H. Schleifer (ed.), The prokaryotes. Springer Verlag, Berlin, Germany.
- 39.Widdel, F., G. W. Kohring, and F. Mayer. 1983. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. III. Characterization of the filamentous gliding Desulfonema limicola gen. nov. sp. nov., and Desulfonema magnum sp. nov. Arch. Microbiol. 134:286-294. [Google Scholar]
- 40.Yano, T., L. Lin-Sheng, E. Weinstein, J. S. The, and H. Rubin. 2006. Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2). J. Biol. Chem. 281:11456-11463. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, Y., and V. N. Gladyshev. 2005. An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics 21:2580-2589. [DOI] [PubMed] [Google Scholar]
- 42.Ziegenhorn, J., M. Senn, and T. Bücher. 1976. Molar absorptivities of β-NADH and β-NADPH. Clin. Chem. 22:151-160. [PubMed] [Google Scholar]
- 43.Zindel, U., W. Freudenberg, M. Rieth, J. R. Andreesen, J. Schnell, and F. Widdel. 1988. Eubacterium acidaminophilum sp. nov., a versatile amino acid-degrading anaerobe producing or utilizing H2 or formate: description and enzymatic studies. Arch. Microbiol. 150:254-266. [Google Scholar]