

Abstract
Pseudomonas putida KT2440 is a model strain for studying bacterial biodegradation processes. However, very little is known about nitrogen regulation in this strain. Here, we show that the nitrogen regulatory NtrC proteins from P. putida and Escherichia coli are functionally equivalent and that substitutions leading to partially active forms of enterobacterial NtrC provoke the same phenotypes in P. putida NtrC. P. putida has only a single PII-like protein, encoded by glnK, whose expression is nitrogen regulated. Two contiguous NtrC binding sites located upstream of the σN-dependent glnK promoter have been identified by footprinting analysis. In vitro experiments with purified proteins demonstrated that glnK transcription was directly activated by NtrC and that open complex formation at this promoter required integration host factor. Transcription of genes orthologous to enterobacterial codB, dppA, and ureD genes, whose transcription is dependent on σ70 and which are activated by Nac in E. coli, has also been analyzed for P. putida. Whereas dppA does not appear to be regulated by nitrogen via NtrC, the codB and ureD genes have σN-dependent promoters and their nitrogen regulation was exerted directly by NtrC, thus avoiding the need for Nac, which is missing in this bacterial species. Based upon these results, we propose a simplified nitrogen regulatory network in P. putida (compared to that in enterobacteria), which involves an indirect-feedback autoregulation of glnK using NtrC as an intermediary.
Global nitrogen regulation is a complex regulatory network involving a number of signal transduction and effector proteins, which has been studied intensively for enterobacteria (reviewed in references 35 and 49). PII proteins play an essential role in nitrogen regulation in different bacteria (24). Their function is modulated by different effectors, by uridylylation by the uridylyltransferase/uridylyl-removing enzyme (GlnD), or by other posttranslational modifications in other bacteria (7, 50, 54). PII proteins control transcription of many nitrogen-regulated genes by regulating the kinase and phosphatase activities of NtrB, the sensor of the global two-component regulatory NtrB/NtrC system, thus regulating the phosphorylation state of the transcriptional activator NtrC. They also control ammonium assimilation through glutamine synthetase by modifying its activity via adenylylation by the adenylyl-transferase enzyme (GlnE), as well as the functions of other target proteins, such as the NifL/NifA regulatory system, controlling the expression of nitrogen fixation genes, or the DraT/DraG system, which leads to nitrogenase switch-off in alphaproteobacteria (18, 24). In enterobacteria, there are two paralogous genes that encode PII proteins, glnB and glnK (57). The functions of GlnB and GlnK are partially redundant, and while GlnB is produced constitutively, GlnK is expressed only under nitrogen-limiting conditions, as transcription of glnK is dependent on NtrC. While the arrangement of the two genes encoding PII proteins is conserved in most alpha- and betaproteobacteria, a number of gammaproteobacteria, such as Azotobacter and Pseudomonas species, have only the glnK ortholog (1). Expression of glnA-ntrBC and glnK-amtB is constitutive in Azotobacter vinelandii (41), but both operons appear to be nitrogen regulated in Pseudomonas putida (30; also see below). Similarly, most gram-positive bacteria possess only a GlnK-like PII protein, while cyanobacteria possess a GlnB-like PII protein (1, 24).
In enterobacteria, most of the operons regulated by nitrogen have promoters recognized by the alternative form of RNA polymerase holoenzyme containing the sigma factor σN. Transcription from σN-dependent promoters is strictly dependent on activation by an enhancer binding protein, which, in the case of nitrogen-regulated promoters, is the phosphorylated form of NtrC (NtrC-P). The mechanism of transcription activation at these promoters involves initial phosphorylation of the NtrC dimer and further oligomerization facilitated by binding of NtrC-P to specific DNA sequences that function as enhancers located at a position relatively distant from the promoter. Subsequently, the enhancer-bound activator interacts with σN RNA polymerase bound to the promoter via formation of a DNA loop of the intervening DNA to catalyze the isomerization of the closed promoter complexes into the transcriptionally productive open complexes in a reaction that requires ATP hydrolysis (59-61). Efficient transcription activation at a number of σN-dependent promoters, some of which are activated by NtrC, also requires the nucleoid-associated protein integration host factor (IHF). IHF binds between the promoter and the enhancer and induces a bend in the DNA upon binding that assists the interaction of the closed complex and the enhancer-bound transcriptional activator (31, 52), while preventing activation by other transcriptional activators that are not bound to the DNA at the appropriate location (47, 58).
A subset of nitrogen-regulated operons in Escherichia coli and Klebsiella species contain promoters that are recognized by σ70 RNA polymerase holoenzyme. Transcription of these operons is regulated by the nitrogen assimilation control protein (Nac), a LysR-type regulator (8, 49). Genes or operons activated by Nac in E. coli are involved in transport of nitrogenated molecules that may serve as nitrogen sources (63). In Klebsiella pneumoniae, utilization of urea and some amino acids is activated by Nac as well as the codBA operon (8, 16, 32, 37). Nac does not respond to nitrogen availability, but its transcription is nitrogen regulated via NtrC (8, 22). Thus, Nac acts as an adapter that couples the transcription of σ70-dependent operons to the nitrogen regulation exerted by the global transcriptional activator of σN-dependent promoters by NtrC (63).
P. putida KT2440 is a model strain for studying bacterial biodegradation processes because of its versatility and ease of manipulation. It has more than 65 sensor-regulator pairs, which make it able to adapt to very different conditions, including nutrient deprivation (44, 55). In spite of this, much less is known about nitrogen regulation in pseudomonads than in other bacteria. A number of studies suggest that the regulatory system in Pseudomonas may share many features with that in enterobacteria. The ntrB, ntrC, and rpoN genes are found in the genomes of different Pseudomonas species. Mutants lacking σN are impaired in utilization of a number of nitrogen sources (34, 56). Similarly, mutational analysis of the ntrC orthologs identified in Pseudomonas indicates that NtrC is the master nitrogen regulator, which is required to activate the expression of a number of genes involved in nitrogen uptake and metabolism (30), including the glnA-ntrBC operon and the atzDEF operon, required for utilization of cyanuric acid as a nitrogen source (26), utilization of nitrite and trinitrotoluene in P. putida JLR11 (12), and nitrogen fixation in Pseudomonas stutzeri (17). However, other features of nitrogen regulation in Pseudomonas are different from those in enterobacteria. First, there is only one gene encoding a PII protein, which was designated glnK because of the similarity of its product to other GlnK PII proteins and its immediacy to the amtB gene, encoding the high-affinity ammonium transporter. Global transcriptome analysis of P. putida suggests that both glnK and amtB are regulated by nitrogen availability via NtrC (30). Second, P. putida lacks an ortholog of Nac, the adapter that allows coregulation by nitrogen of σN- and σ70-dependent promoters in enterobacteria. Finally, a different two-component regulatory system, designated CbrA/CbrB, appears to regulate operons for utilization of amino acids that can be used as carbon and nitrogen sources, which are also regulated by the NtrB/NtrC system (45, 62).
In this paper, we show that E. coli and P. putida NtrC proteins are functionally equivalent. We have characterized the P. putida glnK promoter and its transcriptional activation by NtrC in vitro. Furthermore, we show that P. putida nitrogen-regulated genes orthologous to those activated by Nac in enterobacteria have σN-dependent promoters that are directly activated by NtrC; therefore, nitrogen-regulated expression of these genes in P. putida does not require a second tier of regulation exerted by Nac on σ70-dependent genes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this work and their genotypes are summarized in Table 1. Cells were grown in minimal medium (39) containing 25 mM sodium succinate for P. putida or 11 mM glucose for E. coli. Nitrogen sources were ammonium chloride (1 g liter−1) or l-serine (1 g liter−1) for P. putida and ammonium chloride (1 g liter−1) or l-glutamine (0.15 g liter−1) for E. coli. When required, Luria-Bertani (LB) medium was used as a rich medium. Cultures were grown in culture tubes or flasks with shaking (180 rpm) at 30°C for P. putida and 37°C for E. coli. Antibiotics and other additions were used at the following concentrations, when required: ampicillin (only for E. coli), 100 mg liter−1; carbenicillin (only for P. putida), 500 mg liter−1; rifampin (rifampicin), 20 mg liter−1; tetracycline, 5 mg liter−1; chloramphenicol, 15 mg liter−1; kanamycin, 20 mg liter−1; and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), 25 mg liter−1. All reagents were purchased from Sigma-Aldrich.
TABLE 1.
Bacterial strains, plasmids, and oligonucleotides used in this work
| Name | Relevant characteristic(s) or sequence | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | φ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17 supE44 thi-1 gyrA relA1 | 29 |
| ET8556 | rbs lacZ::IS1 gyrA hutCk ntrC1488 | 38 |
| NCM631 | hsdS gal λDE3::lacI lacUV5::gene1 (T7 RNA polymerase) Δlac linked to Tn10 | 27 |
| P. putida | ||
| KT2440 | mt-2 hsdR1 hsdM+ | 6 |
| KT2440-IHF3 | mt-2 hsdR1 hsdM+ ΔihfA::Km | 40 |
| KT2442 | mt-2 hsdR1 hsdM+ Rifr | 25 |
| MPO201 | mt-2 hsdR1 hsdM+ Rifr ΔntrC::Tc | 26 |
| Plasmids | ||
| pIZ227 | pACYC184 containing lacIq and the T7 lysozyme gene, Cmr | 27 |
| pMPO224 | nifLA-lacZ translational fusion in a broad-host-range vector | García-González et al., unpublished |
| pMPO230 | pUC19-derived plasmid with P. putida wild-type ntrC, Apr | García-González et al., unpublished |
| pMPO231 | Expression vector based on pT7-7 to overproduce NtrC, Apr | García-González et al., unpublished |
| pMPO234 | Broad-host-range trp-lacZ transcriptional fusion vector, based on pBBR1MCS-4, Apr | García-González et al., unpublished |
| pMPO243 | NtrC (with an NdeI site at the first ATG) expressed from PlacUV5 in a pACYC184-derived plasmid, Cmr | García-González et al., unpublished |
| pMPO305 | pUC19-derived plasmid with a glnA-ntrBC operon, Apr | Unpublished |
| pMPO308 | NtrC(D55E) expressed from PlacUV5 in a pACYC184-derived plasmid, Cmr | This work |
| pMPO309 | NtrC(S161F) expressed from PlacUV5 in a pACYC184-derived plasmid, Cmr | This work |
| pMPO310 | NtrC(D55E,S161F) expressed from PlacUV5 in a pACYC184-derived plasmid, Cmr | This work |
| pMPO313 | glnK-lacZ transcriptional fusion in pMPO234 carrying the sequence between positions −371 and +151,a Apr | This work |
| pMPO316 | glnK upstream sequence between positions −371 and +151 in pTE103,a Apr | This work |
| pMPO321 | Expression vector derived from pMPO231 to overproduce NtrC(D55E,S161F) | This work |
| pMPO340 | dppA-lacZ transcriptional fusion in pMPO234 carrying the sequence between positions −319 and +219b | This work |
| pMPO341 | dppA-lacZ transcriptional fusion in pMPO234 carrying the sequence between positions −122 and +219b | This work |
| pMPO342 | codB-lacZ transcriptional fusion in pMPO234 carrying the sequence between positions −251 and +191b | This work |
| pMPO343 | codB-lacZ transcriptional fusion in pMPO234 carrying the sequence between positions −67 and +191b | This work |
| pMPO346 | ureD-lacZ transcriptional fusion in pMPO234 carrying the sequence between positions −268 and +199b | This work |
| pRK2013 | Helper plasmid in conjugation, Kmr Tra+ | 23 |
| pTE103 | In vitro transcription vector derived from pUC8 | 19 |
| Oligonucleotides | ||
| foot0885-1 | GCAAGAAAGCTTGGTGGATGGCACGGGCTT | |
| foot0885-2 | GAAAGTAAGCTTAACGTAAATGGATGTCGC | |
| footcodB1 | GACCGTAAGCTTCGCCGTGGGCTGGGTGAT | |
| footcodB2 | CTCCGGAAGCTTAGTTGGGAGCAGAAAGAG | |
| footglnK1 | CAGCATAAGCTTACAAATCTTGGGGGGCGC | |
| footglnK2 | GCCAGTAAGCTTAAAGCGCCGAAAAACAGC | |
| footureD1 | GCCGTGAAGCTTTTCGATTACTGAGGCTTG | |
| footureD2 | CAAATTAAGCTTCTGGATATAGCAGGTTGC | |
| NtrCasp55-fwd | ATACCGCTCGCCGCAGCCGAAC | |
| NtrCasp55-rev | CCAGGCATGCGAATTTCGGAAATGATCACGTC | |
| NtrCser161-fwd1 | CATACCGCCTTCTCGATTTACCT | |
| NtrCser161-fwd2 | GCCGCCTCAGCCACTTCAACATCACCGTGCT | |
| NtrCser161-rev1 | AGCACGGTGATGTTGAAGTGGCTGAGGCGGC | |
| NtrCser161-rev2 | TGAGGATCTTCGGCTCGACTGC | |
| P0885_fwd | GCCGAATTCAAGTGCAGATGCCATAAG | |
| P0885_fwd_corto | GAAGAATTCCGCAAAACGCCATCATTT | |
| P0885_rev | GAAGGATCCCCCGGTGGTGTACTGCCC | |
| P5234_fwd | GCGGAATTCTGGGGCTGTCCACTGAAA | |
| P5234_rev | GACGGATCCCATGAAACTCTCTCCCGA | |
| PcodB_fwd | TTCGAATTCCTGGCAGGGATTTTTGGC | |
| PcodB_fwd_corto | GCTGAATTCTGCACAGTGCATATGGAT | |
| PcodB_rev | CAAGGATCCATGCCATGCCCAGCTTGC | |
| PEX0885 | GCCTTCGGAGCAGAACACCAGGTTGGATGC | |
| PEXglnK | CAGGAAAACAAACCCGTCTCAAGTCTAAGC | |
| PureD_fwd | AAGGAATTCACCTGTCGATTGCGCAGA | |
| PureD_rev | CCGGGATCCTGGACAAAGCGCAACTGC | |
| sec0885-1 | ACGGCGGTGGATGGCACGGGCTT | |
| sec0885-2 | GTACGAACGTAAATGGATGTCGC | |
| seccodB1 | TAAACCGCCGTGGGCTGGGTGAT | |
| seccodB2 | TTTGTAGTTGGGAGCAGAAAGAG | |
| secglnK1 | AAGGCACAAATCTTGGGGGGCGC | |
| secglnK2 | CCGACAAAGCGCCGAAAAACAGC | |
| secureD1 | ACCGCTTCGATTACTGAGGCTTG | |
| secureD2 | GCAGGCTGGATATAGCAGGTTGC |
Coordinates are related to the transcriptional start of the gene.
Coordinates are related to the to the first G of the −24 box of the promoter.
Construction of plasmids.
The plasmids used in this work are summarized in Table 1. All DNA manipulations were performed according to standard procedures (51). Plasmid DNA preparation and purification kits were purchased from Macherey-Nagel and GE Healthcare, respectively, and used according to the manufacturer's instructions. Plasmid DNA was transferred by transformation (15) to E. coli strains or by triparental mating to P. putida strains (20). E. coli DH5α was used as a host strain in cloning procedures.
Transcriptional lacZ gene fusions to the promoters of interest were constructed by PCR amplification of the promoter region, using genomic DNA from P. putida KT2442 as a template. The oligonucleotides used for PCR amplification were the following: for pMPO313 (glnK fusion), P5234_fwd and P5234_rev; for pMPO340 (dppA [PP0885] long fusion), P0885_fwd and P0885_rev; for pMPO341 (dppA [PP0885] short fusion), P0885_fwd_corto and P0885_rev; for pMPO342 (codB long fusion), PcodB_fwd and PcodB_rev; for pMPO343 (codB short fusion), PcodB_fwd_corto and PcodB_rev; and for pMPO346 (ureD fusion), PureD_fwd and PureD_rev. Oligonucleotide sequences are listed in Table 1. These PCR products were cloned in the transcriptional fusion vector pMPO234. Plasmid pMPO316 was constructed by cloning in pTE103 the same fragment obtained by PCR with the oligonucleotides P5234_fwd and P5234_rev as for the glnK fusion. The NtrC(D55E,S161F) overproduction plasmid pMPO321, based on the pT7-7 vector, was constructed by substitution of wild-type ntrC in pMPO231 with ntrC(D55E,S161F) from pMPO310 (see below). All cloned PCR products were subsequently sequenced.
Construction of ntrC(D55E,S161F).
The D55E mutation was generated by PCR with the mutagenic primer NtrCasp55-rev and the nonmutagenic primer NtrCasp55-fwd, using pMPO230 as the template. The PCR product was digested with SphI and NdeI and cloned in the expression vector pMPO243 digested with SphI and NdeI, thus yielding pMPO308.
The S161F mutation was generated by overlap extension PCR essentially as described previously (13), using pMPO305 as the template and the mutagenic oligonucleotides NtrCser161-rev1 and NtrCser161-fwd2 with the external nonmutagenic oligonucleotides NtrCser161-fwd1 and NtrCser161-rev2, respectively. The final PCR product was cloned in pMPO243 to yield pMPO309.
The double mutant ntrC version encoding the D55E and S161F substitutions was finally generated by replacement of the wild-type 5′ end of ntrC in pMPO309 with the same fragment carrying the D55E mutation from pMPO308, resulting in plasmid pMPO310, which carries ntrC(D55E,S161F).
Purification of NtrC(D55E,S161F).
Two liters of culture of E. coli NCM631 harboring pMPO321 and pIZ227 was grown to an optical density at 600 nm of 0.3 to 0.5 at 37°C and then transferred to a 20°C shaker. After 20 min, IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a final concentration of 0.5 mM, and the culture was incubated at 20°C overnight. Cells were then harvested by centrifugation, resuspended in 20 ml of sonication buffer (50 mM Tris-HCl, pH 8, 10% glycerol, 0.1 mM EDTA, 1 mM dithiothreitol [DTT], and 10 mM NaCl), and broken by sonication. After elimination of cell debris by centrifugation, NtrC(D55E,S161F) was purified from the supernatant by selective precipitation with ammonium sulfate in the range of 30% to 40% saturation, resuspended in storage buffer (50 mM Tris-HCl, pH 8, 20% glycerol, 0.1 mM EDTA, 1 mM DTT, and 10 mM NaCl), and dialyzed against 2 liters of the same buffer at 4°C overnight to remove ammonium sulfate. Purity was estimated visually by sodium dodecyl sulfate-polyacrylamide gel electrophoresis to be ≥90%. Concentration was determined by the Bradford protein assay (10) and is expressed in nM of a dimer. Protein samples were stored at −80°C.
β-Galactosidase assays.
To assay the nifLA-lacZ translational fusion in E. coli, preinocula grown in minimal medium under nitrogen excess conditions (ammonium chloride and l-glutamine) were diluted in the same medium or minimal medium with l-glutamine as the nitrogen source (nitrogen-limiting conditions). After incubation for 2 h, IPTG to a final concentration of 0.1 mM was added to induce expression of P. putida ntrC from the plasmids. Samples were taken 3.5 h later, and β-galactosidase activity was measured as previously described (42).
For transcriptional fusions, preinocula of E. coli or P. putida strains grown in minimal medium under nitrogen excess conditions (ammonium chloride plus l-serine for P. putida and ammonium chloride plus l-glutamine for E. coli) were diluted in minimal medium under nitrogen excess or under nitrogen limitation (l-serine for P. putida and l-glutamine for E. coli) conditions. Samples of the cultures grown to mid-exponential phase were taken 6 h later, and β-galactosidase activity was assayed (42).
Values of β-galactosidase activity are the averages from assays performed with at least three independent cultures.
RNA preparation and primer extension.
Total RNA from P. putida KT2442 or MPO201 grown to mid-exponential phase under nitrogen excess or nitrogen limitation conditions was prepared essentially as previously described (26). Primer extension reactions were performed as previously described (28), with 20 μg of RNA from each condition as the template, using the 32P-end-labeled primers PEXglnK and PEX0885 in reaction mixtures containing Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA). Sequencing reactions were performed with a Thermo Sequenase cycle sequencing kit (USB, Cleveland, OH), according to the manufacturer's instructions. Samples were run on 6% polyacrylamide-urea sequencing gels, and the gels were dried, exposed to radiosensitive screens, and finally scanned with a Typhoon 9410 scanner (GE Healthcare).
DNase I footprinting.
DNase I footprinting assays were run essentially as described previously (48), except for the footprinting buffer (10 mM Tris-acetate, pH 8, 100 mM potassium acetate, 8 mM magnesium acetate, 27 mM ammonium acetate, 5% glycerol, 0.67 mM CaCl2, 1 mM DTT, 5 μg bovine serum albumin). As competitor DNA, 0.33 mg ml−1 salmon sperm DNA for NtrC or 20 μg ml−1 of poly(dI-dC) for IHF footprinting was used in the reaction buffer. Probes for DNase I footprinting were generated by PCR amplification using the following oligonucleotides: for NtrC and IHF footprinting at the glnK promoter, footglnK1 and P5234_rev for the top strand and footglnK2 and P5234_fwd for the bottom strand; for the PP0885 promoter, foot0885-1 and P0885_rev for the top strand and foot0885-2 and P0885_fwd for the bottom strand; for the codB promoter, footcodB1 and PcodB_rev for the top strand and footcodB2 and PcodB_fwd for the bottom strand; and for the ureD promoter, footureD1 and PureD_rev for the top strand and footureD2 and PureD_fwd for the bottom strand. Amplified probes were digested with HindIII and strand specifically labeled with [α-32P]dCTP by Klenow filling in the 5′ overhanging ends. A sequencing reaction performed with a Sequenase 2.0 kit (USB) using an oligonucleotide specific for the labeled strand in each case (secglnK1 and -2 for glnK, sec0885-1 and -2 for dppA, seccodB1 and -2 for codB, and secureD1 and -2 for ureD) was run with the partially digested DNA as a size marker. Gels were processed and analyzed as described for primer extension analysis.
Open complex assays.
Open complex assays to test glnK promoter activation by NtrC were performed essentially as described previously (36). Linearized template DNA was obtained by digestion of pMPO316 to yield a 540-bp fragment containing the upstream intergenic region of glnK. The fragment was labeled with [α-32P]dCTP. Reaction mixtures were made in a final volume of 15 μl in binding buffer with 40,000 cpm 32P-labeled template, 3.4 ng μl−1 denatured salmon sperm DNA, 7.5 μg bovine serum albumin, 4 mM ATP, 100 nM P. putida core RNA polymerase, and 200 nM P. putida σN. This mixture was incubated for 2 min at 30°C, and then reactions were initiated by the addition of 200 nM of NtrC and 0.5 mM of GTP, UTP, or CTP, as an additional nucleotide. When required, 75 nM of E. coli IHF was added at the same time as NtrC. After 20 min of incubation at 30°C, reaction products were mixed with 3 μl of a solution containing 20% glycerol, 0.033% bromophenol blue, 0.033% xylene cyanol, 6.67 mM Tris-HCl, pH 8, 0.67 mM EDTA, and 2 μg of heparin. Reaction mixtures were loaded in 4% polyacrylamide gels (acrylamide-bisacrylamide, 29:1) which had been prerun at 4°C. The gels were run at 4°C, dried, exposed to radiosensitive screens, and finally scanned with a Typhoon 9410 scanner (GE Healthcare).
RESULTS
Construction of a P. putida ntrC constitutive allele.
In order to study the function of P. putida NtrC in the absence of NtrB in vitro, we constructed a derivative of ntrC encoding the substitutions D55E and S161F, equivalent to D54E and S160F in Salmonella enterica NtrC, which give rise to a form of the activator that does not require phosphorylation by NtrB to activate transcription (33). The function of P. putida NtrC(D55E,S161F) in the NtrC− mutant strain of E. coli ET8556 was then characterized by analyzing the expression of a Klebsiella oxytoca translational nifLA-lacZ fusion known to be activated by NtrC in K. oxytoca (43), A. vinelandii (53), and other diazotrophs. As shown in Fig. 1, β-galactosidase activity levels in the medium containing glutamine as the sole nitrogen source indicated that both the mutant and the wild-type alleles of P. putida ntrC complemented NtrC function in E. coli. Under nitrogen excess conditions, the levels of β-galactosidase activity of the nifLA-lacZ fusion in the strain producing wild-type NtrC were 30-fold lower than those under nitrogen-limiting conditions, thus showing that the activity of P. putida NtrC can be regulated in E. coli in response to nitrogen availability, presumably through E. coli NtrB. On the other hand, the strain producing NtrC(D55E,S161F) expressed nifLA under nitrogen excess conditions to 60% of the levels obtained under nitrogen-limiting conditions, which indicated that the activity of NtrC(D55E,S161F) was independent of NtrB activation.
FIG. 1.

Expression of a nifLA-lacZ translational fusion in E. coli ntrC mutant strain ET8556 complemented with P. putida ntrC. Black bars, empty vector control; gray bars, wild-type P. putida ntrC; white bars, P. putida constitutive ntrC(D55E,S161F) mutant. Values are averages from at least three independent measurements. Error bars indicate standard deviations of the means.
Nitrogen regulation of glnK.
Previous global transcriptome analysis showed that expression of glnK, encoding the only PII-like protein in P. putida, is likely to be subject to nitrogen regulation mediated by NtrC (30). With the aim of characterizing the role of NtrC in the activation of glnK, the expression of a glnK-lacZ gene fusion in the wild-type P. putida strain KT2442 and its ΔntrC derivative MPO201 (26) was studied (Table 2) . When serine was used as the sole nitrogen source (nitrogen-limiting conditions), expression of the glnK-lacZ fusion was induced almost 30-fold, thus demonstrating that expression of glnK is nitrogen regulated in P. putida. On the other hand, there was no glnK-lacZ induction (less than twofold) in the ΔntrC mutant strain, thus indicating that NtrC directly or indirectly mediates nitrogen regulation of glnK in the wild-type strain.
TABLE 2.
Expression of glnK-lacZ fusion in P. putida and E. coli
| Fusion plasmid | Structure | Strain | Other plasmid | β-Galactosidase activitya (Miller units ± SD)
|
|
|---|---|---|---|---|---|
| Nitrogen excess | Nitrogen limitation | ||||
| P. putida | |||||
| pMPO313 | glnK-lacZ | KT2442 | 1,167 ± 166 | 33,204 ± 4,453 | |
| pMPO234 | Empty vector | KT2442 | 177 ± 22 | 153 ± 19 | |
| pMPO313 | glnK-lacZ | MPO201 (NtrC−) | 908 ± 126 | 1,477 ± 90 | |
| pMPO234 | Empty vector | MPO201 (NtrC−) | 192 ± 31 | 152 ± 12 | |
| E. coli | |||||
| pMPO313 | glnK-lacZ | ET8000 | 130 ± 35 | 13,783 ± 1,774 | |
| pMPO234 | Empty vector | ET8000 | 741 ± 178 | 1,024 ± 260 | |
| pMPO313 | glnK-lacZ | ET8556 (NtrC−) | pMPO243 (ntrC) | 259 ± 31 | 8,474 ± 201 |
| pMPO234 | Empty vector | ET8556 (NtrC−) | pMPO243 (ntrC) | 535 ± 48 | 626 ± 70 |
| pMPO313 | glnK-lacZ | ET8556 (NtrC−) | pMPO310 [ntrC(D55E,S161F)] | 10,087 ± 1,714 | 10,212 ± 678 |
| pMPO234 | Empty vector | ET8556 (NtrC−) | pMPO310 [ntrC(D55E,S161F)] | 755 ± 101 | 754 ± 139 |
| pMPO313 | glnK-lacZ | ET8556 (NtrC−) | 189 ± 17 | 469 ± 68 | |
| pMPO234 | Empty vector | ET8556 (NtrC−) | 875 ± 148 | 1,637 ± 205 | |
For P. putida, serine was the nitrogen source, and for E. coli, glutamine was the nitrogen source under nitrogen-limited conditions (see Materials and Methods).
Regulation of P. putida glnK expression in the wild-type and ntrC mutant strains of E. coli complemented with either P. putida ntrC or ntrC(D55E,S161F) was also analyzed. As shown in Table 2, glnK-lacZ expression in wild-type E. coli was induced more than 100-fold under nitrogen-limiting conditions, while it was barely induced (2.5-fold) in the ntrC mutant strain. Complementation with P. putida ntrC resulted in 33-fold induction under nitrogen-limiting conditions, while complementation with ntrC(D55E,S161F) resulted in high constitutive expression under all conditions.
Nitrogen regulation of glnK expression was also observed by mapping the glnK transcriptional start site under nitrogen-limiting and nitrogen excess conditions. As shown in Fig. 2A, a single glnK transcript starting at a G was evident only when the total RNA used for the 5′ mapping came from P. putida KT2442 grown under nitrogen-limiting conditions. Upstream from the transcription initiation site, a sequence for a potential σN-dependent promoter bearing the consensus GG-N10-GC was identified (Fig. 2B). Farther upstream, two putative NtrC binding sites were also identified. Both putative sites are on the same face of the DNA helix, since their centers are separated by 33 bp. The most proximal binding site, which is separated from the putative σN-dependent promoter by 73 bp, is centered at position −110 with respect to the transcriptional start site. Both sequence similarity and relative positions of the cis-acting sequences fit well with the arrangement of a regulated σN-dependent promoter directly activated by NtrC (14, 30).
FIG. 2.

Primer extension analysis of the glnK transcript. (A) Primer extension reaction mixture with total RNA prepared from cultures of P. putida KT2442 grown under nitrogen-sufficient (NS, ammonium plus serine) or nitrogen-limiting (S, serine) conditions. GATC lanes show the sequencing ladder (noncoding strand). (B) Sequence of the glnK promoter region, showing the putative NtrC binding sites (open boxes), the σN promoter (sequence underlined with black boxes), and the IHF binding site (dotted box). The transcription initiation site (G in bold with an arrow) and the translation initiation site (underlined ATG) are also highlighted for comparison.
In vitro characterization of NtrC-mediated activation of glnK.
To study P. putida NtrC function in vitro, transcription and translation of ntrC(D55E,S161F) were coupled to the expression vector pT7-7. After induction with IPTG, NtrC(D55E,S161F) was the predominant product in the soluble fraction and was easily purified by selective precipitation to >90% homogeneity, as assessed by Coomassie blue staining of sodium dodecyl sulfate-polyacrylamide gels (data not shown).
Interaction of purified NtrC(D55E,S161F) with the promoter region was analyzed by DNase I footprinting (Fig. 3). For each strand, two windows of protection, spanning from −101 to −125 and from −133 to −159 on the top strand and from −95 to −120 and from −131 to −154 on the bottom strand, were evident. Each protected window covered the corresponding NtrC binding site previously identified by sequence analysis (Fig. 3A and B). Regardless of the labeled strand, a position showing increased sensitivity to DNase I was also very evident within each protected window. These data clearly indicate that NtrC binds to two binding sites in the glnK promoter regulatory region.
FIG. 3.

NtrC DNase I footprint of the glnK promoter region. (A) Top- and bottom-strand footprint patterns. Predicted NtrC binding sites (open boxes), protected regions (black bars), and hypersensitive positions (dots) are marked. NtrC(D55E,S161F) concentrations were 0, 50, 100, 200, and 500 nM for each strand. The coordinates are relative to the glnK transcriptional start site. (B) Schematic representation of the protected regions on the sequence of the glnK promoter, with the same indications as for the footprint pattern.
We examined the NtrC dependence of transcription from the glnK promoter region by analyzing open complex formation using purified components. The formation of open promoter complexes may be stabilized by the addition of the initiating nucleotide, which allows assessment of activation at promoters where open complexes are unstable (21). We therefore tested open complex formation using a labeled DNA fragment covering the glnK promoter region from −371 to +151 by adding the required ATP and an additional nucleotide. As shown in Fig. 4, NtrC stimulated the isomerization of the closed promoter-polymerase complex to the open complex competent for transcription initiation in the presence of IHF, ATP, and an initiator nucleotide. Open complex formation was most efficient in the presence of GTP, which is consistent with the results of the 5′ mapping of the glnK transcript (Fig. 2).
FIG. 4.

Open complex formation at the glnK promoter. The presence (+) or absence (−) of NtrC(D55E,S161F) and/or IHF in each reaction mixture is indicated. ATP (final concentration, 4 mM) was present in all reaction mixtures to promote catalysis of open complex formation by NtrC. Where indicated above each lane, an additional nucleotide (final concentration, 0.5 mM) was added to provide the potential initiating nucleotide.
The addition of IHF was required to detect open complexes under our in vitro experimental conditions. Requirement of the coactivator IHF was also observed in vivo by analyzing expression of the glnK-lacZ gene fusion in strain KT2440 and its derivative IHF-deficient strain KT2440-IHF3. Induction of glnK under nitrogen-limiting conditions in KT2440 resulted in the expression of 46,327 ± 2,733 Miller units. Expression of glnK in the IHF-deficient strain was also induced under nitrogen-limiting conditions, but the expression level in this case was only 10,312 ± 2,872 Miller units, 22% of the level for the wild-type strain. Although the requirement for IHF is not so strong in vivo, these data clearly indicate a direct involvement of IHF in transcriptional activation at the glnK promoter of P. putida. This implies binding of IHF to the promoter regulatory region between the promoter and the activator binding sites. Sequence inspection revealed a T-rich region closely upstream of the GG-N10-GC promoter sequence, which might constitute an IHF binding site. A DNase I footprinting assay of IHF showed its binding to this region, covering from coordinates −38 to −72 from the transcription start site (Fig. 5). This region coincides very well with the previously predicted binding site for IHF in the promoter region of glnK (Fig. 2B).
FIG. 5.

IHF DNase I footprint of the glnK promoter region. The coordinates are relative to the glnK transcriptional start site. IHF concentrations were 0.5, 1, and 2 μM of dimer. The protected regions (black bars) and hypersensitive positions (dots) are marked in the footprint and on the promoter sequence beside it. The σN promoter and the putative IHF binding site identified by sequencing are also marked on the sequence, as described in the legend for Fig. 2.
NtrC directly activates nitrogen-regulated genes that are activated by the Nac regulator in enterobacteria.
Out of the 22 Pseudomonas genomes surveyed, a Nac ortholog showing at least 50% identity to Nac from enterobacteria was detected in only 3 of them. In addition, mutational analysis of the putative Nac ortholog of P. fluorescens strain SBW25 indicated that expression of the hut operon, which is regulated by NtrB/C and CbrA/B, was not affected (62). Therefore, it appears that Pseudomonas strains do not generally have a Nac protein, which serves as an adaptor between NtrC and certain nitrogen-regulated genes. We were thus interested to investigate whether P. putida has genes orthologous to those activated by Nac in enterobacteria and, if so, to determine how their expression is regulated.
In P. putida, we found potential orthologues of the six Nac-activated operons in enterobacteria, but only dpp, ure, and cod were previously identified as being regulated by nitrogen and NtrC (30). In fact, nitrogen regulation and NtrC dependence of P. putida dppA were not very obvious from the genomic microarray analysis, since this gene barely meets the requirements to be unambiguously considered nitrogen regulated by NtrC (30). Analysis of sequences upstream of the first gene of each operon, carried out using the algorithm to identify NtrC binding sites in the P. putida genome (30), revealed the existence of potential binding sites for NtrC in all of them. In addition, putative GG-N10-GC σN-dependent promoter sequences were identified upstream of the respective coding sequences. The putative NtrC binding sites of ureD, dppA, and codB were centered at positions −78, −161, and −72, respectively, from the GG dinucleotide at −24, which is characteristic of σN-dependent promoters.
Transcriptional lacZ fusions to the first gene of each operon were constructed. In the case of dppA and codB, transcriptional fusions that lacked the putative NtrC binding sites in the respective promoters were also constructed. In spite of the substantial basal level of expression of the ureD-lacZ gene fusion under nitrogen excess, its expression was induced in KT2442 under nitrogen-limiting conditions almost 10-fold. In the ΔntrC strain, ureD-lacZ basal expression was slightly higher than that in the wild-type strain, but it was not substantially induced under nitrogen-limiting conditions (1.2-fold induction). This confirmed the NtrC requirement for high levels of expression from the ureD promoter (Fig. 6A).
FIG. 6.

In vivo expression from the ureD, dppA, and codB promoters of P. putida. Expression was measured as β-galactosidase activity of lacZ transcriptional fusions under nitrogen excess or nitrogen-limiting conditions. Values are averages from at least three independent assays. Error bars indicate standard deviations of the means. (A) ureD-lacZ fusion expression in KT2442 (wild-type [wt]) and MPO201 (ΔntrC) strains. Black bars, control empty vector (pMPO234); white bars, ureD-lacZ fusion (pMPO346). (B) codB-lacZ fusion expression under the same conditions as for ureD-lacZ fusion. Black bars, control empty vector (pMPO234); white bars, codB-lacZ complete fusion (pMPO342); gray bars, codB-lacZ fusion lacking the putative NtrC binding site (pMPO343). (C) dppA-lacZ fusion activity under the same conditions as for ureD-lacZ fusion. Black bars, control empty vector (pMPO234); white bars, dppA-lacZ complete fusion (pMPO340); gray bars, dppA-lacZ fusion lacking the putative NtrC binding site (pMPO341).
Similarly, expression of the codB-lacZ gene fusion containing the complete upstream region of the codB promoter was very efficiently induced (50-fold induction) under nitrogen-limiting conditions in the KT2442 strain (Fig. 6B). This induction was severely reduced in the codB-lacZ fusion lacking the putative NtrC binding site (17,848 versus 1,165 Miller units). About 70% of this residual activity still appears to be dependent on NtrC. In the ΔntrC strain, codB-lacZ expression was clearly reduced (fivefold lower than that for the wild-type strain), thus showing the NtrC requirement for activation. However, a residual activation of codB independent of NtrC was still evident. Intriguingly, this activation requires DNA sequences upstream from the promoter.
The expression pattern of dppA was clearly different from those of the other two genes (Fig. 6C). In the first place, expression of the dppA-lacZ gene fusion containing the complete promoter sequence was very high under nitrogen-sufficient conditions, and this expression was independent of NtrC, since the expression level was even higher in the ΔntrC background. Deletion of the putative NtrC binding site upstream of the dppA promoter resulted in a 2.5-fold reduction of expression levels. However, this reduction was also observed in the ΔntrC background, suggesting that the observed decrease in promoter activity is not associated with the loss of the NtrC binding site. Under nitrogen-limiting conditions, the expression levels of the dppA-lacZ fusion containing the complete dppA promoter sequence increased by a factor of ∼2-fold but again the level of dppA expression was not influenced by the presence of NtrC, which indicated a major expression of dppA independent of NtrC-mediated nitrogen regulation.
To shed more light on the regulation of dppA, 5′ mapping of dppA transcripts isolated from cultures grown under nitrogen-sufficient and nitrogen-limiting conditions was performed with strain KT2442 and its ΔntrC derivative. As shown in Fig. 7, a major transcript initiating at a G residue was evident in both strains under all conditions. However, a minor transcript initiating at a C residue 2 nucleotides upstream was induced under nitrogen-limiting conditions. Sequences resembling that of a σN-dependent promoter were found 12 nucleotides upstream from the nitrogen-regulated transcription start site (Fig. 8). These results suggest that there are two overlapping promoters in this region, with the weakest apparently nitrogen regulated and potentially σN dependent. However, from our data, it is not clear that this transcript is dependent on NtrC.
FIG. 7.
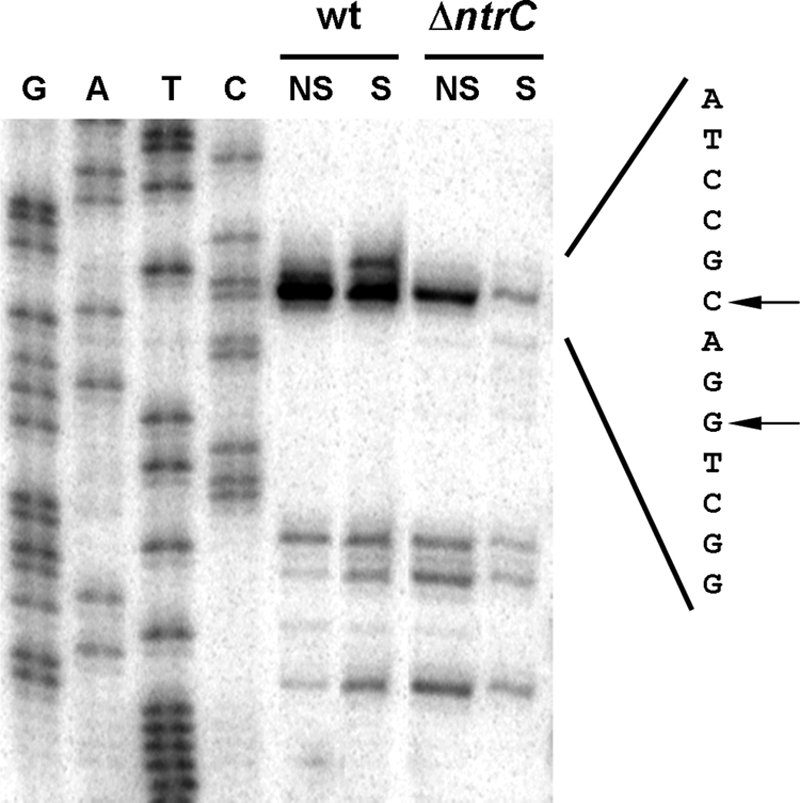
Primer extension analysis of dppA transcript. A primer extension reaction mixture with total RNA was prepared from cultures of P. putida KT2442 (wild type [wt]) and MPO201 (ΔntrC) grown on minimal medium under nitrogen-sufficient (NS, ammonium plus serine) or nitrogen-limiting (S, serine) conditions. GATC lanes indicate the sequencing ladder (noncoding strand). A 13-nucleotide sequence around the transcriptional start (coding strand) is shown. Arrows indicate the mapped 5′ ends of the transcripts.
FIG. 8.

DNase I footprints of dppA, codB, and ureD promoter regions. The strand with the most informative footprint pattern is shown in each case. Predicted NtrC binding sites (open boxes), protected regions (black bars), and hypersensitive positions (dots) are marked. Dotted boxes represent palindromic sequences within the protected regions that may serve as NtrC binding sites, which were not initially identified. Coordinates are relative to the first G of the −24 box of the promoter. (A) DNase I footprint pattern of dppA. NtrC(D55E,S161F) concentrations were 0, 0.2, 0.5, 1, 2, and 4 μM. (B) DNase I footprint pattern of codB. NtrC concentrations were 0, 0.5, 1, 1.5, and 2 μM. (C) DNase I footprint pattern of ureD. NtrC concentrations were 0, 0.2, 0.35, 0.5, and 1 μM. (D) Sequences of the promoter regions covering the putative σN promoter (marked as a sequence underlined with black boxes) and the regions bound by NtrC (marked as boxes).
Binding of NtrC to the ureD, codB, and dppA promoter regions was also tested in order to assess whether the requirement for NtrC for high-level expression of these genes was direct. DNase I footprinting analysis demonstrated binding of NtrC to all probes (Fig. 8). Binding to dppA (Fig. 8A) resulted in a clear protected region from −145 to −170 with respect to the putative σN-dependent promoter, which encompassed the putative NtrC binding site centered at −161. Within the protected region, position −162 showed enhanced sensitivity to DNase I. This was the only protection observed in this promoter region. However, more-extensive protections were evident in the other promoter regions. In the case of codB, a proximal protected region spanning from −63 to −79 that matched the putative NtrC binding site centered at −72 was visible (Fig. 8B). In addition, two protected regions spanning from −109 to −122 and from −130 to −141 on the top strand were evident. The putative NtrC binding site identified by sequence inspection in the ureD promoter region was centered at −78, and a protected region covering this position, from −68 to −83, was evident (Fig. 8C). A second protected region further downstream extended from −62 to −37. The region spanning these two protected regions showed enhanced sensitivity to DNase I (positions −64 to −67), which suggested DNA distortion between the two protected regions. Additional protections were also found farther upstream, from −89 to −114. All seven protected regions revealed palindromic sequences resembling those of NtrC binding sites, although four of them were not initially identified as NtrC binding sites.
DISCUSSION
Our current studies have demonstrated intergeneric complementation of an E. coli ntrC mutant by P. putida ntrC, resulting in nitrogen-regulated expression of the K. oxytoca nifL promoter (Fig. 1). These results indicate that P. putida NtrC can function in this heterologous system and that its activity can be regulated by E. coli NtrB. In addition, amino acid substitutions equivalent to D54E and S160F in S. enterica NtrC (33) give rise to similar effects in P. putida NtrC, resulting in partially constitutive forms (data not shown) which, when combined, function synergistically to result in a more constitutive phenotype (60% of the derepressed transcription levels under nitrogen-sufficient conditions [Fig. 1]). These data clearly indicate that NtrC function is highly conserved between enterobacteria and pseudomonads and that NtrC proteins from both origins are probably fully exchangeable.
Our studies show that transcription of glnK, the only PII-encoding gene in P. putida, is strongly regulated in response to the nitrogen source (Table 2 and Fig. 2). As with the E. coli glnK promoter (2), this regulation is exerted directly by NtrC, as shown by footprinting of the glnKp region (Fig. 3) and in vitro open complex formation at this promoter (Fig. 4). Two NtrC binding sites have been identified in P. putida, compared to the one (and a half) found in E. coli (3, 57). In addition, open complex formation at P. putida glnKp was strongly dependent on IHF, unlike with E. coli glnKp (2), and this requirement for IHF was also observed in vivo. Involvement of IHF together with the presence of two contiguous NtrC binding sites on the same face of the helix suggests that transcriptional activation at P. putida glnKp may be very sensitive to low NtrC-P concentrations, unlike E. coli glnKp expression (2). This difference may be relevant for efficient nitrogen repression in the absence of a constitutively produced GlnB protein (see below).
The function of PII is required under all nitrogen regimens for proteobacteria. Under nitrogen excess conditions, unmodified GlnB stimulates adenylylation of glutamine synthetase. It also inhibits the kinase activity and activates the phosphatase activity of NtrB, thus resulting in dephosphorylation of NtrC and consequently preventing it from activating transcription. This in turn influences the level of expression of ntrC, which is transcribed under the control of the glnAp1 and ntrBp promoters in enterobacteria. Strict control of P. putida glnK expression by NtrC could imply that this organism is limited in PII function under nitrogen-sufficient conditions, which could potentially result in poorer repression under these conditions. However, the amount of GlnK has to be sufficient to keep most NtrC in its inactive form under nitrogen-replete conditions, because otherwise NtrC-P would activate glnK transcription. Thus, we postulate a complex autoregulatory feedback circuit for GlnK production that adjusts the level of GlnK to the minimum concentration needed to maintain nitrogen repression under nitrogen-sufficient conditions. The basal expression of the other genes activated by NtrC-P would also be controlled, their levels depending on the relative sensitivities of their promoters to the activator concentration compared to that of glnKp. This allows efficient nitrogen regulation without the need for an additional, constitutively produced PII protein, unless the two proteins have different target specificities or different sensitivities to effectors. Strict control of the PII concentration under different conditions could also provide an additional level of regulation by PII, if it could interact with a particular target regardless of its uridylylation state or the concentration of potential effectors. GlnK autoregulation can also explain the nitrogen regulation phenotype of a GlnB mutant of E. coli (4, 11). Under nitrogen-sufficient conditions, GlnB represses NtrC-mediated activation of all target genes, including glnK. In the absence of GlnB, GlnK takes over nitrogen repression under nitrogen-sufficient conditions. Lack of the constitutive GlnB repressor function would be compensated by an increase in the levels of GlnK (4), consistent with an autoregulatory feedback loop in which NtrC-P activates glnK expression once GlnK levels become limiting. Intriguingly, although GlnK can efficiently maintain low basal expression levels in most of the nitrogen-repressed operons in the E. coli GlnB− mutant strain, glnA escapes nitrogen repression. However, this may be explained because the glnA promoter region is much more sensitive to low concentrations of NtrC-P than E. coli glnK or the other nitrogen-regulated genes (2). Although such a comparative analysis has not been carried out with P. putida, glnA expression is regulated by nitrogen in this species (30), thus suggesting that the sensitivities of P. putida glnA and glnK to NtrC-P should not be as different as they are in E. coli.
Although there is no Nac ortholog in P. putida, three of the operons activated by Nac in enterobacteria were identified as nitrogen regulated in P. putida (30). Although binding of NtrC to their promoter regions (Fig. 8) indicated that NtrC could directly activate transcription from these promoters, transcription from the dppA promoter region does not appear to depend on NtrC and has a very weak nitrogen regulation, if any. This promoter region contains a single NtrC binding site, while ureD and codB, the genes truly regulated by nitrogen via NtrC, have promoter regions with up to three binding sites. In both cases, the two most distal binding sites are aligned on the same face of the helix (±1 bp) and are separated by two turns of the helix, which suggests that NtrC may efficiently oligomerize at these sites. The third, most proximal NtrC binding sites are located too close to their respective −24/−12 promoters to be considered activation binding sites. However, these sites may work as “governors,” as defined by Atkinson et al. (5), to limit the maximal level of expression when the activator concentration is very high, similarly to the low-affinity NtrC binding sites in the glnAp2 promoter of E. coli.
The additional level of nitrogen regulation in Klebsiella and E. coli mediated by the activation cascade of a number of σ70-dependent genes through the adapter Nac does not seem to operate in Pseudomonas. This level of regulation is apparently not essential for enterobacteria, since some do not have a nac gene. This is the case for S. enterica serovar Typhimurium, which appears to have lost Nac recently, since it cannot control the expression of these σ70-dependent genes in response to nitrogen. However, the promoter for the gene hutC in S. enterica is still susceptible to activation by K. aerogenes Nac (9, 46). In contrast, for P. putida, some of these genes are still regulated by nitrogen but their promoters are σN dependent and are directly regulated by NtrC, thus avoiding an additional tier of regulation.
Thus, it appears that the nitrogen regulatory network in P. putida is a simplified version of that operating in enterobacteria, with only one PII protein and no cascade regulation by the Nac regulatory protein. The differences in the nitrogen regulatory networks that allow this simplification apparently do not reside in the functions of the regulatory factors but are determined by the structures of the target promoters that are regulated by nitrogen.
Acknowledgments
We are grateful to Victoria Shingler for providing P. putida purified proteins for in vitro analysis, Guadalupe Martín Cabello for technical help, and all members of the Santero laboratory for their insights and helpful suggestions.
Work in our laboratories is funded by the Spanish Ministry of Science and Innovation, grants BIO2005-03094, BIO2008-01805, and CSD2007-00005, and by the Andalusian government, grants P05-CVI-131 and P07-CVI-2518.
Footnotes
Published ahead of print on 31 July 2009.
REFERENCES
- 1.Arcondéguy, T., R. Jack, and M. Merrick. 2001. PII signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol. Mol. Biol. Rev. 65:80-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkinson, M. R., T. A. Blauwkamp, V. Bondarenko, V. Studitsky, and A. J. Ninfa. 2002. Activation of the glnA, glnK, and nac promoters as Escherichia coli undergoes the transition from nitrogen excess growth to nitrogen starvation. J. Bacteriol. 184:5358-5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkinson, M. R., T. A. Blauwkamp, and A. J. Ninfa. 2002. Context-dependent functions of the PII and GlnK signal transduction proteins in Escherichia coli. J. Bacteriol. 184:5364-5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkinson, M. R., and A. J. Ninfa. 1998. Role of the GlnK signal transduction protein in the regulation of nitrogen assimilation in Escherichia coli. Mol. Microbiol. 29:431-447. [DOI] [PubMed] [Google Scholar]
- 5.Atkinson, M. R., N. Pattaramanon, and A. J. Ninfa. 2002. Governor of the glnAp2 promoter of Escherichia coli. Mol. Microbiol. 46:1247-1257. [DOI] [PubMed] [Google Scholar]
- 6.Bagdasarian, M., B. Lurz, B. Ruckert, F. C. H. Franklin, M. M. Bagdasarian, J. Frey, and K. N. Timmis. 1981. Specific-purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors for gene cloning in Pseudomonas. Gene 16:237-247. [DOI] [PubMed] [Google Scholar]
- 7.Beckers, G., J. Strosser, U. Hildebrandt, J. Kalinowski, M. Farwick, R. Kramer, and A. Burkovski. 2005. Regulation of AmtR-controlled gene expression in Corynebacterium glutamicum: mechanism and characterization of the AmtR regulon. Mol. Microbiol. 58:580-595. [DOI] [PubMed] [Google Scholar]
- 8.Bender, R. A. 1991. The role of the NAC protein in the nitrogen regulation of Klebsiella aerogenes. Mol. Microbiol. 5:2575-2580. [DOI] [PubMed] [Google Scholar]
- 9.Best, E. A., and R. A. Bender. 1990. Cloning of the Klebsiella aerogenes nac gene, which encodes a factor required for nitrogen regulation of the histidine utilization (hut) operons in Salmonella typhimurium. J. Bacteriol. 172:7043-7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 11.Bueno, R., G. Pahel, and B. Magasanik. 1985. Role of glnB and glnD gene products in regulation of the glnALG operon of Escherichia coli. J. Bacteriol. 164:816-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caballero, A., A. Esteve-Nunez, G. J. Zylstra, and J. L. Ramos. 2005. Assimilation of nitrogen from nitrite and trinitrotoluene in Pseudomonas putida JLR11. J. Bacteriol. 187:396-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camacho, E. M., and J. Casadesus. 2005. Regulation of traJ transcription in the Salmonella virulence plasmid by strand-specific DNA adenine hemimethylation. Mol. Microbiol. 57:1700-1718. [DOI] [PubMed] [Google Scholar]
- 14.Cases, I., D. W. Ussery, and V. de Lorenzo. 2003. The sigma54 regulon (sigmulon) of Pseudomonas putida. Environ. Microbiol. 5:1281-1293. [DOI] [PubMed] [Google Scholar]
- 15.Chung, C. T., S. L. Niemela, and R. H. Miller. 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. USA 86:2172-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins, C. M., D. M. Gutman, and H. Laman. 1993. Identification of a nitrogen-regulated promoter controlling expression of Klebsiella pneumoniae urease genes. Mol. Microbiol. 8:187-198. [DOI] [PubMed] [Google Scholar]
- 17.Desnoues, N., M. Lin, X. Guo, L. Ma, R. Carreno-Lopez, and C. Elmerich. 2003. Nitrogen fixation genetics and regulation in a Pseudomonas stutzeri strain associated with rice. Microbiology 149:2251-2262. [DOI] [PubMed] [Google Scholar]
- 18.Dixon, R., and D. Kahn. 2004. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2:621-631. [DOI] [PubMed] [Google Scholar]
- 19.Elliot, T., and E. P. Geiduschek. 1984. Defining a bacteriophage T4 late promoter: absence of a “−35” region. Cell 36:211-219. [DOI] [PubMed] [Google Scholar]
- 20.Espinosa-Urgel, M., A. Salido, and J. L. Ramos. 2000. Genetic analysis of functions involved in adhesion of Pseudomonas putida to seeds. J. Bacteriol. 182:2363-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eydmann, T., E. Soderback, T. Jones, S. Hill, S. Austin, and R. Dixon. 1995. Transcriptional activation of the nitrogenase promoter in vitro: adenosine nucleotides are required for inhibition of NIFA activity by NIFL. J. Bacteriol. 177:1186-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng, J., T. J. Goss, R. A. Bender, and A. J. Ninfa. 1995. Activation of transcription initiation from the nac promoter of Klebsiella aerogenes. J. Bacteriol. 177:5523-5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Figurski, D. H., and D. R. Helinski. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 76:1648-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forchhammer, K. 2008. P(II) signal transducers: novel functional and structural insights. Trends Microbiol. 16:65-72. [DOI] [PubMed] [Google Scholar]
- 25.Franklin, F. C., M. Bagdasarian, M. M. Bagdasarian, and K. N. Timmis. 1981. Molecular and functional analysis of the TOL plasmid pWWO from Pseudomonas putida and cloning of genes for the entire regulated aromatic ring meta cleavage pathway. Proc. Natl. Acad. Sci. USA 78:7458-7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García-González, V., F. Govantes, O. Porrúa, and E. Santero. 2005. Regulation of the Pseudomonas sp. strain ADP cyanuric acid degradation operon. J. Bacteriol. 187:155-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govantes, F., J. A. Molina-Lopez, and E. Santero. 1996. Mechanism of coordinated synthesis of the antagonistic regulatory proteins NifL and NifA of Klebsiella pneumoniae. J. Bacteriol. 178:6817-6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Govantes, F., J. A. Albrecht, and R. P. Gunsalus. 2000. Oxygen regulation of the Escherichia coli cytochrome d oxidase (cydAB) operon: roles of multiple promoters and the Fnr-1 and Fnr-2 binding sites. Mol. Microbiol. 37:1456-1469. [DOI] [PubMed] [Google Scholar]
- 29.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 30.Hervás, A. B., I. Canosa, and E. Santero. 2008. Transcriptome analysis of Pseudomonas putida in response to nitrogen availability. J. Bacteriol. 190:416-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoover, T. R., E. Santero, S. Porter, and S. Kustu. 1990. The integration host factor stimulates interaction of RNA polymerase with NIFA, the transcriptional activator for nitrogen fixation operons. Cell 63:11-22. [DOI] [PubMed] [Google Scholar]
- 32.Janes, B. K., C. J. Rosario, and R. A. Bender. 2003. Isolation of a negative control mutant of the nitrogen assimilation control protein, NAC, in Klebsiella aerogenes. J. Bacteriol. 185:688-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klose, K. E., D. S. Weiss, and S. Kustu. 1993. Glutamate at the site of phosphorylation of nitrogen-regulatory protein NTRC mimics aspartyl-phosphate and activates the protein. J. Mol. Biol. 232:67-78. [DOI] [PubMed] [Google Scholar]
- 34.Köhler, T., S. Harayama, J. L. Ramos, and K. N. Timmis. 1989. Involvement of Pseudomonas putida RpoN sigma factor in regulation of various metabolic functions. J. Bacteriol. 171:4326-4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leigh, J. A., and J. A. Dodsworth. 2007. Nitrogen regulation in Bacteria and Archaea. Annu. Rev. Microbiol. 61:349-377. [DOI] [PubMed] [Google Scholar]
- 36.Little, R., F. Reyes-Ramirez, Y. Zhang, W. C. van Heeswijk, and R. Dixon. 2000. Signal transduction to the Azotobacter vinelandii NIFL-NIFA regulatory system is influenced directly by interaction with 2-oxoglutarate and the PII regulatory protein. EMBO J. 19:6041-6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu, Q., and R. A. Bender. 2007. Complex regulation of urease formation from the two promoters of the ure operon of Klebsiella pneumoniae. J. Bacteriol. 189:7593-7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacNeil, T., D. MacNeil, and B. Tyler. 1982. Fine-structure deletion map and complementation analysis of the glnA-glnL-glnG region in Escherichia coli. J. Bacteriol. 150:1302-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandelbaum, R. T., L. P. Wackett, and D. L. Allan. 1993. Mineralization of the s-triazine ring of atrazine by stable bacterial mixed cultures. Appl. Environ. Microbiol. 59:1695-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marques, S., M. T. Gallegos, M. Manzanera, A. Holtel, K. N. Timmis, and J. L. Ramos. 1998. Activation and repression of transcription at the double tandem divergent promoters for the xylR and xylS genes of the TOL plasmid of Pseudomonas putida. J. Bacteriol. 180:2889-2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meletzus, D., P. Rudnick, N. Doetsch, A. Green, and C. Kennedy. 1998. Characterization of the glnK-amtB operon of Azotobacter vinelandii. J. Bacteriol. 180:3260-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller, J. H. 1992. A short course in bacterial genetics: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 43.Minchin, S. D., S. Austin, and R. A. Dixon. 1989. Transcriptional activation of the Klebsiella pneumoniae nifLA promoter by NTRC is face-of-the-helix dependent and the activator stabilizes the interaction of sigma 54-RNA polymerase with the promoter. EMBO J. 8:3491-3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson, K. E., C. Weinel, I. T. Paulsen, R. J. Dodson, H. Hilbert, V. A. Martins dos Santos, D. E. Fouts, S. R. Gill, M. Pop, M. Holmes, L. Brinkac, M. Beanan, R. T. DeBoy, S. Daugherty, J. Kolonay, R. Madupu, W. Nelson, O. White, J. Peterson, H. Khouri, I. Hance, P. Chris Lee, E. Holtzapple, D. Scanlan, K. Tran, A. Moazzez, T. Utterback, M. Rizzo, K. Lee, D. Kosack, D. Moestl, H. Wedler, J. Lauber, D. Stjepandic, J. Hoheisel, M. Straetz, S. Heim, C. Kiewitz, J. A. Eisen, K. N. Timmis, A. Dusterhoft, B. Tummler, and C. M. Fraser. 2002. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 4:799-808. [DOI] [PubMed] [Google Scholar]
- 45.Nishijyo, T., D. Haas, and Y. Itoh. 2001. The CbrA-CbrB two-component regulatory system controls the utilization of multiple carbon and nitrogen sources in Pseudomonas aeruginosa. Mol. Microbiol. 40:917-931. [DOI] [PubMed] [Google Scholar]
- 46.Parada, J. L., and B. Magasanik. 1975. Expression of the hut operons of Salmonella typhimurium in Klebsiella aerogenes and in Escherichia coli. J. Bacteriol. 124:1263-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perez-Martin, J., and V. De Lorenzo. 1995. Integration host factor suppresses promiscuous activation of the sigma 54-dependent promoter Pu of Pseudomonas putida. Proc. Natl. Acad. Sci. USA 92:7277-7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porrua, O., M. Garcia-Jaramillo, E. Santero, and F. Govantes. 2007. The LysR-type regulator AtzR binding site: DNA sequences involved in activation, repression and cyanuric acid-dependent repositioning. Mol. Microbiol. 66:410-427. [DOI] [PubMed] [Google Scholar]
- 49.Reitzer, L. 2003. Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 57:155-176. [DOI] [PubMed] [Google Scholar]
- 50.Reuther, J., and W. Wohlleben. 2007. Nitrogen metabolism in Streptomyces coelicolor: transcriptional and post-translational regulation. J. Mol. Microbiol. Biotechnol. 12:139-146. [DOI] [PubMed] [Google Scholar]
- 51.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 52.Santero, E., T. R. Hoover, A. K. North, D. K. Berger, S. C. Porter, and S. Kustu. 1992. Role of integration host factor in stimulating transcription from the sigma 54-dependent nifH promoter. J. Mol. Biol. 227:602-620. [DOI] [PubMed] [Google Scholar]
- 53.Schmitz, R. A., K. Klopprogge, and R. Grabbe. 2002. Regulation of nitrogen fixation in Klebsiella pneumoniae and Azotobacter vinelandii: NifL, transducing two environmental signals to the nif transcriptional activator NifA. J. Mol. Microbiol. Biotechnol. 4:235-242. [PubMed] [Google Scholar]
- 54.Strosser, J., A. Ludke, S. Schaffer, R. Kramer, and A. Burkovski. 2004. Regulation of GlnK activity: modification, membrane sequestration and proteolysis as regulatory principles in the network of nitrogen control in Corynebacterium glutamicum. Mol. Microbiol. 54:132-147. [DOI] [PubMed] [Google Scholar]
- 55.Timmis, K. N. 2002. Pseudomonas putida: a cosmopolitan opportunist par excellence. Environ. Microbiol. 4:779-781. [DOI] [PubMed] [Google Scholar]
- 56.Totten, P. A., J. C. Lara, and S. Lory. 1990. The rpoN gene product of Pseudomonas aeruginosa is required for expression of diverse genes, including the flagellin gene. J. Bacteriol. 172:389-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Heeswijk, W. C., S. Hoving, D. Molenaar, B. Stegeman, D. Kahn, and H. V. Westerhoff. 1996. An alternative PII protein in the regulation of glutamine synthetase in Escherichia coli. Mol. Microbiol. 21:133-146. [DOI] [PubMed] [Google Scholar]
- 58.Wassem, R., E. M. De Souza, M. G. Yates, F. D. Pedrosa, and M. Buck. 2000. Two roles for integration host factor at an enhancer-dependent nifA promoter. Mol. Microbiol. 35:756-764. [DOI] [PubMed] [Google Scholar]
- 59.Wigneshweraraj, S., D. Bose, P. C. Burrows, N. Joly, J. Schumacher, M. Rappas, T. Pape, X. Zhang, P. Stockley, K. Severinov, and M. Buck. 2008. Modus operandi of the bacterial RNA polymerase containing the sigma54 promoter-specificity factor. Mol. Microbiol. 68:538-546. [DOI] [PubMed] [Google Scholar]
- 60.Wyman, C., I. Rombel, A. K. North, C. Bustamante, and S. Kustu. 1997. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science 275:1658-1661. [DOI] [PubMed] [Google Scholar]
- 61.Zhang, X., M. Chaney, S. R. Wigneshweraraj, J. Schumacher, P. Bordes, W. Cannon, and M. Buck. 2002. Mechanochemical ATPases and transcriptional activation. Mol. Microbiol. 45:895-903. [DOI] [PubMed] [Google Scholar]
- 62.Zhang, X. X., and P. B. Rainey. 2008. Dual involvement of CbrAB and NtrBC in the regulation of histidine utilization in Pseudomonas fluorescens SBW25. Genetics 178:185-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimmer, D. P., E. Soupene, H. L. Lee, V. F. Wendisch, A. B. Khodursky, B. J. Peter, R. A. Bender, and S. Kustu. 2000. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc. Natl. Acad. Sci. USA 97:14674-14679. [DOI] [PMC free article] [PubMed] [Google Scholar]