

Abstract
The asc operon of Escherichia coli is one of the cryptic genetic systems for β-d-galactoside utilization as a carbon source. The ascFB genes for β-d-galactoside transport and catabolism are repressed by the AscG regulator. After genomic SELEX screening, AscG was found to recognize and bind the consensus palindromic sequence TGAAACC-GGTTTCA. AscG binding was detected at two sites upstream of the ascFB promoter and at three sites upstream of the prpBC operon for propionate catabolism. In an ascG-disrupted mutant, transcription of ascFB was enhanced, in agreement with the repressor model of AscG. This repression was indicated to be due to interference of binding of cyclic AMP-CRP to the CRP box, which overlaps with the AscG-binding site 1, as well as binding of RNA polymerase to the promoter. Under conditions of steady-state E. coli growth in a rich medium, the intracellular level of AscG stayed constant at a level supposedly leading to tight repression of the ascFB operon. The level of prpR, encoding the activator of prpBCDE, was also increased in the absence of AscG, indicating the involvement of AscG in repression of prpR. Taken together, these data suggest a metabolic link through interplay between the asc and prp operons.
Both laboratory strains and natural isolates of Escherichia coli possess a number of cryptic genes for β-glucoside utilization (5). The most well-characterized bgl operon is the major genetic system for utilization of β-glucoside sugars such as arbutin and salicin. The bglGFB operon includes the genes encoding the enzymes for uptake of β-glucoside sugars and for hydrolysis of the phosphorylated substrates (15, 26, 33, 34). BglG is a positive regulator of this bgl operon (26, 33) which prevents termination of transcription in the presence of β-glucosides. The bgl operon is, however, cryptic and uninducible in most E. coli strains under laboratory culture conditions (3, 23), but it is expressed and functional in some E. coli strains (5, 26, 31). The cryptic chbBCARF (or celABCDF) operon also includes the genes for transport and phosphorylation of cellobiose, arbutin, and salicin and for hydrolysis of phosphorylated substrates (11, 12, 24, 28). The chbR (celD) gene, encoding a repressor-activator dual regulator, is organized within this operon (11, 12, 25). A set of pathogenic E. coli strains carry an additional cryptic bgc operon encoding the enzymes for transport and hydrolysis of β-glucoside sugars (19). The divergently transcribed bgcR gene encodes a GntR-type transcription factor for positive control of the bgc operon. The asc operon (arbutin-salicin-cellobiose) was discovered by selecting for cellobiose utilization in a bgl chb (or bgl cel) double mutant (4, 5, 23). The cryptic asc operon encodes a repressor (ascG), a phosphotransferase system enzyme II (ascF) for the transport of arbutin, salicin, and cellobiose, and a phospho-β-glucosidase (ascB) that hydrolyzes the sugars which are phosphorylated during transport. The ascG and ascFB operons are transcribed from divergent promoters. The ascFB genes are paralogous to the cryptic bglFB genes, and the ascG gene is paralogous to galR.
Various kinds of β-d-glucosides are released from plants, and thus the expression of a genetic system for utilization of β-glucosides is beneficial for bacterial survival in nature. Genes for utilization of β-d-glucosides are considered to be gained by horizontal transfer from gram-positive bacteria, and in most laboratory E. coli strains, they are retained silently by tight repression by genome silencing factors such as the nucleoid protein H-NS (31). For utilization of β-d-glucosides such as arbutin and salicin as a sole carbon source by laboratory E. coli strains, at least one of the cryptic operons must be activated, enhancing the adaptive potential of the strains. The cryptic operons for β-glucoside utilization can be activated by gaining mutations in the signals involved in transcription initiation or in the regulatory genes. In the case of the bglGFB operon, both the cis-acting regulatory sequence and the trans-acting regulatory proteins that bind to the regulatory sequences respond to silencing of the bgl promoter (18, 29, 32, 35). The major class of activating mutations are effective in cis and have been characterized as insertions of insertion elements or point mutations within the regulatory sequences. Mutations in the cryptic chb operon also allow constitutive utilization of β-d-glucosides such as arbutin, salicin, and cellobiose (25).
In an attempt to gain insight into the regulation of the cryptic genes for β-d-glucoside utilization, we performed detailed analyses of the relatively ill-characterized cryptic asc operon, focusing on the function of the regulatory gene ascG. After a systematic search for the regulation targets of AscG, we determined that in addition to the asc operon, the prp operon for catabolism of propionate is under the control of AscG. The physiological relevance of this interplay between the asc and prp operons is discussed.
MATERIALS AND METHODS
Bacterial strains.
E. coli KP7600 (W3110 lacIq lacZΔM15 galK2 galT22) and JD27135 (with a transposon insertion within the ascG coding sequence of KP7600 in the opposite direction from that of ascG [a gift from T. Miki, Fukuoka Dental College]) were used in various experiments for the analysis of the regulatory roles of AscG, including primer extension assays, promoter assays, and Northern and Western blot analyses. E. coli BL21(DE3) [F− ompT hsdSB(rB− mB−) dcm gal λ(DE3)] was used for expression and purification of AscG. Cells were cultured in LB medium or M9-0.4% glucose medium at 37°C. When necessary, 100 μg/ml ampicillin was added to the medium.
Expression and purification of His-tagged AscG protein.
For construction of plasmid pAscG for AscG expression, a DNA fragment corresponding to the AscG coding sequence was amplified by PCR, using E. coli W3110 genomic DNA as a template and a pair of primers designed so as to hybridize upstream or downstream of the AscG coding sequence. After digestion with NdeI and NotI (note that the restriction enzyme sites were included within the primer sequences), PCR products were cloned into pET21a(+) (Novagen) between NdeI and NotI sites. The plasmid construct was confirmed by DNA sequencing. For protein expression, pAscG plasmid was transformed into E. coli BL21(DE3). Transformants were grown in 200 ml of LB medium, and at a cell optical density at 600 nm (OD600) of 0.6, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. After 3 h of incubation, cells were harvested by centrifugation, washed with a lysis buffer (50 mM Tris-HCl, pH 8.0, at 4°C, with 100 mM NaCl), and then stored at −80°C until use.
For protein purification, frozen cells were suspended in 3 ml of lysis buffer containing 100 mM phenylmethylsulfonyl fluoride. Cells were treated with lysozyme and then subjected to sonication for cell disruption. After centrifugation at 15,000 rpm for 20 min at 4°C, the resulting supernatant was mixed with 2 ml of 50% Ni-nitrilotriacetic acid agarose solution (Qiagen) and loaded onto a column. After being washed with 10 ml of the lysis buffer, the column was washed with 10 ml of washing buffer (50 mM Tris-HCl, pH 8.0, at 4°C, with 100 mM NaCl) and then with 10 ml of washing buffer containing 10 mM imidazole. Proteins were eluted with 2 ml of an elution buffer (lysis buffer plus 200 mM imidazole), and AscG peak fractions were pooled, dialyzed against a storage buffer (50 mM Tris-HCl, pH 7.6, at 4°C, 200 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 1 mM dithiothreitol, and 50% glycerol), and stored at −80°C until use. Protein purity was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (PAGE).
Genomic SELEX of AscG-binding sequences.
The genomic SELEX system was used as described previously (36). Genomic DNA of E. coli W3110 was sonicated to generate fragments of 150 to 300 bp in length. The E. coli DNA library was constructed after cloning of these 150- to 300-bp DNA fragments into plasmid pBR322 at the EcoRV site. A collection of 150- to 300-bp DNA fragments could be regenerated by PCR amplification, using an E. coli DNA plasmid library as the template and a set of primers, EcoRV-F and EcoRV-R, which hybridize to the pBR322 vector at EcoRV junctions (for the primer sequences, see Table 1). PCR products thus generated were purified by 5% PAGE. For genomic SELEX screening of AscG-binding sequences, 5 pmol of DNA fragments and 20 pmol of His-tagged AscG were mixed in a binding buffer (10 mM Tris-HCl, pH 7.8, at 4°C, 3 mM magnesium acetate, 150 mM NaCl, 1.25 μg/ml bovine serum albumin) and incubated for 30 min at 37°C. The mixture was applied to a Ni-nitrilotriacetic acid column, and after washing of unbound DNA with binding buffer containing 10 mM imidazole, DNA-AscG complexes were eluted with an elution buffer containing 200 mM imidazole. When necessary, this SELEX cycle was repeated several times. For sequencing of AscG-bound DNA fragments, DNA was isolated from DNA-AscG complexes by PAGE and then PCR amplified. PCR products were cloned into pT7 Blue-T vector (Novagen), using a blunt-end cloning kit (Takara), and transformed into E. coli DH5α. Sequencing analysis was carried out using a T7 primer (Table 1) and an ABI model 3130xl DNA sequencer.
TABLE 1.
Primers used in this study
| Primer group and name | Length (nucleotides) | Sequence |
|---|---|---|
| Primers for amplification of SELEX fragment DNA | ||
| EcoRV-F | 20 | CTTGGTTATGCCCGGTACTGC |
| EcoRV-R | 20 | GCGATGCTGTCGGAATGGAC |
| Primers for sequencing of SELEX fragment DNA | ||
| T7-F primer | 20 | TAATACGACTCACTATAGGG |
| T7-R primer | 21 | GGTTTTCCCAGTCACACGACG |
| Primer for gel shift and DNase footprinting analyses | ||
| FITC-T7 primer | 20 | TAATACGATCACTATAGGG |
| Primers for Northern blot analysis | ||
| ascG-F | 25 | CAGTTTTAACGCCGTGGCAGAGTTG |
| ascG-R | 22 | AAGGAGCAATGAGTGAGTCGCG |
| ascF-F | 23 | ATGCACCGCGTCTTTACGCCAAC |
| ascF-R | 25 | AAGACTTACTTCTTTGGCGACGGTC |
| ascB-F | 23 | TTTCAGCTATTACGCCTCGCGCT |
| ascB-R | 29 | TAAATCTTCCCCATTACTGGCAATCACTT |
| prpR-F | 23 | GGCATCAGCCTGTTCCGGTAGAT |
| prpR-R | 21 | TTTTCAGCCGCCGCCAGAACG |
| prpB-F | 25 | GTCATCGTCCGAATAAAGCGATCGT |
| prpB-R | 22 | ACGGGCAAACAGGTTGTCGAGC |
| prpE-F | 24 | GTGTATTCAGACCATCTGGGGCGA |
| prpE-R | 22 | TGGCGGATCTGATCCAACGACG |
| Primers for primer extension | ||
| FITC anti-GFP | 19 | AGGGTCAGCTTGCCGTAGG |
| FITC ascF | 28 | CCATCGGATTTTATCCTGTTATCAGTAG |
SELEX chip analysis of AscG-binding sequences.
Mapping of SELEX fragments along the E. coli genome was also performed by using a 22,000-feature DNA microarray (Oxford Gene Technology, Oxford, United Kingdom) (38). One genomic SELEX sample obtained with AscG was labeled with Cy3, while another SELEX sample obtained in the absence of AscG addition or with a different transcription factor was labeled with Cy5. After hybridization of samples to the tiled DNA array, the Cy5/Cy3 ratio was measured and the peaks of scanned patterns were plotted against the positions of DNA probes along the E. coli BW25113 chromosome.
Gel mobility shift assay.
Gel mobility shift assays were performed as described previously (20, 21). In brief, probes were generated by PCR amplification of AscG-binding sequences in the SELEX analysis by use of a pair of primers, 5′-fluorescein-isothiocyanate (FITC)-labeled T7-F and T7-R (Table 1), the genomic SELEX plasmids containing the respective AscG recognition sequences as templates, and Ex Taq DNA polymerase (Takara). PCR products with FITC at their termini were purified by PAGE. For gel shift assays, 1.0 pmol each of the FITC-labeled probes was incubated at 37°C for 30 min with various amounts of AscG in 12 μl of gel shift buffer consisting of 10 mM Tris-HCl, pH 7.8, at 4°C, 150 mM NaCl, and 3 mM magnesium acetate. After the addition of a DNA dye solution, the mixture was directly subjected to 6% PAGE. Fluorescently labeled DNA in gels was detected using a Pharos FX Plus system (Bio-Rad).
DNase I footprinting assay.
DNase I footprinting assays were carried out using FITC-labeled DNA fragments as described previously (20, 21). Each 1.0 pmol of FITC-labeled probes was incubated at 37°C for 30 min with various amounts of AscG in DNase I footprinting buffer consisting of 25 μl of 10 mM Tris-HCl (pH 7.8), 150 mM NaCl, 3 mM magnesium acetate, 5 mM CaCl2, and 25 μg/ml bovine serum albumin. After incubation for 30 min, DNA digestion was initiated by the addition of 12.5 ng of DNase I (Takara). After digestion for 30 s at 25°C, the reaction was terminated by the addition of 25 μl of phenol. Digested products were precipitated with ethanol, dissolved in formamide-dye solution, and analyzed by electrophoresis on a 6% polyacrylamide gel containing 7 M urea.
Measurement of promoter activity.
Promoter strength was determined as described previously (16). In brief, green fluorescent protein (GFP) was expressed under the control of a test promoter, while red fluorescent protein (RFP) was expressed under the control of a reference promoter. The promoter assay vector pGRP carries two types of fluorescent protein genes, one for RFP under the control of reference promoter lacUV5 and the other for GFP under the control of a test promoter. The sequences upstream from the initiation codons of ascF and ascG were PCR amplified by use of their respective primers and inserted into the pGRP vector. The primers contained the recognition sequences for EcoT22I, BglII, or BamHI, which could be used for promoter cloning. The PCR products were digested with the respective restriction enzymes and then ligated into pGRP at EcoT22I and BglII sites. The insertions in the promoter assay plasmids thus constructed were confirmed by sequencing. The plasmids used were pGRK259 (pGRPascF), pGRK559 (pGRPascG), and pGRN024 (pGRPprpR).
For measurement of the fluorescence intensity of RFP or GFP expressed in E. coli transformants, each carrying one promoter assay vector (16), cells were grown in LB medium or M9-0.4% glucose medium to an OD600 of 0.3 to 4.0, harvested by centrifugation, and resuspended in phosphate-buffered saline [PBS(−)]. The cell suspensions were diluted with PBS(−) to obtain approximately the same cell density (OD600 of 0.6) for all samples. For the measurement of bulk fluorescence, aliquots of a 0.2-ml cell suspension were added to 0.4-ml flat-bottomed 96-well plates, and the fluorescence was measured with a Wallac 1420 ARVOsx instrument (Perkin-Elmer Life Sciences), where GFP was measured using a 485-nm excitation wavelength and a 535-nm emission wavelength and RFP was measured using a 544-nm excitation wavelength and a 590-nm emission wavelength. The fluorescence intensity of GFP expressed from the test promoter was normalized using the equation (X/Y)/(A/B), in which X and Y indicate the fluorescence intensities of GFP (test promoter) and RFP (lacUV5 promoter), respectively, while A and B represent the fluorescence intensities of GFP (lacUV5 promoter) and RFP (lacUV5 promoter), respectively (16).
Northern blot analysis.
Northern blot analysis of ascF, ascG, prpR, and prpB RNAs was performed essentially as described previously (20, 21). In brief, total RNAs were prepared by the hot phenol method from both E. coli wild-type strain KP7600 and the ascG-disrupted mutant JD27135, separated by agarose gel electrophoresis in the presence of formamide, and transferred to a positively charged nylon membrane (Roche). Digoxigenin (DIG)-labeled probes were prepared by use of a PCR DIG probe synthesis kit (Roche) and a pair of oligonucleotide primers, each complementary to the 5′-proximal or 3′-proximal sequence of test mRNA (see Table 1 for the primer sequences). Hybridization was carried out at 50°C for 16 h in DIG Easy Hyb (Roche) as recommended by the supplier. Hybridized probe on the membrane was detected by CDP-Star ready-to-use reagent (Roche).
Quantitative Western blot analysis.
Antibodies against AscG, cyclic AMP receptor protein (CRP), and PrpR were raised in rabbits by injection of the respective purified proteins. A quantitative Western blot analysis was carried out by a standard method as described previously (9). In brief, E. coli cells grown in 10 ml of either LB or M9-0.4% glucose medium were harvested by centrifugation and resuspended in 0.3 ml lysis buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5% glycerol, and 1 mM dithiothreitol), and then lysozyme was added to a final concentration of 20 μg/ml. Total proteins were subjected to 12% sodium dodecyl sulfate-PAGE and blotted onto polyvinylidene difluoride membranes by use of a semidry transfer apparatus. Membranes were first immunodetected with anti-AscG, anti-CPA, or anti-PrpR, treated with fluorescently labeled secondary antibodies, and then developed with an enhanced chemiluminescence kit (Amersham Pharmacia Biotech). Images were analyzed with an LAS-1000 Plus Lumino image analyzer and Image Gauge (Fuji Film).
RESULTS AND DISCUSSION
AscG is a GalR-type transcription factor consisting of 337 amino acid residues with a molecular mass of 36,944 Da. The gene encoding AscG is transcribed divergently from the ascFB operon and has been proposed to be involved in repression of the cryptic ascFB operon, which is involved in transport and utilization of the β-glucoside sugars arbutin, salicin, and cellobiose (4).
Screening of AscG-binding sequences by genomic SELEX.
To search for AscG-regulated targets within the E. coli genome, attempts were made to identify DNA sequences that are recognized by AscG by use of the genomic SELEX system (36), in which a complete library of E. coli genome DNA fragments is used as substrates instead of synthetic oligonucleotides, with all possible sequences used in the original SELEX method (2, 41, 43). This genetic SELEX screening has been employed successfully for identification of the whole set of regulation targets for transcription factors including AllR (7), CitB (47), Cra (36), LeuO (39), NemR (44), PdhR (21), RstA (20), and RutR (37). A fourfold molar excess of the purified His-tagged AscG protein was added to a mixture of E. coli genomic DNA fragments of 150 to 300 bp in length, and the AscG-DNA complexes were affinity purified. In the early stage of this genomic SELEX cycle, the AscG-bound DNA fragments gave smear bands on PAGE gels, as did the original genome fragment mixture. After two and three SELEX cycles, several discrete bands were identified, indicating that some DNA fragments with high affinity to AscG were enriched. These DNA fragments were recovered from the gel and cloned into pT7 Blue-T plasmid (Novagen) for sequencing.
Among a total of 79 independently isolated clones, two major species were isolated from two different regions, including one spacer region between the ascG and ascF genes and another between prpR and prpB in the E. coli genome (Table 2). Since regulatory proteins affecting transcription initiation generally bind upstream of the respective target genes, the major regulation targets of AscG were indicated to be ascG and/or ascF and prpR and/or prpB. This finding agrees well with the prediction that AscG regulates the promoter for the ascFB operon. However, experimental evidence indicating the involvement of AscG in the regulation of prpR and/or prpB has not been reported. The number of SELEX isolates correlates with the binding affinity of the test transcription factor to the substrate DNA (36, 44). If this is the case with AscG, then the prp operon must be a major regulation target of AscG, in addition to the asc operon.
TABLE 2.
SELEX search for AscG-binding sitesa
| Left gene | SELEX fragment (size [bp])b | Right gene | No. of fragments |
|---|---|---|---|
| ascG (←) | S (244) | ascF (→) | 36 |
| prpR (←) | S (204) | prpB (→) | 22 |
| recR (←) | S (187) (htpG) | adk (→) | 1 |
| ybiS (←) | S (211) (ybiT) | ybiU (←) | 1 |
| Others | 19 | ||
| cspF (→) | AscG box* | quuQ (→) | |
| fimA (→) | AscG box** | fimC (→) |
Genomic SELEX analysis for a search of AscG-binding sequences was performed using purified His-tagged AscG alone in the absence of effectors. The SELEX fragments were also analyzed by a SELEX chip system. A total of 79 DNA SELEX fragments were isolated, among which 36 fragments contained a 244-bp sequence within spacer regions between the ascG and ascF coding frames and 22 fragments contained a 204-bp sequence between the prpR and prpB coding sequences. Each of the other 21 SELEX fragments contained a different sequence originating from a different region of the E. coli genome, among which AscG binding within the coding regions of hptG and ybiT was also detected by the SELEX chip system. After sequence analysis of the entire E. coli genome, an AscG box-like sequence was detected in the cspF-quuQ and fimA-fimC spacer regions. The arrows indicate the direction of transcription of the neighboring genes, and the genes shown in bold represent possible target genes under the control of AscG.
*, TAAACCGGTTTC; **, GAACCGTTTC.
The binding of AscG to the ascG-ascF and prpR-prpB intergenic spaces was confirmed by analysis of the SELEX fragments by use of a tiled array (OGT) (38). In addition to these two major sites, binding of AscG was identified on htpG, encoding the HSP90 chaperone, and ybiT, encoding an ABC-type transporter, by both genomic SELEX and SELEX chip analyses (Table 1).
Identification of AscG-binding DNA sequences.
The binding of AscG protein to the two major sequences identified by the SELEX screening was examined by gel shift assay. As shown in Fig. 1, both SELEX DNA fragments formed AscG complexes in a protein dose-dependent manner. One unexpected finding is that both probes contained multiple sites for AscG binding. Upon increases in the AscG concentration, the AscG complexes with both ascG-ascF (Fig. 1A) and prpR-prpB (Fig. 1B) probes were supershifted, altogether forming two complex bands in the case of ascG-ascF (Fig. 1A) and three complex bands with the prpR-prpB probe (Fig. 1B). This finding indicates that at least two and three binding sites exist in the ascG-ascF and prpR-prpB probes, respectively.
FIG. 1.
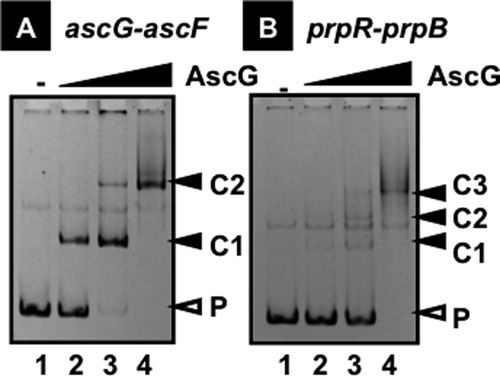
Gel mobility shift assay of AscG-target DNA complex formation. Fluorescently labeled DNA probes (1.0 pmol each) containing a SELEX segment from the ascG-ascF spacer (A) or the prpR-prpB spacer (B) were incubated at 37°C for 30 min in the absence (lane 1) or presence of increasing concentrations of AscG (lane 2, 2.5 pmol; lane 3, 5.0 pmol; and lane 4, 10 pmol). The reaction mixtures were directly subjected to PAGE. P, probe DNA; C1 to C3, AscG-probe DNA complexes.
To identify the recognition sequence on each of the AscG-binding SELEX fragments, we next performed DNase I footprinting assays, using both SELEX fragments as probes. After formation of complexes in the presence of a fixed amount of the fluorescently labeled DNA probe and increasing amounts of AscG, DNase I treatment was carried out for a short period, and the partially digested DNA products were analyzed by PAGE (Fig. 2). In the case of the ascG-ascF probe, two protected regions were detected, one between positions −84 and −59 with respect to the ascF promoter and another between positions −8 and +11 (Fig. 3A), in good agreement with the formation of two bands in gel shift assays (Fig. 1A). On the other hand, in the case of the prpR-prpB probe, clear protection by AscG was observed at a single long region consisting of a 54-bp sequence between prpR positions −11 and −64 (Fig. 3A), indicating that three AscG protomers bind in tandem within this sequence.
FIG. 2.
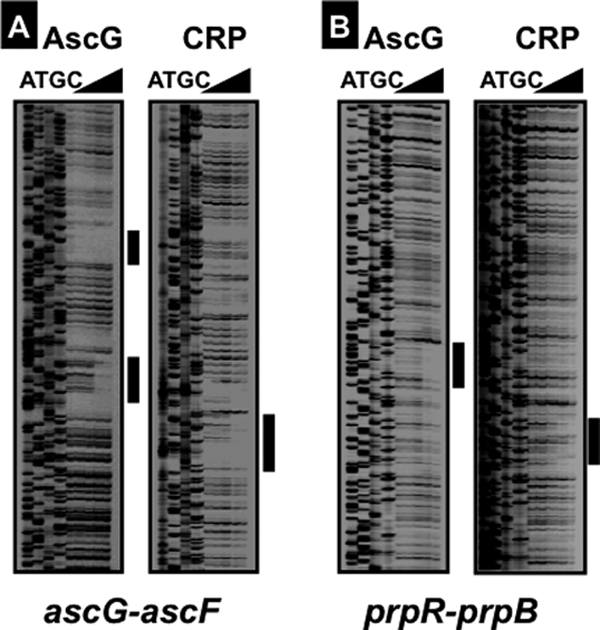
Identification of AscG box sequence. A fluorescently labeled SELEX segment (1.0 pmol) from the ascG-ascF spacer (A) or the prpR-prpB spacer (B) was incubated in the absence or presence of increasing concentrations of purified AscG (2.5, 5.0, 10, and 20 pmol, from right to left) or CRP (0.3, 0.6, 1.25, and 2.5 pmol, from right to left) and then subjected to DNase I footprinting assays. Lanes A, T, G, and C represent the respective sequence ladders.
FIG. 3.
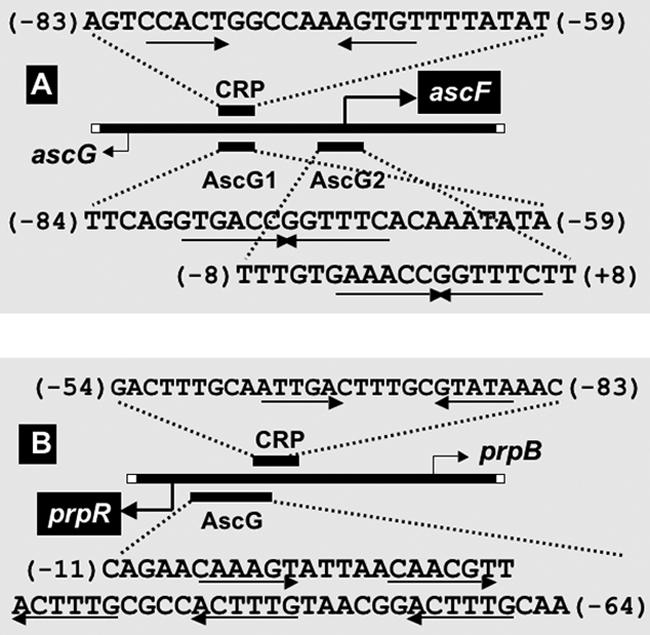
Locations of promoters and AscG- and CRP-binding sites. (A) The transcription initiation sites of ascG and ascF and the binding sites for AscG and CRP are shown along the 256-bp-long ascG-ascF spacer. The transcription initiation sites were determined by primer extension assay (data not shown), while the binding sites for AscG and cAMP-CRP were determined by DNase I footprinting assay as described in the legend to Fig. 2. The numbers in parentheses represent distances from the transcription initiation site of ascF, which is under the control of AscG. Open boxes at the termini of the DNA bar represent the initiation codons of ascG and ascF. (B) The transcription initiation sites of prpR and prpB and the binding sites for AscG and CRP are shown along the 200-bp-long prpR-prpB spacer. The binding sites for AscG and cAMP-CRP were determined by DNase I footprinting assay as described in the legend to Fig. 2. The numbers in parentheses represent distances from the transcription initiation site of prpR, which is under the control of AscG. Boxes at the termini of the DNA bar represent the initiation codons of prpR and prpB.
Prediction of the AscG recognition sequence.
After alignment of the two DNase I protection patterns obtained by the use of two different probes, we could identify the 14-bp-long palindromic sequence TGAAACC-GGTTTCA as the AscG recognition sequence, and it is hereafter referred to as the AscG box (Fig. 4A). The AscG-protected site 1 on the ascF promoter includes this AscG box between positions −80 and −67 with respect to the transcription initiation site of ascF (Fig. 3A). On the other hand, the AscG-protected site 2 includes the complete 14-bp-long palindromic AscG box between positions −4 and +9 (Fig. 4A). Since AscG site 2 overlaps with promoter position −10 and the transcription initiation site of ascF, AscG bound at site 2 may play a major role in repression of ascFB transcription. Since the AscG box sequence in ascF site 2 is identical with the consensus AscG box, the binding affinity of AscG should be strong, presumably leading to strong repression (see below for promoter assay and mRNA analysis). After sequence analysis of the entire E. coli genome, the AscG box-like sequence was identified in the spacer regions of both cspF-quuQ and fimA-fimC (Table 1), raising the possibility that both quuQ (Qin antiterminator) and fimC (pilus chaperone) are under the control of AscG.
FIG. 4.
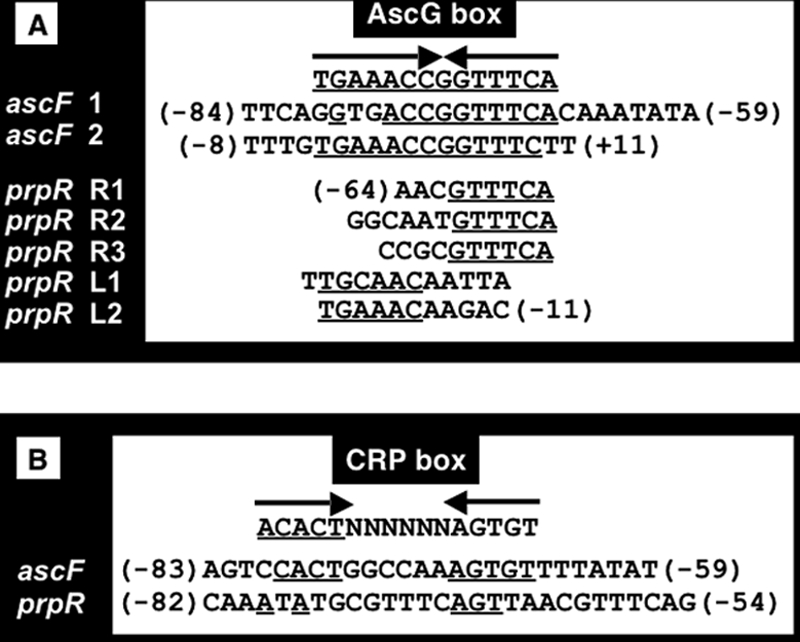
Consensus sequence of AscG box and its location within the asc and prp operons. (A) The sequences represent those protected by AscG within the ascG-ascF and prpR-prpB spacer regions (see Fig. 2 and 3). The consensus sequence of the AscG box was identified after comparison of these AscG-binding sequences. The numbers in parentheses represent distances (bp) from the transcription initiation site of ascF or prpR. The underlined nucleotides represent those identical with the consensus AscG box. (B) The sequences represent those protected by cAMP-CRP within the ascG-ascF and prpR-prpB spacer regions (see Fig. 2 and 3). The numbers in parentheses represent distances (bp) from the transcription initiation site of ascF or prpR.
The cluster of genes required for propionate catabolism was first identified in Salmonella enterica serovar Typhimurium (6, 8), and a closely related cluster was found in E. coli after genome sequencing (30). These genes constitute a locus composed of two divergently transcribed units. One unit is the single gene prpR, which encodes a regulatory protein, PrpR, a member of the RpoN-dependent activator family (22, 40). The second transcriptional unit contains the prpBCDE operon, which encodes the enzymes for propionate catabolism, allowing growth on propionate as a sole carbon and energy source (8). After DNase I footprinting assay of the 200-bp spacer between prpR and prpB (Fig. 2), one 54-bp-long region protected by AscG was detected between positions −64 and −11 with respect to the prpR promoter (Fig. 3). Within this 54-bp sequence, two copies of the left half of the AscG box (L1 and L2) (Fig. 4A) and three copies of its right half (R1, R2, and R3) (Fig. 4A) were identified. Thus, the formation of three complex bands upon gel shift assay (Fig. 1) indicates sequential binding of three AscG protomers within this modified AscG box. Since this AscG-binding sequence overlaps the prpR promoter, AscG should repress transcription initiation of the prpR gene. This AscG-binding site also overlaps the predicted PrpR-binding site for activation of the prpBCDE operon (13), so the binding of AscG may also interfere with the activation of the prpBCDE operon by PrpR.
Identification of CRP-binding DNA sites.
In E. coli, glucose controls the utilization of alternative carbon sources by regulating transcription of the genes involved in their catabolism in response to glucose depletion (1, 10, 14). Since the genes for β-d-glucoside utilization were predicted to be expressed in the absence of glucose and since the AscG box sequence is similar to the CRP box sequence (Fig. 4), we analyzed binding in vitro of cyclic AMP (cAMP)-CRP to the ascF promoter. By gel shift and DNase I footprinting assays, we indeed identified binding of cAMP-CRP between positions −83 and −59 (Fig. 4B), completely overlapping with AscG-binding site 1 (between positions −84 and −59) (Fig. 4A). From the location of the CRP-binding site, CRP was indicated to be involved in activation of the ascFB operon.
Lee et al. (13) proposed the involvement of cAMP-CRP in transcription activation of both the divergently transcribed prpR gene and the prpBCDE operon. We then analyzed the binding site of cAMP-CRP within the ascG-ascF spacer region. After DNase I footprinting assay, one cAMP-CRP-binding site was identified, including a 29-bp sequence between positions −82 and −54 of the prpR promoter or between positions −119 and −92 of the prpBCDE promoter (Fig. 3). A CRP box-like sequence was identified within this sequence (Fig. 4), which overlaps partially with the AscG-binding site (Fig. 3).
Influence of AscG on in vivo promoter activity.
The possible influence of AscG on transcription initiation in vivo from the ascFB, prpR, and prpBCDE promoters was then examined using a two-fluorescent-protein (TFP) promoter assay system. For this purpose, we first identified the hitherto unidentified promoter for the ascFB operon. Using a primer extension assay in the absence of the AscG repressor, a transcription initiation site of ascF was identified at position −95 relative to the initiation codon (data not shown). We then cloned sequences of about 500 bp in length upstream from the initiation codons of the ascF, prpR, and prpB genes (see Fig. 3 and 8 for the gene organization), each including transcription initiation sites and recognition sequences for a transcription factor, i.e., AscG (ascF), CRP (ascF and prpB), or PrpR (prpB), and inserted them into pGRP to construct the promoter assay vectors pGRK259 (pGRPascF), pGRK559 (pGRPascG), and pGRN024 (pGRPprpR). The expression of GFP was under the control of each test promoter, while RFP expression was directed by the reference promoter lacUV5 on the same vector (see Materials and Methods).
FIG. 8.
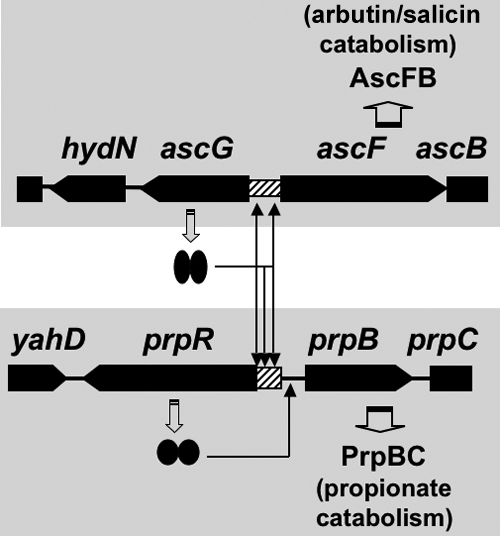
Model of the regulatory roles of AscG. AscG was found to regulate not only the ascFB operon but also the prpR gene, which encodes a positive regulator of the prpBC operon for propionate catabolism. Two AscG-binding sites were identified in the ascF promoter, while three sites were identified in the prpR promoter.
Promoter activity was detected for transformants with pGRK259 (pGRPascF) in both wild-type E. coli KP7600 and the mutant JD27135 with a disrupted ascG gene. The promoter activity in wild-type E. coli increased concomitantly with the advance of cell growth but was always two- to threefold higher for the mutant without AscG protein (Fig. 5A). This finding supports the prediction that AscG represses the expression of the ascFB operon under ordinary culture conditions. Using transformants with pGRN024 (pGRPprpR), the expression of GFP under the control of the prpR promoter was examined (Fig. 5B). Even though the promoter activity of prpR was lower than that of ascF, it was significantly higher in the mutant lacking AscG (Fig. 5B), supporting the prediction that AscG is involved in repression of the prpR promoter. To confirm the results of the promoter assay using the TFP vector, we next performed direct measurement of mRNAs.
FIG. 5.
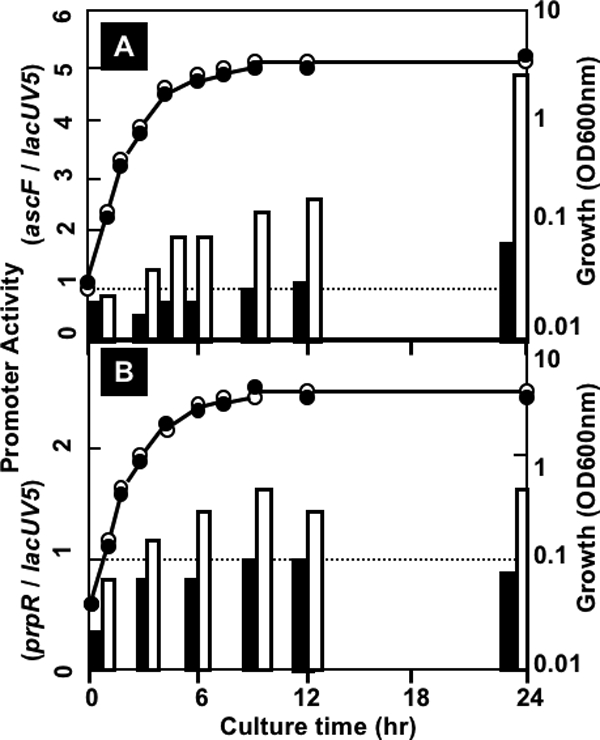
Assay of the acsF and prpR promoters in wild-type and ascG mutant E. coli. The ascF and prpR promoter fragments were inserted into the TFP promoter assay vector pGRP, and the resulting promoter plasmids, pGRPascF (A) and pGRPprpR (B), were transformed into wild-type KP7600 and its ascG-disrupted mutant, JD27135. The promoter activities were determined by measuring the GFP/RFP ratio. Closed bars represent the promoter activities in wild-type KP7600, while open bars represent the promoter activities in the ascG mutant JD27135.
Regulation in vivo of the ascFB and prpBCDE promoters by AscG.
Total RNAs were isolated from both wild-type KP7600 and ascG-disrupted JD27135 and subjected to Northern blot analysis using a fluorescently labeled probe for detection of the 5′-proximal region of ascF mRNA. A single hybridized band was detected (Fig. 6), and its band intensity increased markedly in the ascG mutant, in particular when grown in LB medium (Fig. 6, lanes 1 and 2). When cells were grown in M9-glucose medium, the level of ascF transcription was very low because of the lack of cAMP in the presence of glucose (Fig. 6, lanes 3 and 4). Based on this finding, we concluded that AscG is the repressor of transcription initiation from the promoter of the ascFB operon.
FIG. 6.
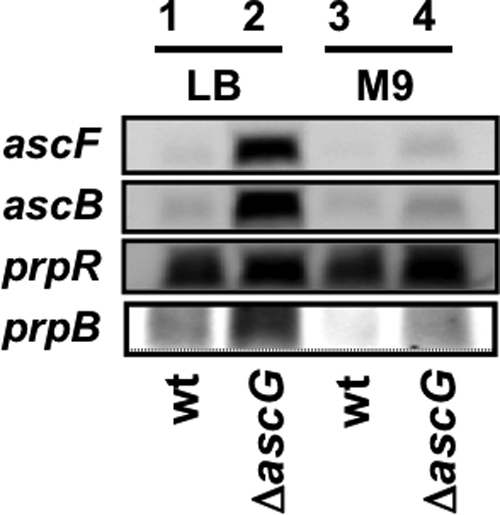
Northern blot analysis of asc and prp operon RNAs. Overnight cultures of wild-type E. coli KP7600 (wt) and its ascG-disrupted mutant JD27135 (ΔascG) in either LB or M9-glucose medium were transferred into the respective fresh medium. After shaking culture at 37°C, cells were harvested in the middle of log phase (OD600, 0.6). Total RNA was isolated as described in Materials and Methods and fractionated by PAGE in the presence of urea. After blotting of RNA onto filters, ascF, ascB, prpR, and prpB RNAs were detected with fluorescently labeled probes. The probes used were prepared by PCR, using the primers described in Table 1. The total amounts of RNA samples were adjusted based on the staining intensity of rRNA with ethidium bromide.
By Northern blot analysis, high-level expression of the prpR gene was detected in wild-type E. coli grown in LB medium (Fig. 6). The level of prpR mRNA increased slightly in the ascG mutant. Taking these observations together with the promoter assay results (Fig. 5), we concluded that AscG plays a repressive role, albeit at a low level, for the prpR gene encoding the activator of the prpBCDE operon. In agreement with the slight increase in prpR expression in the ascG mutant, the mRNA level of prpB, the activation target of PrpR, significantly increased (Fig. 6). The level of prpB mRNA in M9-glucose culture (Fig. 6, lanes 3 and 4) was lower than that in LB culture (Fig. 6, lanes 1 and 2), in agreement with the involvement of CRP in activation of the prpBCDE operon (13).
Competition between AscG and CRP.
The AscG-binding sites identified herein are similar to the CRP box sequence (Fig. 3). We next analyzed binding in vitro of cAMP-CRP to the ascG-ascFB spacer region. By gel shift and DNase I footprinting assays, we indeed identified binding of cAMP-CRP upstream of the ascFB promoter (Fig. 2). Within the protected regions, a sequence similar to the consensus palindromic CRP box sequence, TGTGA-TCACA, was identified (Fig. 3). The binding site (between positions −83 and −59) of cAMP-CRP for activation of the ascFB operon completely overlaps that (between positions −84 and −59) of repressor AscG-binding site 1 (Fig. 3). This immediately suggests that one of the possible mechanisms of ascFB repression by AscG is competition between two regulators, AscG and CRP, for binding to their respective target sites. This possibility was examined by competitive gel shift assay, as employed for AscG binding to the targets identified after genomic SELEX screening (Fig. 1). Protein species bound to the shifted DNA complexes were determined by immunoblot analysis, using specific antibodies against AscG and CRP. Probe DNA-bound CRP was decreased by adding increasing amounts of AscG (Table 3). Likewise, the amount of DNA-bound AscG decreased with an increase in cAMP-CRP. This finding supports the prediction that AscG represses ascFB transcription not only through interference of RNA polymerase binding to the promoter (at AscG site 2) but also through interference of binding of the activator cAMP-CRP (at AscG site1).
TABLE 3.
Competition between AscG and CRP in binding to the ascF promotera
| AscG level (pmol) | Amt (pmol) of added cAMP-CRP | % DNA-bound protein
|
|
|---|---|---|---|
| AscG | CRP | ||
| 2.5 | 0 | 100 | 0 |
| 5.0 | 0 | 100 | 0 |
| 5.0 | 0.3 | 70 | 30 |
| 5.0 | 0.6 | 50 | 30 |
| 0 | 0.3 | 0 | 100 |
| 0 | 0.6 | 0 | 100 |
| 2.5 | 0.6 | 80 | 70 |
| 5.0 | 0.6 | 70 | 50 |
A fluorescently labeled ascG-ascF spacer DNA probe (1.0 pmol) was incubated with the indicated amounts of AscG and/or CRP at 37°C for 30 min in the presence of 0.05 mM cAMP. The reaction mixtures were directly subjected to PAGE. Proteins were blotted onto polyvinylidene difluoride membranes by use of a semidry transfer apparatus. Membranes were first immunodetected with anti-AscG or anti-CRP, treated with fluorescently labeled secondary antibodies, and then developed with an enhanced chemiluminescence kit (Amersham Pharmacia Biotech). The intensities of stained bands were measured with an LAS-1000 Plus Lumino image analyzer and Image Gauge (Fuji Film). The approximate amounts of DNA-bound proteins were estimated from the band intensities.
In the case of the prpB promoter, the cAMP-CRP-binding site does not overlap with the AscG-binding site on the prpR promoter (Fig. 3). Thus, the repression of prpR and prpBCDE transcription by AscG might not be due to interference of the action of cAMP-CRP.
Search for effectors influencing expression of the ascFB operons.
To identify the inducer(s) that enhances or reduces the induction of the ascFB operon, we performed a phenotype microarray assay using the whole set of 20 standard phenotype microarray plates. Significant differences in cell growth between wild-type KP7600 and the isogenic ascG mutant JD27135 were identified in several wells. In the presence of arbutin or salicin as a sole carbon source, cell growth was observed only with the ascR mutant lacking AscG (data not shown). This finding supports the prediction that in wild-type E. coli, AscG represses the genes needed for utilization of arbutin and salicin, but in the absence of AscG, the ascFB genes for the catabolism of these compounds as carbon sources are induced.
Intracellular levels of AscG.
mRNA analysis indicated that in wild-type E. coli, the ascFB genes, encoding the enzymes for utilization of arbutin and salicin, are strictly repressed (Fig. 6). We then determined the intracellular level of AscG in wild-type E. coli grown in LB medium by using quantitative Western blot analysis (9). From exponential growth phase to stationary phase, the intracellular concentration of AscG stayed almost constant, at a level of about 500 to 700 molecules per genome equivalent of DNA (Fig. 7). This finding indicates that a high level of AscG is always present in wild-type E. coli cells and represses transcription of the ascFB operon.
FIG. 7.
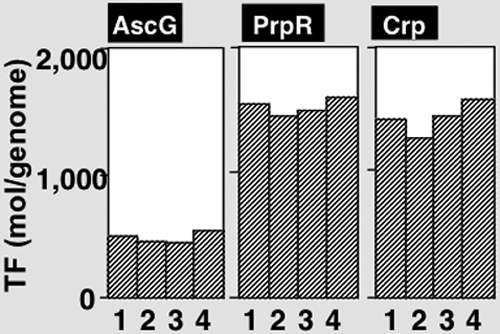
Determination of intracellular concentrations of AscG, CRP, and PrpR. Wild-type E. coli KP7600 was grown in LB medium at 37°C for various times. Cells were harvested at turbidities of 0.3, 0.6, 0.9, and 1.5, and whole-cell lysates were prepared as described in Materials and Methods. Quantitative Western blot analysis was performed by a standard procedure (11), using anti-AscG, anti-CRP, and anti-PrpR antibodies. The intensities of immunoblot bands were measured with an LAS-1000 Plus Lumino image analyzer and Image Gauge (Fuji Film).
Using the same cell lysates, we also determined the intracellular concentrations of CRP and PrpR (Fig. 7). Surprisingly, the level of PrpR (1,500 to 1,700 molecules per genome equivalent of DNA) was as high as that of the most abundant global regulator, CRP, even in the absence of propionate (Fig. 7). At present, it is not clear why such a high level of PrpR is present under normal growth conditions in the absence of propionate. One possibility is that PrpR has an as yet unidentified physiological role, including the regulation of other genes besides the only known target, the prpBCDE operon.
Physiological roles of AscG.
At least four operons for β-d-glucoside utilization have been identified in E. coli, but most of these operons are preserved as cryptic states in most laboratory E. coli strains. Each of these operons is associated with a specific regulatory gene for the control of gene expression. For instance, the genes for the activators BglG and ChbR (CelR) are organized within the respective target operons bglGFB (15, 26, 33, 34) and chbBCARF (or celABCDF) (12, 24, 25, 28). On the other hand, the genes for the regulators AscG and BgcR form single-gene operons, which are located next to the divergently transcribed target operons, namely, ascFB (11, 12) and bgcEFIHA (19), respectively. The modes of action are also different for these regulators. BglG activates the bglGFB operon by preventing transcription termination in the presence of β-glucosides (33), while ChbR (or CelD) is a positive factor for transcription initiation (25). In contrast, AscG is the repressor for keeping the ascFB operon silent under ordinary culture conditions (12; this report). In fact, the level of AscG is maintained at a high level to keep the ascFB genes silent under steady-state cell growth (Fig. 7). In the presence of β-d-glucosides but the absence of useful carbon sources such as glucose in natural environments, however, the expression of these cryptic genes may provide an advantage for growth. These cryptic genes for β-d-glucoside utilization are preserved in a silent state in most wild-type E. coli strains. Silencing of these cryptic operons for β-d-glucoside utilization may be needed to prevent the utilization of toxic aryl-β-d-glucoside substrates (31).
The direct action of AscG as a repressor of the prpR gene could provide a regulatory link of utilization between β-d-glucosides and propionate (Fig. 8). In E. coli, propionate arises as the terminal three-carbon fragment in the oxidation of odd-chain-length fatty acids (45, 46). Propionate is catabolized to pyruvate through alpha oxidation. The 200-bp-long intergenic region between the prpR and prpBCDE operons contains an RpoD-dependent promoter for prpR and an RpoN-dependent promoter for prpBCDE. In addition, the regulatory factors PrpR, cAMP-CRP, and IHF have been suggested to participate in the transcriptional activation of the prpBCDE operon (8, 14, 22, 42). Expression of prpBCDE is dependent on the RpoN RNA polymerase holoenzyme and PrpR (8, 22, 42). PrpR is an NtrC family activator which interacts directly with RpoN RNA polymerase (17). NtrC family activators are generally involved in nitrogen regulation in E. coli, since nitrogen assimilation consumes energy and intermediates of central metabolism (27). Involvement of CRP and AscG indicates another link between carbon and nitrogen metabolism. Taking these observations together, we propose that the catabolism of propionate is under the control of several regulators of the genes for both carbon and nitrogen metabolism.
Acknowledgments
This work was supported by grants-in-aid (17076016, 18310133, and 21241047) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by the Nano-Biology Project of the Micro-Nano Technology Research Center of Hosei University.
We thank T. Miki for the JD27135 mutant strain and T. Shimada, H. Ogasawara, and J. Teramoto (Hosei University) for helpful discussions.
Footnotes
Published ahead of print on 24 July 2009.
REFERENCES
- 1.deCrombrugghe, B., S. Busby, and H. Buc. 1984. Cyclic AMP receptor protein: role in transcription activation. Science 224:831-838. [DOI] [PubMed] [Google Scholar]
- 2.Ellington, A. D., and J. W. Szostak. 1990. In vitro selection of DNA molecules that bind specific ligands. Nature 346:818-822. [DOI] [PubMed] [Google Scholar]
- 3.Hall, B. G., and P. W. Betts. 1987. Cryptic genes for cellobiose utilization in natural isolates of Escherichia coli. Genetics 115:431-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall, B. G., and L. Xu. 1992. Nucleotide sequence, function, activation, and evolution of the cryptic asc operon of Escherichia coli K12. Mol. Biol. Evol. 9:688-706. [DOI] [PubMed] [Google Scholar]
- 5.Hall, B. G., S. Yokoyama, and D. H. Calhoun. 1983. Role of cryptic genes in microbial evolution. Mol. Biol. Evol. 1:109-124. [DOI] [PubMed] [Google Scholar]
- 6.Hammelman, T. A., G. A. O'Toole, J. R. Trzebiatowski, A. W. Tsang, D. Rank, and J. C. Escalante-Semerena. 1996. Identification of a new prp locus required for propionate catabolism in Salmonella typhimurium LT2. FEMS Microbiol. Lett. 137:233-239. [DOI] [PubMed] [Google Scholar]
- 7.Hasegawa, A., H. Ogasawara, A. Kori, and A. Ishihama. 2008. AllR is the allantoin/glyoxylate-sensing master regulator of the genes for degradation and reutilization of purines. Microbiology 154:3366-3378. [DOI] [PubMed] [Google Scholar]
- 8.Horswill, A. R., and J. C. Escalante-Semerena. 1997. Propionate catabolism in Salmonella typhimurium LT2: two divergently transcribed units comprise the prp locus at 8.5 centisomes, prpR encodes a member of the sigma-54 family of activators, and the prpBCDE genes constitute an operon. J. Bacteriol. 179:928-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jishage, M., A. Iwata, S. Ueda, and A. Ishihama. 1996. Regulation of RNA polymerase sigma subunit synthesis in Escherichia coli: intracellular levels of four species of sigma subunit under various growth conditions. J. Bacteriol. 178:2507-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolb, A., S. Busby, H. Buc, S. Garges, and S. Adhya. 1993. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62:749-795. [DOI] [PubMed] [Google Scholar]
- 11.Kricker, M., and B. G. Hall. 1984. Directed evolution of cellobiose utilization in Escherichia coli K12. Mol. Biol. Evol. 1:171-182. [DOI] [PubMed] [Google Scholar]
- 12.Kricker, M., and B. G. Hall. 1987. Biochemical genetics of the cryptic gene system for cellobiose utilization in Escherichia coli K12. Genetics 115:419-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee, S. K., J. D. Newman, and J. D. Keasling. 2005. Catabolite repression of the propionate catabolic genes in Escherichia coli and Salmonella enterica: evidence for involvement of the cyclic AMP receptor protein. J. Bacteriol. 187:2793-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin, E. C. C. 1996. Dissimilatory pathways for sugars, polyols, and carboxylates, p. 307-342. In F. C. Neidhardt, J. L. Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed., vol. 1. ASM Press, Washington, DC. [Google Scholar]
- 15.Mahadeven, S., A. E. Reynolds, and A. Wright. 1987. Positive and negative regulation of the bgl operon of Escherichia coli. J. Bacteriol. 169:2570-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makinoshima, H., A. Nishimura, and A. Ishihama. 2002. Fractionation of Escherichia coli cell populations at different stages during growth transition to stationary phase. Mol. Microbiol. 43:269-279. [DOI] [PubMed] [Google Scholar]
- 17.Merrick, M. J. 1993. In a class of its own—the RNA polymerase sigma factor σ54 (σN). Mol. Microbiol. 10:903-909. [DOI] [PubMed] [Google Scholar]
- 18.Mukerji, M., and S. Mahadevan. 1997. Characterization of the negative elements involved in silencing the bgl operon of Escherichia coli: possible roles for DNA gyrase, H-NS, and CRP-cAMP in regulation. Mol. Microbiol. 24:617-627. [DOI] [PubMed] [Google Scholar]
- 19.Neelakanta, G., T. S. Sankar, and K. Schnetz. 2009. Characterization of a β-d-glucoside operon (bgc) present in septicemic and uropathogenic Escherichia coli strains. Appl. Environ. Microbiol. 75:2284-2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogasawara, H., A. Hasegawa, E. Kanda, T. Miki, K. Yamamoto, and A. Ishihama. 2007. Genomic SELEX search for target promoters under the control of the PhoQP-RstB signal cascade. J. Bacteriol. 187:4791-4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogasawara, H., Y. Ishida, K. Yamada, K. Yamamoto, and A. Ishihama. 2007. PdhR (pyruvate dehydrogenase complex regulator) controls the respiratory electrotransport system in Escherichia coli. J. Bacteriol. 189:5534-5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palacios, S., and J. C. Escalante-Semerena. 2000. prpR, ntrA, and ihf functions are required for expression of the prpBCDE operon, encoding enzymes that catabolize propionate in Salmonella enterica serovar Typhimurium LT2. J. Bacteriol. 182:905-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parker, L. L., and B. G. Hall. 1988. A fourth Escherichia coli gene system with the potential to evolve β-glucoside utilization. Genetics 119:485-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker, L. L., and B. G. Hall. 1990. Characterization and nucleotide sequence of the cryptic cel operon of E. coli K12. Genetics 124:455-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker, L. L., and B. G. Hall. 1990. Mechanisms of activation of the cryptic cel operon of E. coli K12. Genetics 124:473-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prasad, I., and S. Schaffler. 1974. Regulation of the β-glucoside system in Escherichia coli K12. J. Bacteriol. 120:638-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reitzer, L., and B. L. Schneider. 2001. Metabolic context and possible physiological themes of sigma54-dependent genes in Escherichia coli. Microbiol. Mol. Biol. Rev. 65:422-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reizer, J., A. Reizer, and M. H. Saier, Jr. 1990. The cellobiose permease of Escherichia coli K12 consists of three proteins and is homologous to the lactose permease of Staphylococcus aureus. Res. Microbiol. 141:1061-1067. [DOI] [PubMed] [Google Scholar]
- 29.Reynolds, A. E., J. Felton, and A. Wright. 1981. Insertion of DNA activates the cryptic bgl operon in E. coli K-12. Nature 293:625-629. [DOI] [PubMed] [Google Scholar]
- 30.Riley, M., T. Abe, M. B. Arnaoud, M. K. B. Berlyn, F. R. Blattner, R. R. Chaudhuri, J. D. Glasner, T. Horiuchi, I. M. Keseler, T. Kosuge, H. Mori, N. T. Perna, G. Plunkett III, K. E. Rudd, M. H. Serres, G. H. Thomas, N. R. Thomson, D. Wishart, and B. L. Wanner. 2006. Escherichia coli K-12: a cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 34:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sankar, T. S., G. Neelakanta, V. Sangal, G. Plum, M. Achtman, and K. Schnetz. 2009. Fate of the H-NS-repressed bgl operon in evolution of Escherichia coli. PLoS Genet. 5:e1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnetz, K. 1995. Silencing of the Escherichia coli bgl promoter by flanking sequence elements. EMBO J. 14:2545-2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnetz, K., and B. Rak. 1988. Regulation of the bgl operon of Escherichia coli by transcriptional anti-termination. EMBO J. 7:3271-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnetz, K., C. Toloczyki, and B. Rak. 1987. β-Glucoside (bgl) operon of Escherichia coli K12: nucleotide sequence, genetic organization, and possible evolutionary relationship to regulatory components of two Bacillus subtilis genes. J. Bacteriol. 169:2579-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnetz, K., and J. C. Wang. 1996. Silencing of the Escherichia coli bgl promoter: effects of template supercoiling and cell extracts on promoter activity in vitro. Nucleic Acids Res. 24:2422-2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimada, T., N. Fujita, M. Maeda, and A. Ishihama. 2005. Systematic search for the Cra-binding promoters using genomic SELEX systems. Genes Cells 10:907-918. [DOI] [PubMed] [Google Scholar]
- 37.Shimada, T., K. Hirao, A. Kori, K. Yamamoto, and A. Ishihama. 2007. RutR is the uracil/thymine-sensing master regulator of a set of genes for synthesis and degradation of pyrimidines. Mol. Microbiol. 66:744-779. [DOI] [PubMed] [Google Scholar]
- 38.Shimada, T., A. Ishihama, S. J. W. Busby, and D. C. Grainger. 2008. The Escherichia coli RutR transcription factor binds at targets within genes as well as intergenic regions. Nucleic Acids Res. 36:3950-3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada, T., K. Yamamoto, and A. Ishihama. 2009. Involvement of the leucine response transcription factor LeuO in regulation of the genes for sulfa drug efflux. J. Bacteriol. 191:4562-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shingler, V. 1996. Signal sensing by sigma 54-dependent regulators: derepression as a control mechanism. Mol. Microbiol. 19:409-416. [DOI] [PubMed] [Google Scholar]
- 41.Singer, B. S., T. Shtatland, D. Brown, and L. Gold. 1997. Libraries for genomic SELEX. Nucleic Acids Res. 25:781-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsang, A. W., A. R. Horswill, and J. C. Escalante-Semerena. 1998. Studies of regulation of expression of the propionate (prpBCDE) operon provide insights into how Salmonella typhimurium LT2 integrates its 1,2-propanediol and propionate catabolic pathways. J. Bacteriol. 180:6511-6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tuerk, C., and L. Gold. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505-510. [DOI] [PubMed] [Google Scholar]
- 44.Umezawa, Y., H. Ogasawara, T. Shimada, A. Kori, and A. Ishihama. 2008. The uncharacterized YdhM is the regulator of the nemA gene, coding for N-ethylmaleimide reductase. J. Bacteriol. 190:5890-5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weeks, G., M. Shapiro, R. O. Burns, and S. J. Wakil. 1969. Control of fatty acid metabolism. 1. Induction of the enzymes of fatty acid oxidation in Escherichia coli. J. Bacteriol. 97:827-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wegener, W. S., H. C. Reeves, R. Rabin, and S. J. Ajl. 1968. Alternate pathways of propionate metabolism short-chain fatty acids. Bacteriol. Rev. 32:1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto, K., F. Matsumoto, T. Oshima, N. Fujita, N. Ogasawara, and A. Ishihama. 2008. Anaerobic regulation of citrate fermentation by CitAB in Escherichia coli. Biosci. Biotechnol. Biochem. 72:3011-3014. [DOI] [PubMed] [Google Scholar]