

Abstract
Unresolved infection by gram-negative bacteria can result in the potentially lethal condition known as endotoxic shock, whereby uncontrolled inflammation can lead to multiple organ failure and death of the infected host. Previous results have demonstrated that animals deficient in class A scavenger receptor (SRA), a trafficking receptor for bacteria and bacterium-derived molecules, are more susceptible to endotoxic shock. This has been proposed to be a result of impaired SRA-dependent phagocytic clearance of bacteria resulting in stronger proinflammatory stimuli. In this report, we test the hypothesis that there is an obligate reciprocal relationship between SRA-mediated phagocytosis of bacteria and susceptibility to endotoxic shock. Here, we demonstrate that both SRA-dependent and -independent gram-negative bacterial strains elicit SRA-dependent increased cytokine production in vitro and in vivo and increased susceptibility to endotoxic shock in SRA-deficient mice. This is the first evidence showing that SRA-mediated clearance of LPS is functionally distinct from the role of SRA in bacterial phagocytosis and is a formal demonstration that the SRA-dependent cytokine responses and the resultant endotoxic shock are not coupled to SRA-mediated clearance of bacteria.
Class A scavenger receptor (SRA) is a member of the scavenger receptor family that has a broad specificity for polyanionic ligands. In the context of innate immunity to bacterial pathogens, SRA binds a variety of bacterial cell wall components and functions as a phagocytic receptor for a variety of gram-negative and gram-positive bacteria, although the dependence for SRA-mediated phagocytic recognition varies widely for different species of bacteria and even varies among different strains of the same bacterial species (13, 14, 18, 19). Additionally, SRA functions as an endocytic receptor for soluble bacterial cell wall ligands, including lipopolysaccharide (LPS) from gram-negative bacteria (6, 7, 14, 20). The ability of SRA to bind LPS via recognition of the lipid A molecule is characterized to be important for in vivo clearance of LPS and results in an attenuation of the inflammatory response to endotoxin (4, 7, 8, 16, 18). Thus, SRA functions as a trafficking receptor and facilitates both the endocytic clearance of bacterial ligands, like LPS, as well as phagocytic uptake of many bacteria (10, 14, 19). However, in the context of infection by gram-negative bacteria, the role of SRA-mediated phagocytic uptake of bacteria in modulating LPS-driven inflammatory responses has previously never been tested directly.
SRA deficiency in mice causes two major impairments in the innate immune response. SRA knockout (SRA−/−) mice are more susceptible to bacterial infections due to impaired phagocytosis of bacteria (10, 18, 19). Additionally, SRA−/− mice exhibit increased susceptibility to endotoxic shock (8, 16). These two observations have been proposed to be directly related, with the decreased phagocytic clearance of bacteria in the absence of SRA leading to exacerbated activation of TLR4 and resulting in hyperinflammation. Increased susceptibility to lethal endotoxic shock in SRA−/− mice can be recapitulated with purified LPS and is at least partially mediated by increased tumor necrosis factor alpha (TNF-α) production in these animals since TNF-α-neutralizing antibodies can abrogate this phenotype (8). However, the specific relationship between the roles of SRA as a phagocytic receptor for bacteria and an endocytic receptor for LPS is poorly understood, nor is it known whether there is an obligate functional relationship between the role of SRA as a phagocytic receptor for bacteria and the consequential host inflammatory response.
In this work, we use two approaches to demonstrate that the role of SRA in limiting inflammatory responses to LPS is a discrete process from phagocytosis of infecting bacteria. First, we show that SRA+/− cells, which are fully competent at phagocytic clearance of bacteria, display the same enhanced cytokine responses to gram-negative bacteria as those observed in SRA−/− cells. Second, using gram-negative bacteria that are not phagocytosed via SRA, we show that these “SRA-independent” bacteria still elicit increased inflammation in vitro and in vivo in SRA−/− cells and mice. Furthermore, SRA-independent bacteria still cause increased susceptibility to endotoxic shock in SRA−/− mice. These data provide a new understanding of how the role of SRA in endotoxin resistance is not dependent on its function in phagocytosis of bacteria. This report provides a new perspective on SRA function in the innate immune response and on how this multifunctional receptor modulates two distinct pathways during infection by gram-negative bacteria.
MATERIALS AND METHODS
Mice.
C57BL/6 wild-type (WT) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). SRA−/− mice (C57BL/6 background) were a generous gift of T. Kodama (Tokyo University) and M. W. Freeman (Massachusetts General Hospital) (11, 17). TLR4−/− and MyD88−/− mice were generated by Adachi et al. (1). Other mice were generated and screened as previously described (2).
Materials.
LPS was purchased from Sigma Aldrich (St. Louis, MO). DuoSet enzyme-linked immunosorbent assay (ELISA) kits for mouse interleukin-6 (IL-6) and TNF-α were purchased from R&D Systems (Minneapolis, MN). F4/80 antibody was purchased from eBiosciences (San Diego, CA).
Cells.
The bone marrow-derived dendritic cell (BMDC) culture protocol used is a modification of that of Inaba et al. (9) as previously described (2). Escherichia coli DH5α bacteria were transfected with the pTrcHis plasmid (Invitrogen, Carlsbad, CA) to confer ampicillin resistance. E. coli K-12 (chloramphenicol-resistant) bacteria were a gift from Deb Hogan (Dartmouth), and Pseudomonas aeruginosa PA14 bacteria were a gift from George O'Toole (Dartmouth).
Cytokine assays.
For in vitro studies, dendritic cells (DCs) were harvested and washed, and 5 × 105 cells were added to each well of a 48-well tissue culture plate. A total of 100 ng/ml of LPS (Sigma Aldrich) or 5 × 105 of the indicated bacteria were added to treated wells in 400 μl total volume of Hanks balanced salt solution (HBSS). DCs and bacteria were coincubated for 6 h at 37°C. Supernatants were collected for cytokine analysis. For in vivo studies, mice were injected intraperitoneally (i.p.) with either 1 × 109 CFU of the indicated E. coli strains or 5 × 107 CFU of P. aeruginosa bacteria. After 3 h, mice were sacrificed. Blood serum samples were collected by cardiac puncture. Samples were centrifuged at maximum speed on a tabletop microcentrifuge to collect serum samples. Peritoneal cytokine samples were collected by lavage using 3 ml of HBSS. Samples were centrifuged and supernatants collected. Serum and supernatant samples were analyzed by ELISA.
Gentamicin protection assays.
Phagocytosis of live E. coli or P. aeruginosa bacteria was performed as a modified version of previously described protocols (3, 5). Overnight cultures of E. coli or P. aeruginosa were washed twice in 1 ml serum-free HBSS and centrifuged at 12,000 rpm. The bacterial pellets were resuspended in 10 ml HBSS, and the bacterial concentration was determined by spectrophotometric absorbance at 600 nM. A total of 2 × 105 BMDCs from the various mouse genotypes described were incubated with bacteria at a multiplicity of infection of ∼10 for 45 min at 37°C. BMDCs were washed twice in serum-free HBSS and incubated in 100 μg/ml gentamicin for 20 min at 37°C. Cells were then washed and resuspended in 500 μl 0.1% Triton X-100 in phosphate-buffered saline. Ten microliters of lysates was plated onto either LB agar plates containing ampicillin (DH5α), LB agar plates containing chloramphenicol (K-12), or Pseudomonas isolation agar plates (PA14) and incubated overnight at 37°C. Subsequently, colonies were counted, and CFU were calculated based on the fraction of the total sample plated.
In vivo bacterial uptake assay.
A total of 5 × 106 live bacteria (of the strains indicated) were injected i.p. into the indicated genotypes of mice. Mice were sacrificed 1 h postinjection, and peritoneal lavage samples were collected. Peritoneal cells were pelleted, washed twice in serum-free HBSS, and resuspended in 750 μl of HBSS. Five hundred microliters of each suspension was incubated in the presence of gentamicin for 20 min at 37°C to kill noninternalized bacteria. Cells were washed twice in serum-free HBSS and resuspended in 500 μl 0.1% Triton X-100 in phosphate-buffered saline. Fifteen microliters of resuspended cells/bacteria was plated onto LB agar-ampicillin plates and incubated overnight at 37°C. Colonies were counted the following morning, and CFU were calculated based on the fraction of total sample plated.
Peritoneal cell analysis.
For all in vivo phagocytosis studies, analyses of total cell numbers and peritoneal macrophage numbers were done in parallel. Total cell numbers harvested from peritoneal lavage samples were quantified using trypan blue staining and manual cell counts of viable cells using a hemacytometer. Macrophage numbers were assessed by fluorescence-activated cell sorter analysis of F4/80 staining of lavage samples. Total macrophage numbers were calculated based on the percentage of the total population that stained as F4/80 positive.
Lethal challenge experiments.
Mice were lethally challenged by i.p. injection of either 400 μg of LPS, 1 × 109 live E. coli (DH5α or K-12), or 1 × 106 live PA14 bacteria. In these experiments, the results of which are shown in Fig. 6, mice were challenged with 2 × 109 live DH5α E. coli bacteria. Mice were monitored periodically, and upon loss of sternal recumbency for more than 10 s, they were deemed terminal and euthanized.
FIG. 6.

High-dose bacterial challenge-induced lethality is dependent on TLR4 and MyD88 expression. Survival plot of WT, SRA−/−TLR4−/−, and MyD88−/− mice challenged i.p. with 2 × 109 CFU live DH5α bacteria. At lethal doses for WT animals, SRA−/−TLR4−/− and MyD88−/− mice were completely resistant to endotoxic shock. n ≥ 4. Statistical significance (P ≤ 0.05) from WT values is indicated (*).
Data and statistical analyses.
Sample sizes for each experiment varied and are noted in the text. For all graphs, mean and standard deviation are shown. As indicated, one-way analysis of variance with Tukey's multiple comparison post-hoc test, unpaired Student's t test, or Kaplan-Meier analysis was performed to assess the statistical significance of the data. In the figures, statistical significance is represented by an asterisk and indicates P ≤ 0.05.
RESULTS
SRA+/− BMDCs produce elevated cytokines in vitro to DH5α bacteria.
We have previously identified that double heterozygous SRA+/− TLR4+/− BMDCs, but not the respective single heterozygotes, are significantly impaired for uptake of DH5α bacteria in vitro and in vivo (2). Since SRA deficiency is reported to cause increased proinflammatory cytokine production to purified LPS in vitro and in vivo (4, 8), we tested the cytokine responses in our single and double heterozygous BMDCs in response to live gram-negative bacteria. BMDCs were cocultured with DH5α bacteria for 6 h, and supernatants were analyzed for TNF-α and IL-6 production. Consistent with previous reports, SRA-deficient BMDCs produced increased levels of TNF-α and IL-6 in response to the bacteria (Fig. 1). This cytokine increase was absent in SRA−/− TLR4−/− cells, confirming that this phenotype is TLR4 dependent (Fig. 1). Double heterozygous SRA+/− TLR4+/− BMDCs showed a cytokine profile similar to that of SRA−/− cells (Fig. 1), which parallels the observation that both genotypes are impaired for phagocytic uptake of DH5α bacteria (2). Surprisingly, even though SRA+/− BMDCs exhibit WT levels of bacterial phagocytosis (2, 3), we found that these cells produce increased levels of cytokines in response to bacteria, similar to the enhanced cytokine levels seen in SRA+/− TLR4+/− and SRA−/− cells (Fig. 1). This observation is the first inflammatory cytokine defect to be characterized for SRA+/− cells and, importantly, is the first report of a functional dichotomy between SRA-mediated phagocytosis of bacteria and inflammatory cytokine responses.
FIG. 1.

SRA+/− BMDCs exhibit elevated cytokine production in response to E. coli DH5α. A total of 5 × 105 WT, SRA+/−, TLR4+/−, SRA+/− TLR4+/−, SRA−/−, or SRA−/− TLR4−/− BMDCs were cocultured with 5 × 105 DH5α bacteria for 6 hours. Cell supernatants were collected and analyzed by ELISA for TNF-α (A) or IL-6 (B) production. n ≥ 6 for all genotypes; means and standard deviations are shown. Statistical significance (P ≤ 0.05) for values higher than WT (*) and lower than WT (**) is indicated.
Bacteria independent for SRA-mediated phagocytosis elicit elevated cytokines in an SRA-dependent manner.
The dependence upon SRA for bacterial phagocytosis varies widely among bacterial species and strains (14). We exploited this phenomenon by utilizing gram-negative bacterial strains with various SRA phagocytic dependencies to test whether this dictates their ability to elicit cytokines in WT and SRA-deficient BMDCs. We observed the expected deficiency (3) for uptake of DH5α E. coli by SRA−/− cells (Fig. 2A). However, phagocytosis of both the K-12 E. coli strain and the PA14 P. aeruginosa strain was independent of SRA (Fig. 1A). While a minimal role for SRA in phagocytosis of K-12 E. coli has previously been reported (14), this is the first assessment of the contribution of SRA for any P. aeruginosa strain. WT, SRA+/−, and SRA−/− BMDCs were stimulated for 6 hours with the different bacterial strains or LPS and then analyzed for TNF-α (Fig. 2B) and IL-6 (Fig. 2C) production. Despite the varied dependence of the different bacterial strains for SRA-mediated phagocytosis, all strains stimulated increased cytokine production in SRA+/− and SRA−/− BMDCs, which concurred with the effect of LPS stimulation (Fig. 2B and C). These data demonstrate that enhanced cytokine responses in the absence of SRA are independent of SRA phagocytic function.
FIG. 2.

SRA-independent gram-negative bacteria elicit increased cytokines in vitro in the absence of SRA. (A) WT and SRA−/− BMDCs were assessed for relative phagocytosis of E. coli DH5α and K-12 and P. aeruginosa PA14 bacteria by gentamicin protection assay. Data are normalized as the percentage CFU of WT bacterial uptake. n ≥ 9 for all groups. (B and C) A total of 5 × 105 WT, SRA+/−, or SRA−/− BMDCs were cocultured with 5 × 105 CFU of the indicated bacteria, or LPS as a positive control, for 6 hours. Supernatants from these cultures were collected and analyzed by ELISA for TNF-α (B) and IL-6 (C). n ≥ 9; means and standard deviations are shown. Statistical significance (P ≤ 0.05) from WT values is indicated (*).
Differential SRA-dependent phagocytosis in vivo.
In order to test the in vivo relevance of our findings, we assessed phagocytic uptake of the different bacteria by peritoneal cells in mice. One hour following injection of live bacteria, peritoneal cells were harvested and treated in a gentamicin protection assay to assess relative bacterial uptake levels. SRA+/− mice exhibited no deficit for the in vivo uptake of DH5α E. coli, while SRA−/− mice displayed a significant defect in this regard (Fig. 3A) (2). In corroboration with our in vitro results, SRA−/− animals showed no impairment for the in vivo uptake of either K-12 (Fig. 3B) or PA14 (Fig. 3C). This is the first demonstration of the relative SRA dependence in vivo for the phagocytosis of different gram-negative bacterial strains and allowed us to subsequently test the relationship between phagocytosis and cytokine elicitation in vivo. To confirm that the different experimental conditions resulted in similar numbers of peritoneal phagocytes, the total cells harvested for each genotype and condition were quantified; no significant differences among the groups were observed (Fig. 3D, left panel). Additionally, the peritoneal cell isolates were stained for F4/80 to assess the quantity of macrophages present in each sample. No significant differences for macrophage numbers were observed among the experimental groups (Fig. 3D, right panel).
FIG. 3.

SRA dependence for phagocytosis varies among different bacterial strains in vivo. (A) WT, SRA+/−, or SRA−/− mice were injected i.p. with 5 × 106 live DH5α E. coli cells. Peritoneal phagocytes were subsequently harvested by lavage, and relative phagocytosis was assessed by gentamicin protection assay. SRA−/− mice were found to be significantly impaired in phagocytosis of DH5α (P ≤ 0.01) from both WT and SRA+/−mice. (B and C) No statistically significant difference was observed between WT and SRA−/− mice injected with 5 × 106 live K-12 E. coli (B) or PA14 P. aeruginosa (C) bacteria. (D) Total cell harvests from peritoneal lavage samples and total macrophage numbers were quantified. No statistical differences among the groups were found. For phagocytosis assays, data are normalized as the percentage CFU of WT bacterial uptake. n ≥ 8 for all groups. The mean and individual mouse values are represented on the graphs. Statistical significance (P ≤ 0.05) from WT values is indicated (**).
SRA−/− mice produce increased TNF-α in vivo to both SRA-dependent and -independent bacteria.
In order to assess the in vivo relationship between SRA-dependent bacterial phagocytosis and cytokine production, we tested in vivo cytokine production in response to both SRA-dependent and SRA-independent bacteria. WT or SRA−/− mice were injected i.p. with the indicated bacteria, and serum and peritoneal lavage samples were collected and analyzed for TNF-α production (Fig. 4). SRA−/− mice showed significant elevation in both serum and peritoneal TNF-α levels compared to WT mice in response to DH5α (Fig. 4A), K-12 (Fig. 4B), and PA14 (Fig. 4C) bacteria. Thus, in response to in vivo gram-negative bacterial infection, increased cytokine production by SRA-deficient animals is independent of SRA-mediated phagocytic clearance of bacteria.
FIG. 4.
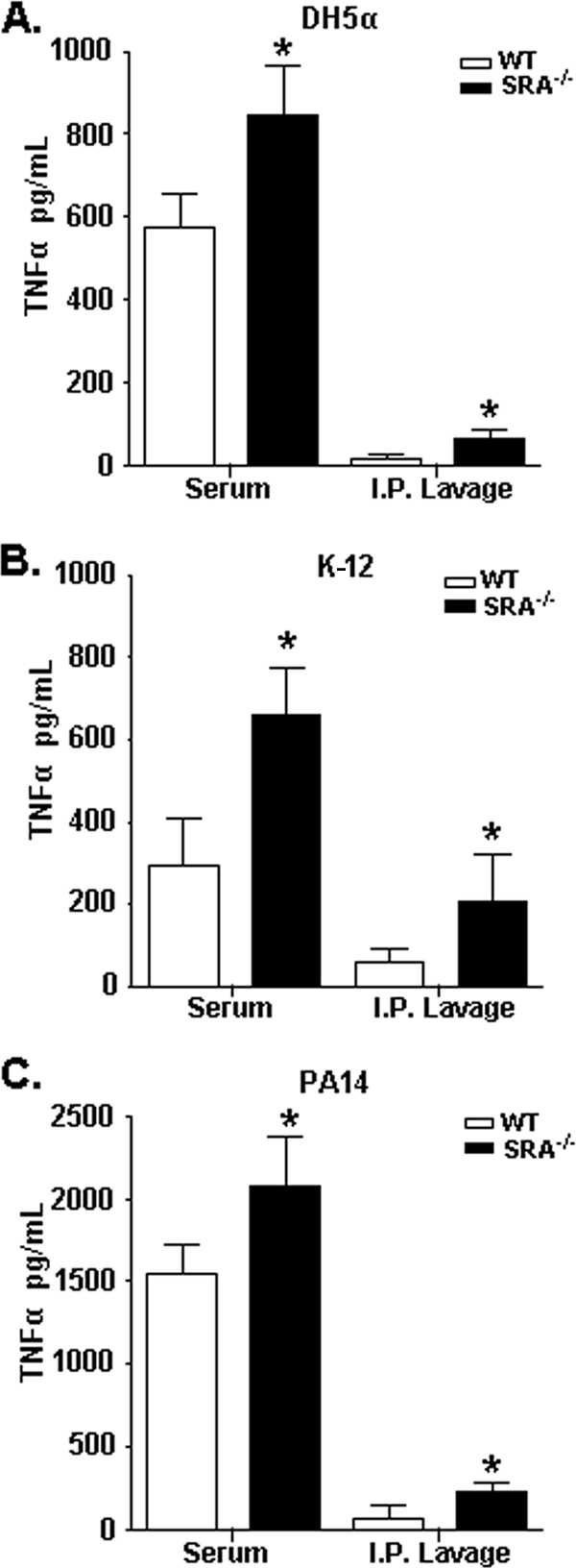
SRA−/− mice produce increased cytokines in vivo to both SRA-dependent and -independent bacteria. WT and SRA−/− mice were injected i.p. with 1 × 109 live DH5α bacteria (A), 1 × 109 live K-12 bacteria (B), or 5 × 107 live PA14 bacteria (C). After 3 hours, serum and peritoneal lavage samples were collected and analyzed by ELISA for TNF-α levels. n ≥ 4; means and standard deviations are shown. Statistical significance (P ≤ 0.05) from WT values is indicated (*).
SRA-independent bacteria elicit increased susceptibility to endotoxic shock in SRA-deficient mice.
To examine whether the increased susceptibility to endotoxic shock in SRA−/− mice is dependent upon the loss of SRA-mediated phagocytosis of bacteria, we challenged WT and SRA−/− mice with the various bacterial strains. As a positive control, SRA-deficient mice challenged i.p. with LPS showed the expected increase in lethality (Fig. 5A). Lethal challenge studies with DH5α (Fig. 5B), K-12 (Fig. 5C), and PA14 (Fig. 5D) bacteria showed that SRA−/− mice were more susceptible than WT mice to endotoxic shock for all the bacteria tested. These are the first reports of endotoxic shock studies of SRA−/− mice using either E. coli or P. aeruginosa bacterial challenge models. Importantly, these data formally demonstrate that SRA-mediated resistance to endotoxin in vivo is functionally independent from SRA-mediated phagocytosis of bacteria.
FIG. 5.

SRA−/− mice are more susceptible to lethal endotoxic shock induced by both SRA-dependent and -independent bacteria. (A) As a positive control, WT and SRA−/− mice were challenged i.p. with 400 μg LPS. (B to D) Survival plots of WT and SRA−/− mice challenged i.p. with 1 × 109 CFU live DH5α bacteria (B), 1 × 109 CFU live K-12 bacteria (C), or 1 × 106 CFU live PA14 bacteria (D). For all graphs, n ≥ 9. Under every condition, SRA−/− mice were found to be significantly more susceptible to the indicated challenge than WT mice. Statistical significance (P ≤ 0.05) from WT values is indicated (*).
Lethal endotoxic shock in SRA-deficient mice is dependent on TLR4 and MyD88.
To demonstrate that the gram-negative bacterial toxicity in SRA-deficient animals is mediated by increased proinflammatory signaling induced by LPS, we injected WT, SRA−/− TLR4−/− double knockout, and MyD88−/− knockout mice with a high-dose DH5α challenge. At a dose (1 × 109 CFU) that was fully lethal for WT animals, SRA−/− TLR4−/− and MyD88−/− knockout mice were completely protected from the toxic effects of this challenge (Fig. 6). These results provide formal in vivo evidence that lethality to gram-negative bacterial challenge is mediated by proinflammatory signaling of the TLR4/MyD88-dependent signaling axis. These data strongly support a model where, even in the context of infection by bacteria that are not recognized by SRA during phagocytic clearance, SRA still provides a critical role in modulation of the host inflammatory response and endotoxic shock.
DISCUSSION
During infection by gram-negative bacteria, the immune response is challenged with the task of eliminating the bacteria as well as inflammatory bacterial products, like LPS, that are shed into the extracellular space (12). When gram-negative bacterial infections remain unresolved in the infected host, these infections can result in lethal hyperinflammatory responses known as endotoxic shock (8). Resistance to endotoxic shock is critically regulated by the ability of the infected organism to eliminate endotoxin and thereby limit the proinflammatory stimulation elicited by this molecule. Because SRA is uniquely involved in both phagocytic bacterial clearance and modulation of inflammation via endocytic scavenging of soluble LPS, it is a key molecule in controlling the inflammatory response to endotoxin and infection by gram-negative bacteria. However, the relationship between bacterial phagocytosis and LPS clearance by SRA has not previously been defined.
In this report, we used two alternative and complementary approaches to demonstrate a functional dichotomy between SRA-mediated phagocytosis of bacteria and the associated LPS-driven inflammatory response. First, we utilized a genetic approach to show that SRA+/− cells exhibit increased cytokine responses to gram-negative bacteria in the absence of a phagocytic defect in these cells. These data are the first report of an inflammatory defect in cells heterozygous for SRA and underscore the importance of SRA-mediated endotoxin clearance in dampening the inflammatory response to gram-negative bacteria. Additionally, by using gram-negative bacteria that have no SRA dependence for their phagocytic uptake, we show that these bacteria still elicit hyperinflammation in vitro and in vivo and enhanced toxic shock in the context of SRA deficiency. This uncoupling of SRA-mediated phagocytic clearance of bacteria from SRA-dependent regulation of inflammatory signaling demonstrates a highly important immune function for SRA even in the context of bacterial infection by microbes that can evade phagocytic recognition by this receptor.
SRA has been well characterized for its ability to bind and clear soluble LPS via recognition of the lipid A moiety of endotoxin (7). Until recently, SRA-mediated recognition of LPS has been presumed to be a mechanism by which SRA recognizes and mediates phagocytic uptake of gram-negative bacteria. However, recently published work using Neisseria meningitidis has shown that SRA recognizes this particular gram-negative bacterium in an endotoxin-independent manner (13, 15). In conjunction with these studies, our data challenge the notion that LPS recognition by SRA is critically involved in the phagocytic function of SRA for gram-negative bacteria. Our findings that SRA can regulate proinflammatory cytokine production irrespective of its role in bacterial phagocytosis are consistent with a model in which LPS and the bacterial phagocytic ligands may possibly bind to distinct sites of the SRA receptor. From a teleological perspective, this model provides an intriguing mechanistic interpretation of how endocytic and phagocytic functions of SRA can function simultaneously without compromising either phagocytic clearance of infecting bacteria or the regulation of the inflammatory response to infection.
Heretofore, increased susceptibility to endotoxic shock and infection in SRA−/− mice has been described only for gram-negative bacteria that are phagocytosed in an SRA-dependent manner (8, 10, 16, 18). Under endotoxic shock conditions, SRA limits TLR4-driven proinflammatory cytokine responses by clearing LPS from the extracellular environment (8). This phenomenon was previously thought to be intrinsically related to decreased phagocytic clearance of bacteria in the absence of SRA. We have utilized in vitro and in vivo models to demonstrate that SRA-mediated endocytosis of LPS is independent of phagocytic uptake of bacteria during gram-negative bacterial infection. These data provide a novel understanding of the role of SRA in limiting the inflammatory responses to endotoxin. Furthermore, these data illustrate for the first time that SRA utilizes two distinct and independent trafficking pathways to mediate innate immune responses during infection by gram-negative bacteria.
Acknowledgments
This research was supported by National Institutes of Health (NIH) grants COBRE P20RR016437 and R01 AI067405, a Dartmouth DCCTS Pilot Grant, a Cystic Fibrosis Foundation-RDP grant (B.B.), and NIH Training Grant T32 AI07363 (E.A.).
We thank Kevin Hart, George O'Toole, and Deb Hogan (Dartmouth) for their help and advice.
Editor: A. Camilli
Footnotes
Published ahead of print on 10 August 2009.
REFERENCES
- 1.Adachi, O., T. Kawai, K. Takeda, M. Matsumoto, H. Tsutsui, M. Sakagami, K. Nakanishi, and S. Akira. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143-150. [DOI] [PubMed] [Google Scholar]
- 2.Amiel, E., A. Alonso, S. Uematsu, S. Akira, M. E. Poynter, and B. Berwin. 2009. Pivotal advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J. Leukoc. Biol. 85:595-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amiel, E., S. Nicholson-Dykstra, J. J. Walters, H. Higgs, and B. Berwin. 2007. Scavenger receptor-A functions in phagocytosis of E. coli by bone marrow dendritic cells. Exp. Cell Res. 313:1438-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker, M., A. Cotena, S. Gordon, and N. Platt. 2006. Expression of the class A macrophage scavenger receptor on specific subpopulations of murine dendritic cells limits their endotoxin response. Eur. J. Immunol. 36:950-960. [DOI] [PubMed] [Google Scholar]
- 5.Duncan, M. J., G. Li, J. S. Shin, J. L. Carson, and S. N. Abraham. 2004. Bacterial penetration of bladder epithelium through lipid rafts. J. Biol. Chem. 279:18944-18951. [DOI] [PubMed] [Google Scholar]
- 6.Dunne, D. W., D. Resnick, J. Greenberg, M. Krieger, and K. A. Joiner. 1994. The type I macrophage scavenger receptor binds to gram-positive bacteria and recognizes lipoteichoic acid. Proc. Natl. Acad. Sci. USA 91:1863-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hampton, R. Y., D. T. Golenbock, M. Penman, M. Krieger, and C. R. Raetz. 1991. Recognition and plasma clearance of endotoxin by scavenger receptors. Nature 352:342-344. [DOI] [PubMed] [Google Scholar]
- 8.Haworth, R., N. Platt, S. Keshav, D. Hughes, E. Darley, H. Suzuki, Y. Kurihara, T. Kodama, and S. Gordon. 1997. The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J. Exp. Med. 186:1431-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inaba, K., M. Inaba, M. Deguchi, K. Hagi, R. Yasumizu, S. Ikehara, S. Muramatsu, and R. M. Steinman. 1993. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. USA 90:3038-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishiguro, T., M. Naito, T. Yamamoto, G. Hasegawa, F. Gejyo, M. Mitsuyama, H. Suzuki, and T. Kodama. 2001. Role of macrophage scavenger receptors in response to Listeria monocytogenes infection in mice. Am. J. Pathol. 158:179-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunjathoor, V. V., M. Febbraio, E. A. Podrez, K. J. Moore, L. Andersson, S. Koehn, J. S. Rhee, R. Silverstein, H. F. Hoff, and M. W. Freeman. 2002. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277:49982-49988. [DOI] [PubMed] [Google Scholar]
- 12.Mattsby-Baltzer, I., K. Lindgren, B. Lindholm, and L. Edebo. 1991. Endotoxin shedding by enterobacteria: free and cell-bound endotoxin differ in Limulus activity. Infect. Immun. 59:689-695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peiser, L., M. P. De Winther, K. Makepeace, M. Hollinshead, P. Coull, J. Plested, T. Kodama, E. R. Moxon, and S. Gordon. 2002. The class A macrophage scavenger receptor is a major pattern recognition receptor for Neisseria meningitidis which is independent of lipopolysaccharide and not required for secretory responses. Infect. Immun. 70:5346-5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peiser, L., P. J. Gough, T. Kodama, and S. Gordon. 2000. Macrophage class A scavenger receptor-mediated phagocytosis of Escherichia coli: role of cell heterogeneity, microbial strain, and culture conditions in vitro. Infect. Immun. 68:1953-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peiser, L., K. Makepeace, A. Pluddemann, S. Savino, J. C. Wright, M. Pizza, R. Rappuoli, E. R. Moxon, and S. Gordon. 2006. Identification of Neisseria meningitidis nonlipopolysaccharide ligands for class A macrophage scavenger receptor by using a novel assay. Infect. Immun. 74:5191-5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plüddemann, A., J. C. Hoe, K. Makepeace, E. R. Moxon, and S. Gordon. 2009. The macrophage scavenger receptor a is host-protective in experimental meningococcal septicaemia. PLoS Pathog. 5:e1000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki, H., Y. Kurihara, M. Takeya, N. Kamada, M. Kataoka, K. Jishage, H. Sakaguchi, J. K. Kruijt, T. Higashi, T. Suzuki, T. J. van Berkel, S. Horiuchi, K. Takahashi, Y. Yazaki, and T. Kodama. 1997. The multiple roles of macrophage scavenger receptors (MSR) in vivo: resistance to atherosclerosis and susceptibility to infection in MSR knockout mice. J. Atheroscler. Thromb. 4:1-11. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki, H., Y. Kurihara, M. Takeya, N. Kamada, M. Kataoka, K. Jishage, O. Ueda, H. Sakaguchi, T. Higashi, T. Suzuki, Y. Takashima, Y. Kawabe, O. Cynshi, Y. Wada, M. Honda, H. Kurihara, H. Aburatani, T. Doi, A. Matsumoto, S. Azuma, T. Noda, Y. Toyoda, H. Itakura, Y. Yazaki, T. Kodama, et al. 1997. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 386:292-296. [DOI] [PubMed] [Google Scholar]
- 19.Thomas, C. A., Y. Li, T. Kodama, H. Suzuki, S. C. Silverstein, and J. El Khoury. 2000. Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J. Exp. Med. 191:147-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Lenten, B. J., A. M. Fogelman, J. Seager, E. Ribi, M. E. Haberland, and P. A. Edwards. 1985. Bacterial endotoxin selectively prevents the expression of scavenger-receptor activity on human monocyte-macrophages. J. Immunol. 134:3718-3721. [PubMed] [Google Scholar]