

Abstract
In most human immunodeficiency virus type 1 (HIV-1)-infected individuals who achieve viral loads of <50 copies/ml during highly active antiretroviral therapy (HAART), low levels of plasma virus remain detectable for years by ultrasensitive methods. The relative contributions of ongoing virus replication and virus production from HIV-1 reservoirs to persistent low-level viremia during HAART remain controversial. HIV-1 vaccination of HAART-treated individuals provides a model for examining low-level viremia, as immunizations may facilitate virus replication and sequence evolution. In a phase 1 trial of modified vaccinia virus Ankara/fowlpox virus-based HIV-1 vaccines in 20 HIV-infected young adults receiving HAART, we assessed the prevalence of low-level viremia and sequence evolution, using ultrasensitive viral load (<6.5 copies/ml) and genotyping (five-copy sensitivity) assays. Viral evolution, consisting of new drug resistance mutations and novel amino acid changes within a relevant HLA-restricted allele (e.g., methionine, isoleucine, glutamine, or arginine for leucine at position 205 of RT), was found in 1 and 3 of 20 subjects, respectively. Sequence evolution was significantly correlated with levels of viremia of between 6.5 and <50 copies/ml (P = 0.03) and was more likely to occur within epitopes presented by relevant HLA alleles (P < 0.001). These findings suggest that ongoing virus replication contributes to low-level viremia in patients on HAART and that this ongoing replication is subject to CD8+ T-cell selective pressures.
Highly active antiretroviral therapy (HAART) inhibits human immunodeficiency virus type 1 (HIV-1) replication, decreasing plasma viremia below the detection limit (<50 copies/ml) of clinically used viral load assays (32). Most patients receiving standard HAART, however, have low levels of viremia (median, 3.1 copies/ml) detectable by more sensitive assays (11, 17, 22, 28, 31, 34), the source of which remains unclear and controversial. Low-level viremia may arise from viral expression from cellular reservoirs established before HAART (2, 21-23, 25, 29, 31, 34, 46) or from low levels of complete cycles of virus replication (7, 46). Evidence supporting the former includes the stable persistence of low-level viremia during long-term HAART (2, 11, 23, 25, 28, 31, 34), intermingling of HIV-1 sequences from free plasma virus with persistent replication-competent viral sequences in resting memory CD4+ T cells or proviral DNA in peripheral blood mononuclear cells (PBMC) (2, 23, 34, 46), and a lack of decay in low-level viremia with treatment intensification of standard three-drug HAART regimens (10, 24). The contribution of ongoing complete cycles of virus replication during long-term suppressive HAART is supported by findings of sequence evolution with new drug-resistant variants in a subset of patients during seemingly effective HAART (7, 46), decreases in plasma viremia with treatment intensification (22), and transient increases in episomal cDNA following treatment intensification with an integrase inhibitor (6). Quantitatively, the magnitude of residual viremia is directly correlated with pretherapy, steady-state plasma viral load and proviral DNA levels, in support of virus expression from chronically infected cells (28, 31). Low-level viremia and even transient increases in viral loads to above 50 copies/ml (known as “blips”) are often not directly associated with evolution of plasma virus (2, 25, 30, 33, 34) or with decreased effectiveness of the HAART regimen (21, 26, 28, 31) but may replenish latent reservoirs and therefore preclude viral clearance with HAART (38).
Studies aimed at understanding the source(s) of residual viremia in patients on stable HAART have examined the effects of intensification of durable HAART regimens on viremia below 50 copies/ml (10, 22, 24) or on cell-associated forms of viral DNA (6). In an earlier study, the addition of the reverse transcriptase (RT) inhibitor abacavir to a two-drug durable HAART regimen resulted in a lowering of residual viremia (22). More recently, a study of treatment intensification of durable three-drug HAART regimens with protease inhibitors or nonnucleoside RT inhibitors (NNRTI) did not reduce residual viremia (10). However, two studies of treatment intensification of three-drug HAART regimens with the integrase inhibitor raltegravir produced different findings. In one study, 30 days of raltegravir produced no detectable change in residual viremia, assessed using a single-copy viral load assay (24). In a second study, however, intensification with raltegravir was associated with a transient increase in episomal viral cDNA at 2 weeks, suggesting de novo infection of and reverse transcription in susceptible target cells, although no change in total DNA burden was observed (6). Thus, the question remains of whether ongoing low-level viremia during HAART is dominated by low-level virus release from latently infected cellular reservoirs, complete cycles of virus replication de novo in susceptible target cells, or both.
Therapeutic HIV-1 vaccines are being evaluated to boost HIV-1-specific immunity that wanes during HAART (12). If successful, such immune boosting carries the promise of eventual treatment discontinuation, although that goal was tempered recently by findings of increased morbidity with treatment discontinuation (1, 8, 14, 15). To our knowledge, there are no studies to date on the effects of HIV-1 vaccines on the frequency and evolution of low-level viremia in HAART-treated patients.
We hypothesized that immune activation through HIV-1 vaccines may amplify low-level viremia and facilitate sequence evolution through enhanced virus expression from latently infected CD4+ T cells (13, 19, 35) and by increasing target cell availability for virus replication (19). We therefore examined the prevalence and sequence evolution of low-level viremia during a clinical trial of recombinant poxvirus vaccinations (modified vaccinia virus Ankara/fowlpox virus [MVA/FPV]) in infected young adults on durable suppressive HAART to investigate the dynamics of low-level viremia.
MATERIALS AND METHODS
Study cohort.
The study cohort was described previously in a report detailing the safety and immunogenicity of the vaccine products (18). Twenty HIV-1-infected young adults from 19 to 24 years of age (median age, 23 years) who were on HAART received up to four vaccinations while enrolled in a phase 1 clinical trial of MVA- and FPV-based HIV-1 vaccines containing gag, env, the RT gene, nef, tat, and rev (Pediatric AIDS Clinical Trials Group P1059) (18). The MVA vaccinations were administered at weeks 0 and 4, and the FPV vaccine was administered at weeks 8 and 24. Plasma samples were collected for ultrasensitive HIV-1 load testing and genotyping at nine time points during the trial, including screening, entry, and weeks 2, 4, 6, 24, 26, 40, and 72. Ultrasensitive HIV-1 load and genotyping assays were part of the parent clinical trial and were included in the consent form approved by the institutional review boards at all sites enrolling subjects.
Sample preparation.
Plasmas were separated from PBMC by centrifugation and were stored at −70°C until use. PBMC were isolated using Ficoll-Hypaque centrifugation, fractionated to enrich for resting CD4+ T cells, and cultured at limiting dilutions, using previously published methods (42). Individual HLA serotyping, gamma interferon (IFN-γ)-specific enzyme-linked immunospot (ELISPOT) assays, and CD4 lymphoproliferative response assays with cryopreserved PBMC were performed as previously described (18).
Quantitative assessment of low-level viremia.
The viral load was quantified from 1 ml of plasma by use of a modified ultrasensitive RNA assay in which the limit of detection was further validated to detect viremia at 6.5 copies/ml instead of the previously reported limit of 12.5 copies/ml (36).
Genotyping of HIV-1 RT during low-level viremia.
HIV genotyping of RT from low-level viremia variants was performed using our previously published methods with a sensitivity of 5 copies/ml (25, 34). Briefly, viral RNA was extracted from virions pelleted at 23,500 × g for 2 h at 4°C from four 1-ml aliquots of thawed plasma, using a QIAamp viral RNA isolation kit (Qiagen, Valencia, CA). Following DNase treatment to ensure removal of cell-associated DNA (Invitrogen, Carlsbad, CA), eight independent one-step RT-PCRs were set up for the DNase-treated RNA, followed by a nested PCR with previously published primers (49). The PCR products were purified using a QIAquick PCR purification kit (Qiagen, Valencia, CA) and directly sequenced bidirectionally using gene-specific primers.
Analysis of HIV-1 RT sequences.
Sequences were aligned using Bioedit and cleaned using a previously published method (CleanCollapse [http://sray.med.som.jhmi.edu/SCRoftware/CleanCollapse]) that removes sporadic changes likely to represent PCR-induced errors (25). Evolution has been reported to be overestimated systematically without such adjustments (44).
Nonsynonymous substitutions at known drug resistance sites (Stanford HIV Drug Resistance Database [http://hivdb.stanford.edu]) were preserved, as were the novel changes detected at amino acid 205 of HIV-1 RT in the sequences amplified from episodes of low-level viremia after vaccination of four subjects. Using this approach, only four sporadic nonsynonymous substitutions were removed from the total data set of 209 sequences spanning amino acids 40 to 219 of HIV-1 RT. These sporadic changes, by definition, occurred only once in the data set; therefore, our estimates are lower bounds for evolution occurring in this cohort.
Phylogenetic analysis parameters were estimated using ModelTest, version 3.7 (37), and these parameters were used as input to PhyML, version 3.0, to generate maximum likelihood trees, using the SPR search option and starting from a BioNJ tree (20). For bootstrap analysis, maximum likelihood trees were inferred from 1,000 permutations of the original data set. Genetic diversity in each plasma sampling from each subject was calculated by the Kimura 2 parameter distance model, using MEGA4 (45). Nonsynonymous amino acid changes detected postvaccination within HIV-1 RT were also analyzed with respect to relevant HLA-restricted CD8+ T-cell epitopes in the Los Alamos Database (http://www.hiv.lanl.gov) for subjects who maintained suppression during the trial. The two substitutions observed at position 205 of HIV-1 RT (L205M/Q) at week 40 for the one subject who discontinued HAART at week 24 were also included in this analysis, since these mutations have not been reported previously for subtype B HIV-1 infection.
Analysis of CD4+ T cells infected with replication-competent HIV-1.
The frequencies of CD4+ T cells carrying replication-competent virus and sequences of HIV-1 RT from replication-competent isolates were assessed using previously published methods (33). Representative HIV-1 RT sequences from replication-competent clones recovered at the prevaccination visits were used to confirm the patient specificity of the HIV-1 RT sequences amplified during low-level viremia.
Immunogenicity studies.
HLA serotypes, HIV-1-specific immune responses, and immune activation analyses were assessed as part of the parent trial, as previously described (18). These measurements were used to examine potential immunological differences among subjects experiencing sequence evolution during the trial compared to those who did not.
Statistical analysis.
Most analyses consist of descriptive summaries of the various measurements and changes from baseline in those values. The summaries are provided as medians and interquartile ranges (IQR), unless otherwise specified. The changes from prevaccination to subsequent weeks were tested using the Wilcoxon sign rank test. The proportion of subjects with low-level viremia above 6.5 copies/ml at each study visit was computed and compared to baseline by the McNemar test.
The study did not have a control group that did not receive vaccines. However, subjects were divided into several groups based on criteria specific to each data analysis. Exploratory data analyses were performed to compare groups with respect to raw data or changes from baseline. No adjustments for multiple comparisons were made. Differences in baseline levels of immune activation (percent CD8+ CD38+ HLA-DR+ T cells), frequencies and magnitudes of low-level viremia, and frequencies of resting CD4+ T cells with replication-competent virus (infectious units per million) for subjects who had viremia genotypes versus those who did not were assessed using the Wilcoxon two-sample test. Rates of viremic episodes of between 6.5 and 50 copies/ml postvaccination for subjects with and without sequence evolution in RT were compared using the Wilcoxon two-sample test. Similarly, the Wilcoxon two-sample test was used to compare the magnitudes of low-level viremia for subjects who were viremic above 6.5 copies/ml and of HIV-1 specific CD8+ T-cell responses to Pol in the subjects experiencing HIV-1 RT sequence evolution to the same measurements for those with no evolution. The relationship between HIV-1 RT mutation events and HLA serotypes was assessed using a logistic model for the probability that mutations occupy epitopes recognized by HLA type (27). For each individual, a measure of recognition capacity was computed as the number of (nonredundant) residues occupied by epitopes associated with the individual's HLA type, gathered from the Los Alamos HIV Immunology Database (http://www.hiv.lanl.gov).
Nucleotide sequence accession numbers.
The nucleotide sequences determined in this study have been submitted to GenBank (accession numbers GQ424058 to GQ424103 and GQ428780 to GQ428988).
RESULTS
Study population.
The demographics, antiretroviral treatment histories, and virologic data for the patients are summarized in Table 1. The subjects received durable successful HAART for a median of 3.3 years (range, 0.6 to 6.4 years) before receipt of HIV-1 vaccines, and 11 of the 20 (55%) subjects controlled viremia on their first HAART regimen. As previously reported, 75% of the 20 participants acquired HIV-1 infection through high-risk behaviors, and the remainder acquired infection perinatally (18). Three participants (patients 5, 11, and 15) developed rebound viremia during the trial; patients 5 and 15 rebounded due to discontinuation of their HAART regimens by the week 24 visit, and the third participant (patient 11) rebounded at week 60 while on HAART (18).
TABLE 1.
Patient demographics, HAART regimens, and extent of sampling of low-level viremia genotypesa
| Group and subjectb | Age at study entry (yrs)/sex | Pre-HAART antiretroviral drug exposure | HAART regimen at study entry (yr) | No. of vaccines received | No. of visits with amplifiable genotypes/no. of time points tested (%) | Prevaccination genotype(s) |
|---|---|---|---|---|---|---|
| Subjects with amplifiable low-level viremia genotypes experiencing sequence evolution following vaccination | ||||||
| 7 | 20/F | None | AZT, 3TC, NFV (0.9) | 4 | 7/8 (88) | Wild type |
| 8 | 24/F | AZTc | AZT, 3TC, NVP (5.7) | 2 | 7/8 (88) | Wild type |
| 11†* | 23/M | AZT, 3TC, DDI, ABC, IDV, EFV | D4T, TDF, LPV/r (2.3) | 4 | 8/8 (100) | Wild type, D67G K70R V018I K219E mutant |
| 17† | 23/F | AZT, DDI, 3TC, D4T, DDC, NVP, ABC, RTV, AMP, LPV/r | TDF, ATZ/r, EFV (1.7) | 3 | 5/9 (56) | M41L, D67N, T69D, M184V, T215Y/C |
| Subjects with amplifiable low-level viremia genotypes with no sequence evolution following vaccination | ||||||
| 4 | 23/F | AZT, 3TC | AZT, 3TC, NVP (4.3) | 4 | 5/9 (56) | Y188C |
| 5*# | 23/M | None | AZT, 3TC, EFV (2.6) | 4 | 1/5 (20) | NA |
| 9 | 24/M | None | AZT, 3TC, EFV (6.4) | 4 | 4/9 (44) | Wild type |
| 12 | 20/M | AZT, 3TC, ABC, LPV/r | ABC, 3TC, ATV (0.6) | 3 | 6/8 (75) | Wild type |
| 13† | 22/F | AZT, DDI, D4T, ADF, 3TC, IDV, APV, NFV, SQV, DDC, hydroxyurea | AZT, TDF, FTC, ATZ/r (1.1) | 4 | 4/8 (50) | V108I, M184V, T215S/D |
| 15*# | 24/M | None | DDI, FTC, EFV (3.8) | 3 | 1/5 (20) | Wild type |
| 16 | 21/F | None | DDI, FTC, EFV (4.2) | 3 | 3/7 (43) | Wild type |
| 19† | 19/F | None | DDI, EFV, NFV (4.5) | 3 | 6/8 (75) | Wild type and M41L, M184V, and T215F mutants |
| Subjects without amplifiable low-viremia genotypes | ||||||
| 1 | 22/M | DDI, 3TC, TDF, EFV | TDF, FTC, LPV/r (0.9) | 1 | 1/6 (17)d | NA |
| 2 | 24/F | None | AZT, 3TC, NFV (5.1) | 4 | 0/9 (0) | NA |
| 3 | 24/M | None | AZT, 3TC, EFV (4.7) | 4 | 0/9 (0) | NA |
| 6 | 23/M | None | AZT, 3TC, EFV (5.3) | 4 | 0/9 (0) | NA |
| 10 | 21/M | None | DDI/FTC/EFV (4.3) | 4 | 0/9 (0) | NA |
| 14 | 20/F | AZT, 3TC, LPV/r | TDF, FTC, EFV (2.8) | 4 | 0/7 (0) | NA |
| 18 | 23/M | AZT, 3TC, NFV | TDF, FTC, ATV/r(1.4) | 3 | 0/3 (0) | NA |
| 20 | 22/M | None | DDI, FTC, EFV (4.3) | 3 | 0/9 (0) | NA |
F, female; M, male; AZT, zidovudine; 3TC, lamivudine; NFV, nelfinavir; EFV, efavirenz; DDI, didanosine; FTC, emtracitabine; TDF, tenofovir; LPV/r, lopinavir boosted with ritonavir; ATV, atazanavir; NVP, nevirapine; ABC, abacavir; IDV, indinavir; D4T, stavudine; ADF, adefovir; APV, amprenavir; SQV, saquinavir; DDC, zalcitibine; ATZ/r, atazanavir boosted with ritonavir; RTV, ritonavir; AMP, amprenavir; NA, not applicable.
†, perinatally infected subject; *, patient developed rebound viremia while on HAART; #, self-discontinued HAART.
Received 30 days during pregnancy for prevention of mother-to-child transmission.
Only the plasma sample at the week 72 visit yielded sequences; all three variants comingled with replication-competent viral clones cultured at the prevaccine visits.
Prevalence of low-level viremia before and after recombinant HIV-1 poxvirus vaccination.
We assessed the prevalence of low-level viremia pre- and post-HIV-1 vaccination in young adults with durable control of virus replication via HAART. Viral loads measured in the three subjects experiencing rebound viremia (subjects 5, 11, and 15) were excluded from analysis at the time of rebound and at subsequent time points. Nineteen of the 20 subjects had plasma samples analyzed at the two visits prior to HIV-1 vaccination. Low-level viremia at levels greater than 6.5 copies/ml was detected in 21% (4 of 19 samples) and 25% (5 of 20 samples) of the samples at the screening and entry visits, respectively. The nine episodes of detectable low-level viremia prevaccination were in seven participants, with two of the seven subjects having viremia detectable at both prevaccine visits (Table 2).
TABLE 2.
Plasma viral levels and recovery of amplifiable genotypes before and after recombinant HIV-1 poxvirus vaccinations
| Group and subject | Level of viremia (copies/ml)/genotyping success at indicated wk of studya
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Screening | 0 | 2 | 4 | 6 | 24 | 26 | 40 | 72 | |
| Subjects with amplifiable low-level viremia genotypes experiencing sequence evolution following vaccination | |||||||||
| 7 | 40/+ | 45/+ | ND/ND | 10/+ | 50/+ | 17/+ | 30/− | <6.5/+ | <6.5/+ |
| 8 | <6.5/− | <6.5/+ | <6.5/+ | <6.5/+ | 12/+ | ND/+ | 25/+ | <6.5/+ | 23/+ 172e |
| 11 | 9/+ | 7/+ | <6.5/+ | <6.5/+ | <6.5/+ | 18/+ | 13/+ | 16/+ | 87,121/+ |
| 17 | <6.5/+ | <6.5/+ | <6.5/− | 8/+ | <6.5/− | <6.5/− | <6.5/+ | 8/+ | <6.5/− |
| Subjects with amplifiable low-level viremia genotypes without sequence evolution following vaccination | |||||||||
| 4 | <6.5/+ | <6.5/+ | <6.5/+ | <6.5/+ | 10/+ | <6.5/− | <6.5/− | <6.5/− | <6.5/− |
| 5b | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/+ | 4,719e/+ | 2,756e/+ | 4,297e/+ | 2,850e/+ |
| 9 | <6.5/− | <6.5/+ | <6.5/+ | <6.5/− | <6.5/− | 19/+ | <6.5/+ | <6.5/− | <6.5/− |
| 12 | <6.5/+ | <6.5/+ | <6.5/− | <6.5/− | <6.5/+ | <6.5/+ | <6.5/ND | <6.5/+ | <6.5/+ |
| 13 | <6.5/+ | <6.5/− | <6.5/+ | <6.5/+ | <6.5/+ | <6.5/− | <6.5/ND | 13/− | <6.5/− |
| 15b | <6.5/− | <6.5/+ | <6.5/− | <6.5/− | <6.5/− | 12,682e/+ | ND/ND | 3,862e/+ | 12,207e/+ |
| 16 | <6.5/+ | 11/− | <6.5/+ | <6.5/ND | <6.5/− | <6.5/− | <6.5/ND | 36/+ | 62/− |
| 19 | <6.5/+ | 7/+ | 17/+ | <6.5/+ | <6.5/− | 37/+ | ND/ND | <6.5/+ | <6.5/− |
| Subjects without amplifiable low-level viremia genotypes | |||||||||
| 1 | 14/− | <6.5/ND | ND/ND | 22/− | <6.5 (148)e/− | 7/− | ND/ND | <6.5/− | 7/+ |
| 2 | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | 436 (497)e/− | 9/− | 19/− | <6.5/− |
| 3 | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− |
| 6 | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | ND/− |
| 10 | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | 14/− | <6.5/− |
| 14 | ND/ND | <6.5/ND | <6.5/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | 24/− | <6.5/− |
| 18 | 25/ND | <6.5/ND | ND/ND | ND/ND | 10/− | 25/− | ND/ND | 15/ND | 15/− |
| 20 | <6.5/− | 15/− | <6.5/− | <6.5/− | <6.5/− | <6.5/− | ND/− | <6.5/− | <6.5/− |
| Total proportion with genotyping successc | 8/18 | 9/17 | 7/17 | 7/18 | 7/20 | 6/18 | 4/12 | 7/17 | 4/17 |
| Total proportion with low-level viremiad | 4/19 | 5/20 | 2/17 | 3/19 | 5/20 | 7/17 | 4/14 | 8/18 | 5/17 |
ND, not done.
Self-discontinued HAART.
Number of subjects with low-level viremia genotypes detected/number of subjects assessed.
Total number of subjects with low-level viremia between 6.5 and 50 copies/ml/number of subjects assessed.
Viral load measurement was done by standard clinical viral load assay.
Following vaccination, the prevalence of low-level viremia ranged from 12% (2/17 samples) at week 2 postvaccination to as high as 44% (8/18 samples) at the week 40 visit but did not differ significantly from that at study entry for each time point tested (P ≥ 0.51) (Table 2). The median viral load at study entry for the five subjects with detectable viremia of >6.5 copies/ml was 11 copies/ml (IQR, 7 to 15 copies/ml), and this value was 19 copies/ml for the time points with the highest median low-level viremia postvaccination (weeks 24 [IQR, 17 to 37 copies/ml], 26 [IQR, 11 to 27.5 copies/ml], and 72 [IQR, 11 to 42.5 copies/ml]).
Extent of recovery and sequence evolution of low-level viremia genotypes before and after recombinant HIV-1 poxvirus vaccination.
Longitudinal analyses of low-level viremia genotypes were carried out to examine sequence evolution in plasma virus. Genotyping was performed on all available plasma samples (n = 158 [88%]) from the 180 scheduled study visits. Eighty-nine percent of the 158 samples genotyped were from the 18 subjects who remained on HAART during the trial, 97% (137/141 samples) of whom had viral loads of <50 copies/ml at the time of genotyping. Low-level viremia genotypes were recovered from a median of 56% (range, 20 to 100%) of the time points analyzed, for 60% (12/20 subjects) of the study participants (Tables 1 and 2). For 8 of the 20 study subjects, 98% of the 61 plasma samples analyzed were negative. For one subject (subject 1), the plasma sample obtained at the last study visit at week 72 yielded genotypes (Tables 1 and 2). We were unable to identify any factors that may have contributed to the selective sequence amplification for 12 study subjects. The 8 subjects whose plasma samples were negative did not differ significantly at baseline from the 12 subjects with amplifiable genotypes with respect to median viral loads below 50 copies/ml, baseline frequencies of CD4+ T cells harboring replication-competent HIV-1, levels of immune activation, or sequence variation at the primer binding site. Prior to vaccination, 3 of 15 (20%) plasma samples tested for the eight subjects with no amplifiable genotypes were positive for viral loads of >6.5 copies/ml, compared to 6 of 24 (25%) samples from the 12 subjects with amplifiable genotypes. The median viral load at study entry was <6.5 copies/ml for both groups (IQR, <6.5 to <6.5 copies/ml and <6.5 to 7.0 copies/ml, respectively [P = 0.39]). The median frequencies of latently infected CD4+ T cells were 0.2 (IQR, 0.1 to 0.2) and 0.3 (IQR, 0.1 to 1.1), respectively (P = 0.33), and the median percentages of CD8+ CD38+ HLA-DR+ T cells were 18% (IQR, 14 to 25%) and 29% (IQR, 18 to 33%) (P = 0.19) for the subjects with and without amplifiable low-level viremia genotypes, respectively. Analysis of HIV-1 RT sequences derived from replication-competent viral clones cultured at the prevaccine time points showed that only one subject (subject 2) (Table 2) had a substitution at the 3′ end that may have affected binding of the primers used for reverse transcription (data not shown). Together, these data argue against differences in baseline levels of cell-associated, replication-competent infection, immune activation, or residual viremia that may influence the level of viral expression from persistently infected cells or the potency of inhibition below 50 copies/ml. Alternatively, this selective amplification may represent the stochastic nature of amplification near the limit of detection.
Eleven of the 12 study participants with amplifiable genotypes at more than one time point postvaccination also had genotypes recovered prevaccination. Phylogenetic analysis demonstrated patient-specific clustering of plasma genotypes and commingling with corresponding replication-competent clones recovered prevaccination. (This was also the case for the one subject [subject 1] who had genotypes amplified only at the last study visit, at week 72 [Fig. 1].) For 6 of these 11 (54.5%) subjects (subjects 4, 7, 8, 9, 12, and 17), postvaccination HIV RT sequences were identical to the prevaccine sequences, which persisted for a median of 28 weeks (range, 8 to 74 weeks), consistent with long-term persistence of a dominant plasma sequence in low-level viremia during HAART (Fig. 1). In three of these six subjects (subjects 7, 8, and 12), the persistent plasma sequence shared sequence identity with replication-competent virus cultured from resting CD4+ T cells at study entry (Fig. 1), in contrast to one other report that persistent plasma sequences in low-level viremia are genetically distinct from proviral DNA sequences in resting CD4+ T cells (2).
FIG. 1.

Maximum likelihood phylogenetic tree of HIV-1 RT sequences as a function of time postvaccination. Representative prevaccine replication-competent viral isolates, subtype B, C, and D reference sequences for HIV-1 RT, and the vaccine sequence (vaccine) are shown. The plasma sequences amplified during episodes of rebound viremia postvaccination are shown with a strike through the symbols. Wild-type variants, drug-resistant variants, and variants with amino acid substitutions at position 205 of HIV-1 RT are indicated. Bootstrap values of >80% are shown.
Evolution of the plasma sequence was observed during the study for four subjects. New drug resistance mutations occurring in low-level viremia during the nevirapine-based HAART regimen were observed in one subject (subject 8). Multiple unique amino acid substitutions in HIV-1 RT position 205 were seen at week 40 for three subjects (subjects 7, 11, and 17). Viral variants substituted at HIV-1 RT position 205 were also transiently detected at week 40 during rebound viremia in a fourth subject who discontinued HAART (subject 15) (Fig. 1 and Fig. 2).
FIG. 2.

Amino acid alignment, relative to the consensus region of HIV-1HXB2 RT, showing nonsynonymous substitutions detected during low-level viremia postvaccination within relevant CD8+ T-cell epitopes. Homology to the consensus sequence is represented by dots. (a) Subject 8 developed the nonsynonymous substitutions V108I and G112S in an HLA-A*02 epitope (red box), and the I135T substitution within an HLA-A*02/B*51 epitope (blue box) later became fixed in the free plasma virus present at low levels. The selection of substitutions at position 205 occurred within an HLA-A*02 epitope (red box) for three subjects (subject 7 [b], subject 15 [d], and subject 17 [e]). For the one subject (subject 11) (c) who showed temporal clustering of low-level viremia genotypes during the trial and subsequently failed therapy while maintained on HAART, and who is HLA-A*6801 positive (which is also an HLA-A*03 supertype), the L205M position also lies outside the HLA-A*68 (green box) and HLA-A*03 (red box) epitopes.
In the one subject (subject 8) who developed new NNRTI resistance mutations through sequence evolution, the V108I mutation was first detected 4 weeks after receipt of the first MVA vaccination. Additional NNRTI resistance mutations (K103N and V106A) were also detected in low-level viremia at the week 6 visit following the second vaccine dose (Fig. 1 and 2). The presence of new NNRTI-resistant variants was not associated with breakthrough viremia in this subject, despite a regimen of nevirapine-based HAART. In fact, prevaccine wild-type low-level viremia was maintained at all subsequent postvaccination visits (weeks 24, 26, 40, and 72, when the viral load was 172 copies/ml) (Fig. 1). In this subject, another amino acid change reported to confer decreased susceptibility to NNRTIs (I135T) (5) was first detected in low-level viremia linked to the V108I mutation at the week 4 visit and subsequently fixed in the low-level viremia through the last study visit at week 72 (Fig. 2). In addition to being associated with decreased susceptibility to NNRTIs, the I135T mutation is also reported to reside within an epitope presented by a relevant host HLA allele (http://www.hiv.lanl.gov).
Preexisting NNRTI drug resistance mutations in low-level viremia were also detected in subject 4, who was on a primary nevirapine-based HAART regimen. In this subject, all four sequences amplified at the two prevaccine visits contained the substitution Y188C, which confers intermediate to high-level nevirapine resistance (Stanford HIV Drug Resistance Database [http://hivdb.stanford.edu]). The Y188C variant remained detectable at weeks 4 and 6 postvaccination but was not associated with breakthrough viremia following HIV-1 vaccinations (Fig. 1). This subject maintained plasma viremia at <50 copies/ml throughout the study, and no additional plasma sequences were amplified at study visits from week 24 through week 72.
Unique nonsynonymous or amino acid-changing substitutions which were not previously described for subtype B HIV-1 at position 205 of HIV-1 RT (methionine [M], isoleucine [I], glutamine [Q], or arginine for leucine [L]) were detected in four study participants (subjects 7, 11, 15, and 17) (Fig. 1 and 2; Table 2). For all four subjects, these mutations were detected at the week 40 visit (Fig. 1). For three of the four subjects, the nonsynonymous changes were detected at viral loads of <50 copies/ml, and for one subject (subject 15) they were detected during rebound viremia following HAART discontinuation. A leucine-to-methionine (L205M) substitution was most common and was found in all four subjects (11/15 sequences) (Fig. 2). For two subjects (subjects 7 and 15), L205I/R/Q substitutions were also detected, suggesting immunologic pressure at this site.
One subject developed rebound viremia while on HAART (18). In this one perinatally infected subject (subject 11), rebound viremia developed at week 60, despite no reported change in HAART adherence. A time-dependent shift in low-level viremia genotypes with a decrease in viral diversity was observed as early as week 6 following vaccination (Fig. 1). For this subject, a 3.5-fold increase in HIV-1-specific CD8+ T-cell responses to HIV-1 Pol was detected at the same time (week 6 following MVA vaccinations) (see Fig. 4). These responses were also sustained above baseline levels (sevenfold) at week 26 following the fourth vaccine dose and preceded the amino acid substitution at position 205 of HIV-1 RT (L205M). Moreover, the L205M substitution was detected in multiple viral lineages (Fig. 1 and Fig. 2) at week 40, suggestive of vaccine-induced responses contributing to selection and possible immune escape before viral breakthrough at week 60.
FIG. 4.
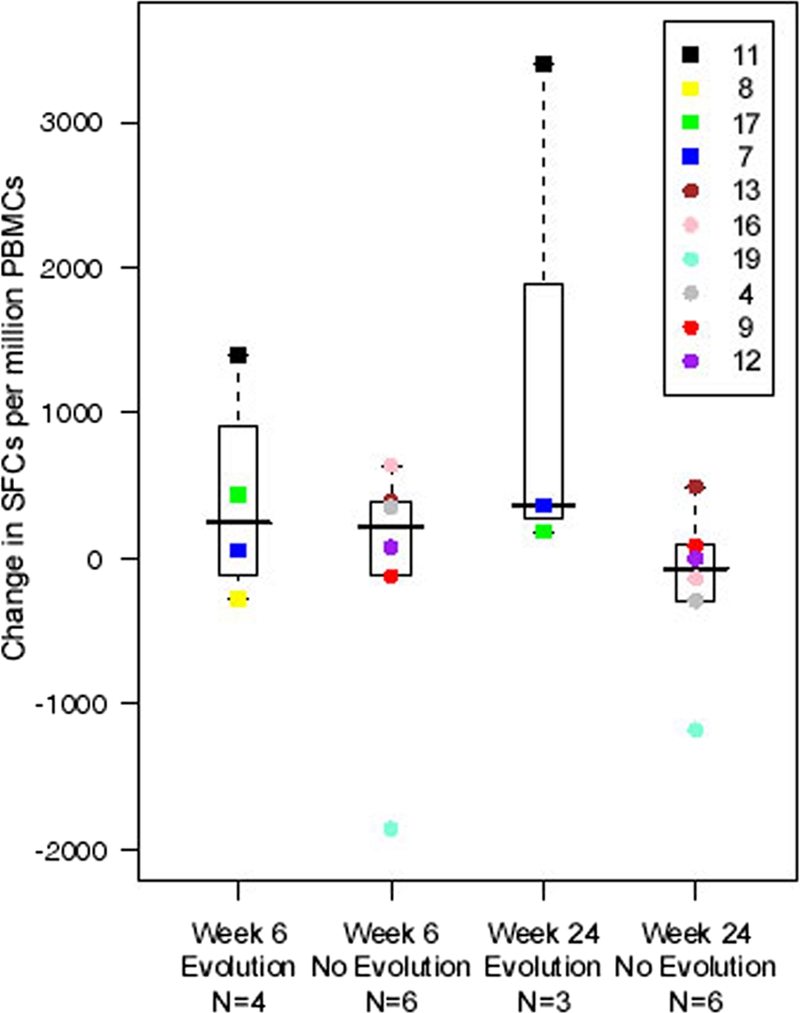
Changes from baseline in HIV-1-specific CD8+ IFN-γ ELISPOT responses to overlapping peptides spanning amino acids 1 to 556 of HIV-1 Pol at weeks 6 and 24 or 26 for the subjects experiencing sequence evolution in low-level viremia compared to those without detectable sequence evolution. The one subject (subject 15) who also developed substitutions at amino acid position 205 at week 40 in rebounding virus was excluded from this analysis. One subject (subject 8) did not receive any additional vaccine doses after week 4 and therefore did not have immune studies performed at week 24 or 26 of the study. The colored dots indicate individual subject data. The black symbols represent data from the one subject (subject 11) who developed rebound viremia during the trial while still on HAART.
Association of HIV-1 sequence evolution during recombinant poxvirus vaccinations with frequency of episodes of viremia below 50 copies/ml and with HIV-1-specific immune responses.
Given that significant increases in HIV-1-specific CD8+ T-cell responses to Pol were detected postvaccination among the study participants (18), we analyzed the low-viremia genotypes for nonsynonymous substitutions within relevant CD8+ T-cell epitopes following HIV-1 vaccination (http://www.hiv.lanl.gov/content/hiv-db/mainpage.html). There were 45 nonsynonymous substitutions (median of 3.5 substitutions per subject) observed in the 111 postvaccination sequences spanning amino acid positions 40 to 219 of HIV-1 RT from 7 of the 10 subjects (70%) with amplifiable genotypes and maintained on HAART (Table 3). Of these 45 substitutions, 56% (25/45 changes) occurred within defined CD8+ T-cell RT epitopes, and 56% of these (14/25 changes) were in HLA-A*02 epitopes. Using logistic regression modeling for correlated binary responses (27), we found that nonsynonymous mutations in HLA epitopes were significantly more likely to occur among individuals with the restricting HLA alleles (P < 0.001) (Table 3).
TABLE 3.
Nonsynonymous substitutions in HIV-1 RT detected in low-level viremia postvaccination sequences relative to prevaccination sequences and in relationship to defined CD8+ T-cell epitopes restricted by HLA serotypes
| Group and subject | Nonsynonymous amino acid substitution(s) | HLA serotypea | HLA-binding residues in HIV-1 RTb |
|---|---|---|---|
| Subjects with amplifiable low-level viremia genotypes and sequence evolutionc following vaccination | |||
| 7 | L205M | A*02 | KIEELRQHL |
| L205M | B*40 | IEELRQHLL | |
| F214L | A*02 | LLRWGLTTPDKK | |
| K166T, E169G | Not associated | NA | |
| 8 | V108I, G112S | A*02 | TVLDVGDAYFSV |
| I135T | A*02 | KYTAFTIPSI | |
| I135T | B*51 | TAFTIPSI | |
| K103N, V106A | Not associated | NA | |
| 11 | K154E | B*7 | WKGSPAIFQSSMT |
| K166T | A*68 | AIFQSSMTK | |
| K166T | B*7 | AIFQSSMTK | |
| K166T | B*35 | AIFQSSMTK | |
| K173E, I178M | A*30 | KQNPEIVIYQY | |
| I178M | B*35 | NPEIVIYQY | |
| T215I, K219E | A*68 | FTTPDKKHQK | |
| I135M, E194G, E203K, E204G, L205M | Not associated | NA | |
| Not associated | NA | ||
| 17 | V178Md | B*15 | KQNPDIVIY |
| V179I, Y181C, V184M | A*02 | VIYQYMDDLYV | |
| L205I/M/R, Y215F,d D218E, K219Q | A*02 | KIEELRQHLLRWGLTTPDKK | |
| Y215F,d D218E, K219Q | A*68 | LTTPDKKHQK | |
| K70R, V75S, K101E, K122E, G190A | Not associated | NA | |
| Subjects with amplifiable low-level viremia genotypes without evolution following vaccination | |||
| 4 | N54T, N123D, C188Y | Not associated | NA |
| 16 | T165I | B*7 | WKGSPAIFQSSMT |
| E123D | Not associated | NA | |
| 19 | S162A/D | B*7 | WKGSPAIFQSSMT |
| V179I | A*30/B*15 | AQNPDIVIYQY | |
| K43E, V118I, E203D | Not associated | NA |
HLA serotypes of study subjects with nonsynonymous changes within the relevant HLA epitopes postvaccination.
CD8+ T-cell epitopes in HIV-1 RT encompassing the observed nonsynonymous substitutions, as defined in the Los Alamos HIV Immunology Database (http://www.hiv.lanl.gov/content/index). Substitutions at reference sites are underlined; those in the four subjects experiencing sequence evolution are also shown in bold. To reduce redundancy, overlapping peptides were combined into one sequence. NA, not applicable; indicates a substitution in a region not likely recognized by the subject's HLA serotypes.
New drug resistance mutations or novel changes at amino acid position 205 of RT.
Preexisting substitutions relative to consensus HIV-1 RT sequences.
To explore whether substitutions at position 205 were associated with decreased epitope recognition due to CD8+ T-cell escape, we compared recognition of the HIV-1 RT HLA-A*02 KIEELRQHL wild-type epitope to that of an autologous L205M mutant peptide in IFN-γ ELISPOT assays. However, none of the four subjects demonstrated postimmunization responses to wild-type or mutant peptides above background levels with a control peptide (an HLA-A*02-restricted epitope in human T-cell leukemia virus type 1 Tax) (data not shown).
The five subjects whose data are depicted in Fig. 2 had evidence of either new drug resistance or unique amino acid substitutions in the RT region not appearing in the nearly 914 subtype B sequences in the Los Alamos HIV Sequence Database. Because one of these subjects (subject 15) discontinued HAART and had levels of viremia increasing to >1,000 copies/ml, pre- and postvaccination viremia levels for the four remaining subjects (subjects 7, 8, 11, and 17) with unexpected sequence evolution were compared to those for the six subjects with no sequence evolution (subjects 4, 9, 12, 13, 16, and 19). For this analysis, rates of quantifiable viremia pre- and postvaccination were calculated for each subject by dividing the number of viremic episodes above 6.5 but below 50 copies/ml by the numbers of visits pre- and postvaccination, respectively, at which viremia was measured. Since blood samples were obtained immediately before vaccine administration, the visit at study entry (week 0) was a prevaccine visit. For the four subjects with sequence evolution, 26 postvaccination visits (median, 6.5 per subject) produced quantifiable low-level viremia, while 41 visits (median, 7 per subject) produced quantifiable viremia for the six subjects without sequence evolution (Table 2). The median rate of quantifiable viremia did not change significantly for each group pre- and postvaccination (P = 0.1 for subjects experiencing evolution and 0.81 for those who did not), but rates were significantly greater for subjects experiencing sequence evolution than for subjects who did not. The rate of quantifiable viremia postvaccination was 54% (IQR, 39 to 62%) for those experiencing evolution versus 14% (IQR, 14 to 29%) for those who did not (P = 0.03) (Fig. 3). However, this did not reflect a change from prevaccination frequencies for these subjects. Prevaccination, 50% of the 8 samples from the subjects experiencing evolution were viremic, compared to 17% of 12 samples from subjects not experiencing evolution. The median change in the rate of quantifiable viremia pre- and postvaccination was −2.4% (IQR, −38.1% to 39.3%) (P = 1.0) for subjects experiencing evolution and 7.1% (IQR, −16.7% to 14.3%) (P = 0.81) for those who did not. When only the highest viral load postvaccination was considered for each subject, median viral loads were 21.5 copies/ml (IQR, 13.0 to 37.5 copies/ml) and 16.0 copies/ml (IQR, 10.0 to 37.0 copies/ml) for those with evolution and those without, respectively (P = 0.92).
FIG. 3.
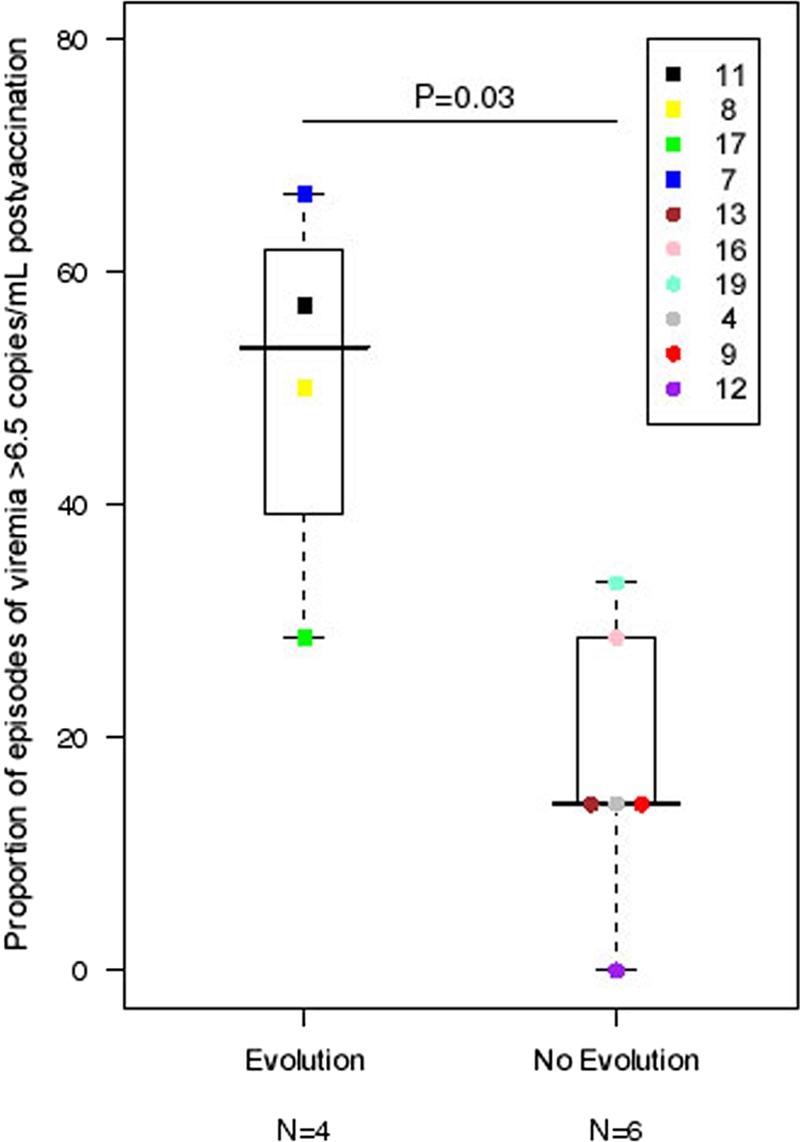
Frequency of episodes of low-level viremia above 6.5 copies/ml for the subjects experiencing sequence evolution (n = 4) at viral loads of <50 copies/ml compared to the frequency for those who did not (no evolution) following HIV-1 recombinant poxvirus vaccinations. Sequence evolution was defined as either new drug resistance mutations (subject 8) or novel changes at amino acid position 205 of HIV-1 RT while still being suppressed on HAART (subjects 7, 11, and 17). The one subject (subject 15) who also developed substitutions at amino acid position 205 of RT at week 40 in rebounding virus was excluded from this analysis. Median values and IQR are indicated.
We compared HIV-1-specific CD8+ T-cell responses to Pol (defined as the sum of responses to the Pol1, Pol2, and Pol3 peptides spanning amino acids 1 to 556 of HIV-1 Pol) (18) for the four subjects experiencing RT sequence evolution with those for subjects with no evolution. For this analysis, which was not adjusted for multiple comparisons, the median and IQR for the HIV-1 Pol responses for the two groups were compared at weeks 0, 6, and either 24 or 26. At baseline, the median HIV-1-specific CD8+ T-cell response to Pol was 547.3 spot-forming units (SFC) per million PBMC (IQR, 315.5 to 759 SFC/million PBMC) for those experiencing sequence evolution, compared to 376 SFC/million PBMC (IQR, 140.5 to 692.5 SFC/million PBMC) for those with no evolution, and these were not statistically different (P = 0.75). At week 6, the median changes from baseline were similar for those with and without HIV-1 RT sequence evolution (P = 0.75) (Fig. 4), but at week 24 or 26, the median change was 365.5 SFC/million PBMC (IQR, 183 to 3,400 SFC/million PBMC) for the three subjects who experienced evolution and also received at least one FPV booster dose and −69.3 SFC/million PBMC (IQR, −291 to 87.5 SFC/million PBMC) for the six subjects not experiencing evolution (P = 0.09) (Fig. 4). Notably, the one subject (subject 11) who failed to maintain suppression of virus replication while on HAART also experienced sustained increases in HIV-1-specific CD8+ T-cell responses to Pol peptides postvaccination (Fig. 4). Furthermore, in this subject, a decrease in viral diversity was observed during low-level viremia by week 24 postvaccination and prior to viral rebound at week 60. At screening and at entry, prevaccine low-level viremia diversity levels were 0.026 and 0.027, respectively, and remained high through week 6 of the study (0.031), after which diversity levels decreased to 0.005, 0.01, 0.02, and 0.00 at weeks 24, 26, 40, and 72, respectively (Fig. 1).
DISCUSSION
The pathogenesis of persistent low-level viremia in patients on HAART is unclear, but it is important to understand to improve control of viral replication in patients on long-term HAART. Evidence exists for both ongoing virus expression from latent cellular reservoirs (or drug sanctuary sites) (2, 10, 21-23, 25, 26, 29, 31, 34, 46) and low-level virus replication during HAART (6, 7, 22, 46). While these two mechanisms may not be mutually exclusive, it is likely that interpatient differences in the potency of suppression of virus replication below 50 copies/ml shift the contributions of virus release from long-lived infected cells and complete cycles of replication to sustaining residual viremia in the context of HIV-1 immunizations.
In this first study to examine serially the effects of immune activation with recombinant MVA/FPV-based HIV-1 vaccinations on low-level viremia in HAART-treated patients, we found evidence for both complete cycles of virus replication, with sequence evolution, in a subset of patients receiving durable successful HAART (current clinical criterion of a plasma viral load of <50 copies/ml) and virus expression from a latent reservoir. In particular, sequence evolution in free plasma virus was significantly more likely to occur within relevant CD8+ T-cell epitopes of HIV-1 RT and consisted of novel amino acid changes at a non-drug-resistance site, position 205 of HIV-1 RT, as well as new relevant drug resistance mutations. Furthermore, we found that sequence evolution following HIV-1 vaccinations was significantly associated with more episodes of quantifiable low-level viremia, suggesting ongoing cycles of virus replication that may be amenable to CD8+ T-cell selective pressures. At baseline, the subjects were heterogeneous with respect to the predicted potency of their HAART regimens (41) and the presence of drug resistance mutations, but their respective HAART regimens had controlled plasma viremia to clinically undetectable levels for years before study entry. Furthermore, plasma viral loads remained low postvaccination even in the presence of relevant new drug resistance mutations or evidence of sequence evolution at non-drug-resistance sites during low-level viremia, except for in one subject. Importantly, for most subjects in this study, administration of MVA/FPV HIV-1 therapeutic vaccines was not associated with temporal shifts or sequence changes within persistent wild-type or drug-resistant plasma viral sequences in very low viremia or with new drug resistance mutations for up to 18 months postvaccination. The 40% of study participants without amplifiable variants prevaccination also did not have amplifiable genotypes postvaccination and thus did not provide evidence of either enhanced virus production or replication following HIV-1 vaccination. Virus replication was even controlled for the individuals for whom NNRTI-resistant variants were detected postvaccination, despite their remaining on the same nevirapine-based HAART regimen as that at study entry. While they are important for the safety profile of these vaccines (18), these observations also support the clinical significance of HAART to limit HIV-1 replication in most patients. In particular, the current clinical criterion of <50 copies/ml indicates that HAART has significantly reduced the effective virus population size, thereby slowing virus replication and sequence evolution in most individuals (39).
Because recent studies of treatment intensification have shown variable effects on low-level viremia (6, 10, 22, 24), study-to-study variation in the baseline level of viremia must be considered. In one study, baseline viremia was quite low, at 1 to 2 copies/ml, most likely reflecting the potency of the regimen, and no evidence of additional suppression was generated by the addition of raltegravir (24). In contrast, abacavir intensification of an efavirenz and indinavir two-drug regimen in subjects with a baseline viral load of 10 copies/ml produced a lowering of the viral load to <2.5 copies/ml (22). Our data demonstrate discernible sequence evolution in HAART-suppressed subjects experiencing low-level viremia of >6.5 copies/ml. Together, these findings suggest that a threshold level of viremia below 50 copies/ml may exist, above which productive infection may be a greater contributor to sustained viremia than low-level virus release from long-lived cellular reservoirs. Under this model, the sequence evolution we observed during therapeutic HIV-1 poxvirus vaccination, in or near epitopes recognizable based on HLA serotypes, would be facilitated by steady-state, low-level, productive infection. To test this will require further study of patients receiving newer, more potent HAART regimens than those used by the subjects in our study. Nevertheless, the data suggest that the current therapeutic target of control of viremia to <50 copies/ml, while clinically meaningful, is insufficient for distinguishing contributions of low-level virus expression from those of productive infection. A new therapeutic target for viremia may be necessary for clinical trials assessing HIV-1 treatment approaches involving newer drug combinations, therapeutic vaccinations, or treatment intensification and deintensification strategies.
Among nonimmunized individuals, substitution at position 205 of HIV-1 RT is exceedingly rare, appearing in only a single HIV-1 subtype C isolate (L205S) among 914 HIV-1 sequences in the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov). Amino acid position 205 of HIV-1 RT lies within an HLA-A*02 cytotoxic T-lymphocyte epitope, KIEELRQHL (48), and three of four subjects with L205 changes were HLA-A*02 positive (subjects 7, 15, and 17) in our study. While the fourth (subject 11) was HLA-A*6801 and -A*3001 positive, and not HLA-A*02 positive, the L205M substitution was also flanked by HLA-A*68 (FTTPDKKHQK) (9) and HLA-A*03 (DLEIGQHRTK) (48) epitopes. It is notable that HLA-A*6801 could share binding to these regions as an A*03 supertype (Fig. 2). Additionally, in the one subject (subject 8) who developed new drug resistance mutations in low-level viremia postvaccination, the first NNRTI resistance mutation detected (V108I) lay within an HLA-A*02-restricted epitope (VLDVGDAYFSV) (47) that was linked to an isoleucine-to-threonine substitution at amino acid position 135 of HIV-1 RT (I135T), known to confer decreased susceptibility to NNRTI (5). This substitution also represents an amino acid change within relevant HLA-A*02 and -B*51 epitopes (KYTAFTIPSI and TAFTIPSI, respectively) (40, 43) for this individual, who was indeed HLA-A*02 and -B*51 positive (Fig. 2). While the NNRTI resistance mutations were transient, the I135T substitution became fixed and persisted through week 72 of the trial, suggesting selection due to either immune or drug pressure. Moreover, in the one subject who developed rebound viremia while on HAART, decreasing sequence diversity in low-level viremia was associated with sustained CD8+ T-cell responses to HIV-1 Pol postvaccination, also suggesting specific HIV-1 immune selective pressure on residual replication during HAART. This idea is supported by reports of selection of immunity-escaping HIV-1 variants at clinically undetectable viral loads in long-term nonprogressors (4, 16).
Together, these findings imply that viral populations replicating at low levels may be targeted by HIV-1-specific immune responses in HAART-treated patients. We were unable to demonstrate that CD8+ T-cell responses to the peptide containing position 205 of HIV-1 RT were responsible for sequence evolution, though a finely detailed analysis of CD8+ T-cell recognition with overlapping peptides was not performed. Finer mapping of immunological responses, including interleukin-2-mediated responses (3), to this region of HIV-1 RT may help to elucidate the role of CD8+ T-cell selective pressures in L205 substitutions.
This study has some limitations. These include restriction of genetic analysis to the HIV-1 RT region, the sensitivities of the low-level viremia genotyping and viral load assays, the lack of a placebo group to fully assess sequence evolution at viremia levels of <50 copies/ml within HLA-restricted CD8+ T-cell epitopes, and the small study population. Nevertheless, we identified in vivo selection and convergent sequence evolution associated with novel mutations within HLA epitopes in residual viremia among a subset of individuals receiving recombinant HIV-1 MVA/FPV vaccinations who had viral loads of <50 copies/ml on durable successful HAART. Moreover, this selection occurred in association with higher frequencies of episodes of low-level viremia above 6.5 copies/ml. The data support contributions of both low-level virus release (2, 10, 24, 28, 31, 34, 46) and productive infection (6, 22, 46), which suggests that a low-level threshold may exist where the balance shifts for these two contributors. Although reinforcing the benefits of HAART suppression of viremia levels of <50 copies/ml, this potential model suggests that a new therapeutic target for viremia may be necessary for clinical trials assessing newer treatment approaches for HIV-1, including vaccines. Furthermore, the potential for virus replication and sequence evolution within epitopes presented by relevant HLA sites observed in this study has implications for therapeutic HIV-1 vaccines. Both of these implications warrant further study.
Acknowledgments
This clinical trial (ClinicalTrials.gov identifier NCT00107549) was supported by the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Network of the National Institute of Allergy and Infectious Diseases (NIAID) (grant U01 A1068632), by the general clinical research center units funded by the National Center for Research Resources and the International and Domestic Pediatric and Maternal HIV Clinical Trials Network of the National Institute of Child Health and Human Development (grant N01-HD-3-3345), and by the Pediatric AIDS Clinical Trials Group (PACTG) (group 1059). The study on virus evolution in low-level viremia was supported by NIAID grants R01 A155312 and R01 A1062446, awarded to D. Persaud. Immunology studies were supported by an IMPAACT Immunology Laboratory grant and NIAID grant RO1 32391 to K. Luzuriaga.
We acknowledge the contributions of the PACTG1059 study team, clinical trials specialist Elizabeth Sheeran, and database manager Barbara Heckman. We also acknowledge Linda Lambrecht from the University of Massachusetts, who performed the IFN-γ ELISPOT assays; Roxann Ashworth from the Institute of Genetics, Johns Hopkins School of Medicine, for her independent analyses of the HIV-1 RT sequence chromatograms; and Estelle Piwowar-Manning for her contributions to the ultrasensitive viral load quantitation. We also express great appreciation to the clinical trial sites and the young adults who participated in this trial.
Footnotes
Published ahead of print on 15 July 2009.
REFERENCES
- 1.Ananworanich, J., A. Gayet-Ageron, M. Le Braz, W. Prasithsirikul, P. Chetchotisakd, S. Kiertiburanakul, W. Munsakul, P. Raksakulkarn, S. Tansuphasawasdikul, S. Sirivichayakul, M. Cavassini, U. Karrer, D. Genne, R. Nuesch, P. Vernazza, E. Bernasconi, D. Leduc, C. Satchell, S. Yerly, L. Perrin, A. Hill, T. Perneger, P. Phanuphak, H. Furrer, D. Cooper, K. Ruxrungtham, and B. Hirschel. 2006. CD4-guided scheduled treatment interruptions compared with continuous therapy for patients infected with HIV-1: results of the Staccato randomised trial. Lancet 368:459-465. [DOI] [PubMed] [Google Scholar]
- 2.Bailey, J. R., A. R. Sedaghat, T. Kieffer, T. Brennan, P. K. Lee, M. Wind-Rotolo, C. M. Haggerty, A. R. Kamireddi, Y. Liu, J. Lee, D. Persaud, J. E. Gallant, J. Cofrancesco, Jr., T. C. Quinn, C. O. Wilke, S. C. Ray, J. D. Siliciano, R. E. Nettles, and R. F. Siliciano. 2006. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 80:6441-6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey, J. R., T. M. Williams, R. F. Siliciano, and J. N. Blankson. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J. Exp. Med. 203:1357-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey, J. R., H. Zhang, B. W. Wegweiser, H. C. Yang, L. Herrera, A. Ahonkhai, T. M. Williams, R. F. Siliciano, and J. N. Blankson. 2007. Evolution of HIV-1 in an HLA-B*57-positive patient during virologic escape. J. Infect. Dis. 196:50-55. [DOI] [PubMed] [Google Scholar]
- 5.Brown, A. J., H. M. Precious, J. M. Whitcomb, J. K. Wong, M. Quigg, W. Huang, E. S. Daar, R. T. D'Aquila, P. H. Keiser, E. Connick, N. S. Hellmann, C. J. Petropoulos, D. D. Richman, and S. J. Little. 2000. Reduced susceptibility of human immunodeficiency virus type 1 (HIV-1) from patients with primary HIV infection to nonnucleoside reverse transcriptase inhibitors is associated with variation at novel amino acid sites. J. Virol. 74:10269-10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buzon, M., M. Llibre, J. Gatell, P. Domingo, R. Paredes, S. Palmer, M. Sharkey, M. Stevenson, B. Clotet, and J. Martinez-Picado. 2009. Transient increase in episomal viral cDNA following raltegravir intensification of a stable HAART regimen, abstr. 423a. Abstr. 16th Conf. Retrovir. Opportun. Infect.
- 7.Cohen Stuart, J. W., A. M. Wensing, C. Kovacs, M. Righart, D. de Jong, S. Kaye, R. Schuurman, C. J. Visser, and C. A. Boucher. 2001. Transient relapses (“blips”) of plasma HIV RNA levels during HAART are associated with drug resistance. J. Acquir. Immune Defic. Syndr. 28:105-113. [DOI] [PubMed] [Google Scholar]
- 8.Danel, C., R. Moh, M. L. Chaix, D. Gabillard, J. Gnokoro, C. J. Diby, T. Toni, L. Dohoun, C. Rouzioux, E. Bissagnene, R. Salamon, and X. Anglaret. 2009. Two-months-off, four-months-on antiretroviral regimen increases the risk of resistance, compared with continuous therapy: a randomized trial involving West African adults. J. Infect. Dis. 199:66-76. [DOI] [PubMed] [Google Scholar]
- 9.De Groot, A. S., B. Jesdale, W. Martin, A. C. Saint, H. Sbai, A. Bosma, J. Lieberman, G. Skowron, F. Mansourati, and K. H. Mayer. 2003. Mapping cross-clade HIV-1 vaccine epitopes using a bioinformatics approach. Vaccine 21:4486-4504. [DOI] [PubMed] [Google Scholar]
- 10.Dinoso, J. B., S. Y. Kim, A. M. Wiegand, S. E. Palmer, S. J. Gange, L. Cranmer, A. O'Shea, M. Callender, A. Spivak, T. Brennan, M. F. Kearney, M. A. Proschan, J. M. Mican, C. A. Rehm, J. M. Coffin, J. W. Mellors, R. F. Siliciano, and F. Maldarelli. 2009. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 106:9403-9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dornadula, G., H. Zhang, B. VanUitert, J. Stern, L. Livornese, Jr., M. J. Ingerman, J. Witek, R. J. Kedanis, J. Natkin, J. DeSimone, and R. J. Pomerantz. 1999. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 282:1627-1632. [DOI] [PubMed] [Google Scholar]
- 12.Dorrell, L. 2006. Therapeutic immunization for the control of HIV-1: where are we now? Int. J. STD AIDS 17:436-441. [DOI] [PubMed] [Google Scholar]
- 13.Douek, D. C., J. M. Brenchley, M. R. Betts, D. R. Ambrozak, B. J. Hill, Y. Okamoto, J. P. Casazza, J. Kuruppu, K. Kunstman, S. Wolinsky, Z. Grossman, M. Dybul, A. Oxenius, D. A. Price, M. Connors, and R. A. Koup. 2002. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417:95-98. [DOI] [PubMed] [Google Scholar]
- 14.El-Sadr, W. M., J. D. Lundgren, J. D. Neaton, F. Gordin, D. Abrams, R. C. Arduino, A. Babiker, W. Burman, N. Clumeck, C. J. Cohen, D. Cohn, D. Cooper, J. Darbyshire, S. Emery, G. Fatkenheuer, B. Gazzard, B. Grund, J. Hoy, K. Klingman, M. Losso, N. Markowitz, J. Neuhaus, A. Phillips, and C. Rappoport. 2006. CD4+ count-guided interruption of antiretroviral treatment. N. Engl. J. Med. 355:2283-2296. [DOI] [PubMed] [Google Scholar]
- 15.Emery, S., J. A. Neuhaus, A. N. Phillips, A. Babiker, C. J. Cohen, J. M. Gatell, P. M. Girard, B. Grund, M. Law, M. H. Losso, A. Palfreeman, and R. Wood. 2008. Major clinical outcomes in antiretroviral therapy (ART)-naive participants and in those not receiving ART at baseline in the SMART study. J. Infect. Dis. 197:1133-1144. [DOI] [PubMed] [Google Scholar]
- 16.Feeney, M. E., Y. Tang, K. A. Roosevelt, A. J. Leslie, K. McIntosh, N. Karthas, B. D. Walker, and P. J. Goulder. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927-8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furtado, M. R., D. S. Callaway, J. P. Phair, K. J. Kunstman, J. L. Stanton, C. A. Macken, A. S. Perelson, and S. M. Wolinsky. 1999. Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N. Engl. J. Med. 340:1614-1622. [DOI] [PubMed] [Google Scholar]
- 18.Greenough, T. C., C. K. Cunningham, P. Muresan, M. McManus, D. Persaud, T. Fenton, P. Barker, A. Gaur, D. Panicali, J. L. Sullivan, and K. Luzuriaga. 2008. Safety and immunogenicity of recombinant poxvirus HIV-1 vaccines in young adults on highly active antiretroviral therapy. Vaccine 26:6883-6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grossman, Z., M. Polis, M. B. Feinberg, Z. Grossman, I. Levi, S. Jankelevich, R. Yarchoan, J. Boon, F. de Wolf, J. M. Lange, J. Goudsmit, D. S. Dimitrov, and W. E. Paul. 1999. Ongoing HIV dissemination during HAART. Nat. Med. 5:1099-1104. [DOI] [PubMed] [Google Scholar]
- 20.Guindon, S., and O. Gascuel. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696-704. [DOI] [PubMed] [Google Scholar]
- 21.Havlir, D. V., R. Bassett, D. Levitan, P. Gilbert, P. Tebas, A. C. Collier, M. S. Hirsch, C. Ignacio, J. Condra, H. F. Gunthard, D. D. Richman, and J. K. Wong. 2001. Prevalence and predictive value of intermittent viremia with combination HIV therapy. JAMA 286:171-179. [DOI] [PubMed] [Google Scholar]
- 22.Havlir, D. V., M. C. Strain, M. Clerici, C. Ignacio, D. Trabattoni, P. Ferrante, and J. K. Wong. 2003. Productive infection maintains a dynamic steady state of residual viremia in human immunodeficiency virus type 1-infected persons treated with suppressive antiretroviral therapy for five years. J. Virol. 77:11212-11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hermankova, M., S. C. Ray, C. Ruff, M. Powell-Davis, R. Ingersoll, R. T. D'Aquila, T. C. Quinn, J. D. Siliciano, R. F. Siliciano, and D. Persaud. 2001. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 286:196-207. [DOI] [PubMed] [Google Scholar]
- 24.Jones, J., D. McMahon, A. Wiegand, M. Kearney, S. Palmer, S. McNulty, J. Metcalf, J. Coffin, J. Mellors, and F. Maldarelli. 2009. No decrease in residual viremia during raltegravir intensification in patients on standard ART, abstr. 423b. Abstr. 16th Conf. Retrovir. Opportun. Infect.
- 25.Kieffer, T. L., M. M. Finucane, R. E. Nettles, T. C. Quinn, K. W. Broman, S. C. Ray, D. Persaud, and R. F. Siliciano. 2004. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 189:1452-1465. [DOI] [PubMed] [Google Scholar]
- 26.Lee, K. J., D. Shingadia, D. Pillay, A. S. Walker, A. Riordan, E. Menson, T. Duong, G. Tudor-Williams, and D. M. Gibb. 2007. Transient viral load increases in HIV-infected children in the U.K. and Ireland: what do they mean? Antivir. Ther. 12:949-956. [PubMed] [Google Scholar]
- 27.Liang, K. Y., and S. L. Zeger. 1986. Longitudinal data-analysis using generalized linear-models. Biometrika 73:13-22. [Google Scholar]
- 28.Maldarelli, F., S. Palmer, M. S. King, A. Wiegand, M. A. Polis, J. Mican, J. A. Kovacs, R. T. Davey, D. Rock-Kress, R. Dewar, S. Liu, J. A. Metcalf, C. Rehm, S. C. Brun, G. J. Hanna, D. J. Kempf, J. M. Coffin, and J. W. Mellors. 2007. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 3:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nettles, R. E., T. L. Kieffer, P. Kwon, D. Monie, Y. Han, T. Parsons, J. Cofrancesco, Jr., J. E. Gallant, T. C. Quinn, B. Jackson, C. Flexner, K. Carson, S. Ray, D. Persaud, and R. F. Siliciano. 2005. Intermittent HIV-1 viremia (blips) and drug resistance in patients receiving HAART. JAMA 293:817-829. [DOI] [PubMed] [Google Scholar]
- 30.Nettles, R. E., T. L. Kieffer, R. P. Simmons, J. Cofrancesco, Jr., R. D. Moore, J. E. Gallant, D. Persaud, and R. F. Siliciano. 2004. Genotypic resistance in HIV-1-infected patients with persistently detectable low-level viremia while receiving highly active antiretroviral therapy. Clin. Infect. Dis. 39:1030-1037. [DOI] [PubMed] [Google Scholar]
- 31.Palmer, S., F. Maldarelli, A. Wiegand, B. Bernstein, G. J. Hanna, S. C. Brun, D. J. Kempf, J. W. Mellors, J. M. Coffin, and M. S. King. 2008. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 105:3879-3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. [DOI] [PubMed] [Google Scholar]
- 33.Persaud, D., S. C. Ray, J. Kajdas, A. Ahonkhai, G. K. Siberry, K. Ferguson, C. Ziemniak, T. C. Quinn, J. P. Casazza, S. Zeichner, S. J. Gange, and D. C. Watson. 2007. Slow human immunodeficiency virus type 1 evolution in viral reservoirs in infants treated with effective antiretroviral therapy. AIDS Res. Hum. Retrovir. 23:381-390. [DOI] [PubMed] [Google Scholar]
- 34.Persaud, D., G. K. Siberry, A. Ahonkhai, J. Kajdas, D. Monie, N. Hutton, D. C. Watson, T. C. Quinn, S. C. Ray, and R. F. Siliciano. 2004. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J. Virol. 78:968-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Persaud, D., Y. Zhou, J. M. Siliciano, and R. F. Siliciano. 2003. Latency in human immunodeficiency virus type 1 infection: no easy answers. J. Virol. 77:1659-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piwowar-Manning, E. M., T. A. Henderson, L. Brisbin, and J. B. Jackson. 2003. A modified ultrasensitive assay to detect quantified HIV-1 RNA of fewer than 50 copies per milliliter. Am. J. Clin. Pathol. 120:268-270. [DOI] [PubMed] [Google Scholar]
- 37.Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817-818. [DOI] [PubMed] [Google Scholar]
- 38.Ramratnam, B., R. Ribeiro, T. He, C. Chung, V. Simon, J. Vanderhoeven, A. Hurley, L. Zhang, A. S. Perelson, D. D. Ho, and M. Markowitz. 2004. Intensification of antiretroviral therapy accelerates the decay of the HIV-1 latent reservoir and decreases, but does not eliminate, ongoing virus replication. J. Acquir. Immune Defic. Syndr. 35:33-37. [DOI] [PubMed] [Google Scholar]
- 39.Rouzine, I. M., A. Rodrigo, and J. M. Coffin. 2001. Transition between stochastic evolution and deterministic evolution in the presence of selection: general theory and application to virology. Microbiol. Mol. Biol. Rev. 65:151-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shankar, P., H. Sprang, and J. Lieberman. 1998. Effective lysis of HIV-1-infected primary CD4+ T cells by a cytotoxic T-lymphocyte clone directed against a novel A2-restricted reverse-transcriptase epitope. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 19:111-120. [DOI] [PubMed] [Google Scholar]
- 41.Shen, L., S. Peterson, A. R. Sedaghat, M. A. McMahon, M. Callender, H. Zhang, Y. Zhou, E. Pitt, K. S. Anderson, E. P. Acosta, and R. F. Siliciano. 2008. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 14:762-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siliciano, J. D., and R. F. Siliciano. 2005. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol. Biol. 304:3-15. [DOI] [PubMed] [Google Scholar]
- 43.Sipsas, N. V., S. A. Kalams, A. Trocha, S. He, W. A. Blattner, B. D. Walker, and R. P. Johnson. 1997. Identification of type-specific cytotoxic T lymphocyte responses to homologous viral proteins in laboratory workers accidentally infected with HIV-1. J. Clin. Investig. 99:752-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith, D. B., J. McAllister, C. Casino, and P. Simmonds. 1997. Virus ‘quasispecies’: making a mountain out of a molehill? J. Gen. Virol. 78:1511-1519. [DOI] [PubMed] [Google Scholar]
- 45.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. [DOI] [PubMed] [Google Scholar]
- 46.Tobin, N. H., G. H. Learn, S. E. Holte, Y. Wang, A. J. Melvin, J. L. McKernan, D. M. Pawluk, K. M. Mohan, P. F. Lewis, J. I. Mullins, and L. M. Frenkel. 2005. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J. Virol. 79:9625-9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Burg, S. H., M. R. Klein, C. J. van de Velde, W. M. Kast, F. Miedema, and C. J. Melief. 1995. Induction of a primary human cytotoxic T-lymphocyte response against a novel conserved epitope in a functional sequence of HIV-1 reverse transcriptase. AIDS 9:121-127. [PubMed] [Google Scholar]
- 48.Walker, B. D., C. Flexner, K. Birch-Limberger, L. Fisher, T. J. Paradis, A. Aldovini, R. Young, B. Moss, and R. T. Schooley. 1989. Long-term culture and fine specificity of human cytotoxic T-lymphocyte clones reactive with human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 86:9514-9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ziemniak, C., A. George-Agwu, W. J. Moss, S. C. Ray, and D. Persaud. 2006. A sensitive genotyping assay for detection of drug resistance mutations in reverse transcriptase of HIV-1 subtypes B and C in samples stored as dried blood spots or frozen RNA extracts. J. Virol. Methods 136:238-247. [DOI] [PubMed] [Google Scholar]