

Abstract
The cathepsin family of endosomal proteases is required for proteolytic processing of several viruses during entry into host cells. Mammalian reoviruses utilize cathepsins B (Ctsb), L (Ctsl), and S (Ctss) for disassembly of the virus outer capsid and activation of the membrane penetration machinery. To determine whether cathepsins contribute to reovirus tropism, spread, and disease outcome, we infected 3-day-old wild-type (wt), Ctsb−/−, Ctsl−/−, and Ctss−/− mice with the virulent reovirus strain T3SA+. The survival rate of Ctsb−/− mice was enhanced in comparison to that of wt mice, whereas the survival rates of Ctsl−/− and Ctss−/− mice were diminished. Peak titers at sites of secondary replication in all strains of cathepsin-deficient mice were lower than those in wt mice. Clearance of the virus was delayed in Ctsl−/− and Ctss−/− mice in comparison to the levels for wt and Ctsb−/− mice, consistent with a defect in cell-mediated immunity in mice lacking cathepsin L or S. Cathepsin expression was dispensable for establishment of viremia, but cathepsin L was required for maximal reovirus growth in the brain. Treatment of wt mice with an inhibitor of cathepsin L led to amelioration of reovirus infection. Collectively, these data indicate that cathepsins B, L, and S influence reovirus pathogenesis and suggest that pharmacologic modulation of cathepsin activity diminishes reovirus disease severity.
As obligate intracellular parasites, viruses must coopt basic cellular processes to enter host cells and deliver their genomes to the appropriate intracellular site for replication (45). Viral entry steps include attachment of the virus to the cell surface, penetration of the virus into the cell interior, disassembly of the viral capsid, and activation of the viral genetic program. These events are essential for the virus to transition from the extracellular environment to the cellular compartment in which viral transcription and replication occur. Entry steps also play key roles in viral pathogenesis, as these events often determine cell tropism within the infected host.
Mammalian orthoreoviruses (reoviruses) are important models for studies of virus cell entry and the pathogenesis of viral disease. Reoviruses form nonenveloped, double-shelled particles that contain a segmented, double-stranded RNA genome (70). Virtually all mammals, including humans, serve as hosts for reovirus infection (84). However, reovirus causes disease primarily in the very young (44, 77, 79). Newborn mice infected with reovirus sustain injury to a variety of organs, including the brain, heart, and liver (5, 56, 84). Mechanisms of reovirus-induced disease, including cellular determinants of viral spread and tropism, are only partially understood.
Reovirus entry into cells is initiated by the attachment of virions to cell surface receptors via the σ1 protein (41, 85) and internalization into cells by receptor-mediated endocytosis (6, 7, 24, 76). In cellular endosomes, virions undergo stepwise disassembly, forming discrete intermediates, the first of which is the infectious subvirion particle (ISVP) (7, 14, 74, 76). ISVPs are generated by proteolytic removal of the σ3 protein and cleavage of the μ1 protein to form particle-associated fragments δ and ϕ. Following formation of ISVPs, σ1 is shed and the μ1 cleavage fragments undergo conformational rearrangement, yielding the ISVP* (11, 12). ISVP*s penetrate endosomes to deliver transcriptionally active viral cores into the cytoplasm (54, 55).
Endocytic proteases cathepsins B and L catalyze reovirus virion-to-ISVP disassembly in murine fibroblasts, although cathepsin L is the major mediator of this process (23). These proteases are expressed in most organs, including the intestine, brain, heart, and liver (78). In P388D cells, a macrophage-like cell line, cathepsin S, mediates the uncoating of some reovirus strains (28). Cathepsin S expression is largely restricted to cells and tissues of the immune system (16), which may be important during enteric infection, as reovirus replication in the intestine occurs in mononuclear cells of Peyer's patches (25, 51). Cathepsin S is known to be expressed in mononuclear cells, including alveolar macrophages in the lung (72, 73) and microglial cells in the brain (61).
Cathepsins B, L, and S are responsible for unique, tissue-specific activities (65). Cathepsin B modulates pathological trypsinogen activation (30) and apoptosis induced by tumor necrosis factor alpha (29). Cathepsin L is required for hair follicle cycling and epidermal homeostasis (68). By virtue of its activity at neutral pH (9, 39), cathepsin S is thought to participate in remodeling of the extracellular matrix (72, 89). Functions of cathepsins B, L, and S intersect in the regulation of adaptive immunity. Cathepsin L cleaves the invariant chain in cortical thymic epithelial cells (52) and is hypothesized to mediate efficient endosomal protein fragmentation to ensure diverse peptide generation in the thymus (33, 43). Through these functions, cathepsin L serves to facilitate positive selection of CD4+ T cells (15, 35). Cathepsin S cleaves the invariant chain in peripheral antigen-presenting cells, leading to CD4+ T-cell activation (53). Both cathepsin L (32) and cathepsin S (67) participate in NK1.1+ T-cell selection in the thymus through proteolytic processing in thymocytes and antigen-presenting cells, respectively. As a result, cathepsin L-deficient (Ctsl−/−) and cathepsin S-deficient (Ctss−/−) mice have impairments in both CD4+ and NK1.1+ T-cell activities. Cathepsin S also processes antigen in endosomes for cross-presentation via the major histocompatibility complex class I pathway (71). Like cathepsins L (34) and S (63), cathepsin B processes endocytosed antigen for display by major histocompatibility complex class II molecules (46, 48). However, cathepsin B-deficient mice (Ctsb−/−) do not display overt immunodeficiency (65).
Underscoring the importance of endosomal cathepsin proteases in host functions, viruses have usurped these enzymes to allow entry into the cytoplasm. In addition to reovirus, cathepsins catalyze proteolytic events required for membrane fusion of several important pathogens. Ebola virus requires both cathepsin B and cathepsin L for efficient cell entry (13), while severe acute respiratory syndrome coronavirus requires cathepsin L but also can utilize cathepsins B and S (36, 75). Hendra (58) and Nipah (57) viruses utilize cathepsin L for fusion protein processing, most likely at the stage of virion assembly (47). Despite the importance of cathepsins in viral growth, nothing is known about the function of these proteases in the pathogenesis of viral disease.
To determine the role of cathepsin proteases in viral virulence, we studied reovirus disease by using mice lacking a single cathepsin. Mice deficient for cathepsin B, L, or S were monitored for survival, disease symptoms, and viral replication following reovirus infection. We found that following peroral inoculation of reovirus, cathepsin deficiency leads to decreased viral replication in sites of secondary replication. However, Ctsl−/− and Ctss−/− mice succumb to doses of virus nonlethal to wild-type (wt) and Ctsb−/− animals. Although viremia is not affected by cathepsin deficiency, we observed alterations in disease pathogenesis in the hearts, livers, and brains of cathepsin-deficient animals. Furthermore, treatment of wt mice with an inhibitor of cathepsin L reduces disease severity. These studies demonstrate that cathepsin activity plays a key role in viral pathogenesis and identify a new target for antiviral drug development.
MATERIALS AND METHODS
Cells and viruses.
L929 cells were maintained in Joklik's minimal essential medium (Lonza) supplemented to contain 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). T3SA+ is a reassortant virus containing the S1 gene segment of strain T3C44MA and the remaining nine gene segments of strain T1L (4). Virus was purified after growth in L929 cells by CsCl gradient centrifugation (27). Viral titers were determined by a plaque assay (83). ISVPs were generated by treatment of 5 × 106 virions per ml with 20 μg of N-α-tosyl-l-lysine chloromethylketone-treated α-chymotrypsin type VII from bovine pancrease (Sigma-Aldrich) per ml at 37°C for 1 h.
Treatment of virions with purified cathepsins.
Purified reovirus virions at a concentration of 1012 particles per ml, as determined by optical density at 260 nm, in reaction buffer B-L (50 mM sodium acetate [pH 5.0], 15 mM MgCl2, 100 mM NaCl, 5 mM dithiothreitol) were treated with 400 μg/ml bovine spleen cathepsin B (Calbiochem-Novabiochem) or 100 μg/ml purified recombinant human cathepsin L (10) at 37°C for various intervals. Alternatively, purified virions at a concentration of 1012 particles per ml in reaction buffer S (50 mM sodium acetate [pH 6.0], 15 mM MgCl2, 100 mM NaCl) were treated with 300 μg/ml purified recombinant human cathepsin S (Calbiochem) at 37°C for various intervals. Proteolysis was terminated by incubation of reaction mixtures on ice and addition of 1 mM (final concentration) E64 (Sigma-Aldrich), a pan-cysteine-containing protease inhibitor (3). A 30-μl aliquot of each reaction mixture was incubated with 7 μl of 6× sample buffer (350 mM Tris [pH 6.8], 9.3% dithiothreitol, 10% sodium dodecyl sulfate, 0.012% bromophenol blue) at 100°C for 5 min. Samples were loaded into wells of 10% polyacrylamide gels and electrophoresed. After electrophoresis, gels were fixed and stained using colloidal Coomassie blue (Invitrogen).
Mice.
C57BL/6J mice (wt) were obtained from Jackson Laboratory. Ctsb−/− (21, 30) and Ctsl−/− (68) mice, each backcrossed to a C57BL/6 background at least eight times to ensure that all strains studied were of similar genetic backgrounds, were provided by D. Hanahan (San Francisco, CA). Ctss−/− mice, backcrossed at least 10 times to a C57BL/6 background (67), were provided by H. Chapman (San Francisco, CA).
Infection of mice.
Newborn mice, 2 to 4 days old, weighing approximately 2 g were inoculated perorally or intracranially with purified reovirus virions diluted in phosphate-buffered saline (PBS). Peroral inoculations (50 μl) were delivered intragastrically (69). Intracranial inoculations (5 μl) were delivered to the left cerebral hemisphere by using a Hamilton syringe and 30-gauge needle (80). For analysis of virulence, mice were monitored for weight loss and symptoms of disease for 21 days after inoculation and euthanized when found to be moribund (defined by rapid or shallow breathing, severe lethargy, or paralysis). For analysis of viral replication, mice were euthanized at various intervals following inoculation, and organs were harvested into 1 ml of PBS before freezing, thawing, and homogenization by sonication (1, 5, 18), using a midrange setting on a VirSonic 100 sonicator (VirTis). Organ sizes did not noticeably differ between genotypes of mice. For analysis of viremia, mice were euthanized and decapitated at various intervals following inoculation. Whole blood was collected from the neck into a 1 ml syringe containing 100 μl Alsever's solution (Sigma) and frozen, thawed, and sonicated. Viral titers in organ and blood homogenates were determined by a plaque assay (83). Animal husbandry and experimental procedures were performed in accordance with Public Health Service policy and approved by the Vanderbilt University School of Medicine Institutional Animal Care and Use Committee.
Treatment of mice with an inhibitor of cathepsin L.
Mice were inoculated intraperitoneally with approximately 100 μg/g average litter weight of CLIK-148 in dimethyl sulfoxide (DMSO) or DMSO alone in a volume of 10 μl. One hour following treatment, mice were inoculated perorally with PBS or reovirus T3SA+. Mice were subsequently treated with CLIK-148 daily for 7 days. Analysis of virulence was conducted for 21 days or mice were euthanized at 8 days postinoculation and organs resected for determination of viral titers by a plaque assay. Pups that had obvious injury from intraperitoneal injections or that died within 6 days postinoculation were eliminated from the study. CLIK-148 specificity in vitro and in vivo has previously been established (26, 37, 50, 88).
Statistical analysis.
For survival experiments, curves were obtained using the Kaplan-Meier method and compared using the log rank test. For experiments in which viral titers in an organ or blood sample were determined, the Mann-Whitney test was used to calculate two-tailed P values. This test is appropriate for experimental data that display a non-Gaussian distribution (66). Mann-Whitney analysis lacks the power of the t test, and therefore, statistical significance is achieved less frequently with this method. All statistical analyses were performed using GraphPad Prism software.
Histology.
Newborn mice, 2 to 4 days old, weighing approximately 2 g were inoculated perorally with purified reovirus virions diluted in PBS. At various intervals following inoculation, mice were euthanized, organs were resected, and a wedge of liver was removed for titer determination by a plaque assay. Remaining organs were incubated in 10% formalin at room temperature for 24 h, followed by incubation in 70% ethanol at room temperature. Fixed organs were embedded in paraffin, and 5-μm sections were prepared. Consecutively obtained sections were stained with hematoxylin and eosin for evaluation of histopathologic changes or processed for immunohistochemical detection of reovirus protein (56). The histology was reviewed by a pathologist blinded to the conditions of the experiment.
Quantitation of serum hepatic enzymes.
Newborn mice, 2 to 4 days old, weighing approximately 2 g were inoculated perorally with purified reovirus virions diluted in PBS. At various intervals following inoculation, mice were euthanized and decapitated. Blood samples were collected and allowed to coagulate, and sera were separated by centrifugation. Sera were stored at −20°C, protected from light, and submitted in batches to Charles River Research Animal Diagnostic Services (Wilmington, MA). A small wedge of liver was resected concurrently with blood collection for correlative titer determination by a plaque assay.
Growth of reovirus in vitro in the presence of cathepsin inhibitors.
Monolayers of L cells (2 × 105 cells) in 24-well plates were preincubated in medium supplemented to contain 10 to 100 μM CLIK-148 or 200 μM E64 for 4 h. The medium was removed, and cells were adsorbed with T3SA+ virions or ISVPs at a multiplicity of infection of 2 PFU per cell. After incubation at 4°C for 1 h, the inoculum was removed, cells were washed with PBS, and 1 ml fresh medium supplemented with CLIK-148 or E64 was added. After incubation at 37°C for 0 or 24 h, cells were frozen and thawed twice, and viral titers in cell lysates were determined by a plaque assay.
RESULTS
Processing of reovirus virions by purified cathepsin proteases.
Cathepsins B (23), L (23), and S (28) can catalyze reovirus disassembly in vitro and in certain types of cells. Reovirus strain T3SA+ is a reassortant virus that contains the S1 gene segment from strain T3C44-MA on the genetic background of T1L (4). Following peroral inoculation of newborn mice, T3SA+ replicates in the intestine and disseminates systemically from that site to the brain, heart, and liver (5). To determine whether T3SA+ is susceptible to cathepsins B, L, and S, we incubated purified virions with each enzyme over a time course and resolved the resultant digestion mixtures by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Following incubation for 24 h with each protease, we observed complete degradation of σ3 and cleavage of μ1 to δ (Fig. 1). Therefore, treatment of T3SA+ virions with cathepsin B, L, or S results in formation of ISVPs.
FIG. 1.
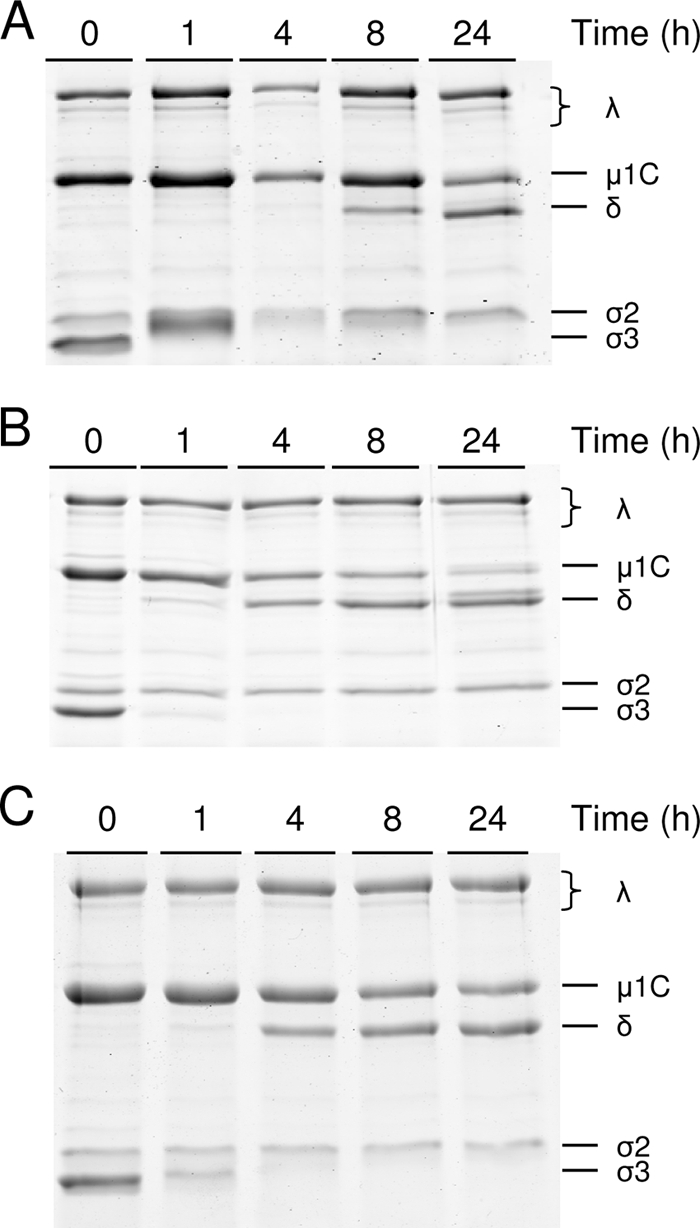
Treatment of reovirus virions with cathepsins B, L, and S. Purified virions of T3SA+ were treated with 400 μg/ml cathepsin B (A), 100 μg/ml cathepsin L (B), or 300 μg/ml cathepsin S (C) at 37°C for the times shown. Equal numbers of viral particles were loaded into wells of 10% polyacrylamide gels and electrophoresed. Viral proteins are labeled on the right.
Cathepsin deficiency differentially affects survival following reovirus infection.
To determine the function of cathepsin proteases in reovirus disease, we inoculated wt, Ctsb−/−, Ctsl−/−, and Ctss−/− mice perorally with 107 PFU of reovirus T3SA+. Mice were monitored for 21 days after infection for morbidity and mortality. The cathepsin-null mice displayed differential survival patterns in comparison to wt animals. Following a 21-day observation interval, the survival rate of Ctsb−/− animals was greater than that of wt mice; 82% of Ctsb−/− mice survived, in comparison to 61% for wt mice (Fig. 2A). In contrast, the survival rates of Ctsl−/− and Ctss−/− mice were decreased in comparison to that of wt animals; only 7% and 39% of Ctsl−/− and Ctss−/− mice, respectively, survived T3SA+ infection. However, the mean survival time for each strain of cathepsin-deficient mice was increased in comparison to that for wt animals (Table 1). The disease phenotypes in all strains did not notably differ, with all strains of mice displaying weakness and lethargy during the period of illness. All pups of wt and cathepsin-deficient strains inoculated with PBS and observed under identical conditions survived and did not display symptoms (data not shown). As a surrogate marker for disease severity, Ctsb−/− mice displayed approximately equivalent levels of weight gain in comparison to wt animals at the time of peak illness (Fig. 2B). However, Ctsl−/− and Ctss−/− mice had more weight loss than wt mice, and subsequent weight gain, indicative of recovery of these juvenile animals, was delayed. These findings suggest that despite the increased mortality of Ctsl−/− and Ctss−/− mice, these animals display slower kinetics of disease development. Thus, expression of cathepsins B, L, and S influences reovirus pathogenesis.
FIG. 2.

Survival of wt and cathepsin-deficient mice following peroral inoculation. C57BL/6 wt and cathepsin-deficient mice, 2 to 4 days (d) old, were inoculated perorally with 107 PFU T3SA+. Mice (n = 14 to 21) were monitored for survival (A) and weight gain (B). (A) *, P values of <0.01, as determined by a log rank test, in comparison to the wt level. (B) Results are expressed as mean weight of all living infected animals. Statistical significance (P values of <0.05 as determined by Student's t test) in comparison to the level for wt mice was achieved for Ctsb−/− mice between days 2 and 8; for Ctsl−/− mice at days 1, 13, and 14; and for Ctss−/− mice at days 4 to 8, 11, and 14.
TABLE 1.
Mean survival time following reovirus infectiona
| Genotype | No. of mice | Mortality (%)b | Mean survival time (days)c | Pd |
|---|---|---|---|---|
| wt | 21 | 38.1 | 9.0 ± 1.2 | |
| Ctsb−/− | 17 | 17.6 | 18.3 ± 0.9 | 0.0017 |
| Ctsl−/− | 15 | 93.3 | 12.6 ± 0.5 | 0.0041 |
| Ctss−/− | 13 | 61.5 | 13.3 ± 0.7 | 0.0098 |
C57/Bl6 wt and cathepsin-deficient mice, 2 to 4 days old, were inoculated perorally with 107 PFU T3SA+. Mice were monitored for survival for 21 days.
Percent animals dead after 21 days.
Mean survival time, as defined by the average day of death for animals that succumbed to infection, in number of days ± standard error of the mean.
P value, as determined by Student's t test, in comparison to the level for wt mice.
Peak reovirus titers are diminished in cathepsin-deficient mice.
To understand differences in susceptibility to reovirus infection among the cathepsin-deficient mice, we inoculated 3-day-old mice perorally with a lower dose of T3SA+, 102 PFU, and quantified viral growth in selected organs. In comparison to wt mice, Ctsb−/− mice demonstrated equivalent growths of reovirus in the intestine, the site of primary replication, at all time points tested (Fig. 3A). Although titers were lower at day 4 in the livers of Ctsb−/− mice, peak titers at day 8 were equivalent to those in wt mice. In contrast, peak titers in the hearts and brains of Ctsb−/− mice were lower than those in wt mice, reaching statistical significance at day 12 in the brain. In concordance with these results, Ctsb−/− mice exhibited greater weight gain than wt mice at day 12 (Fig. 4), suggesting that lower titers in the heart and brain are associated with diminished disease and lead to enhanced survival. In these experiments, neither wt nor Ctsb−/− mice displayed signs of illness or succumbed to infection.
FIG. 3.

Reovirus titers in organs of wt and cathepsin-deficient mice following peroral inoculation. C57BL/6 wt and Ctsb−/− (A), Ctsl−/− (B), and Ctss−/− (C) mice, 2 to 4 days (d) old, were inoculated perorally with 102 PFU T3SA+. Organs were resected at the times shown and homogenized by freeze-thawing and sonication. Viral titers in organ homogenates were determined by a plaque assay. Results are presented as mean viral titers in whole organs of 6 to 20 mice. Error bars represent standard errors of the means. *, P values of <0.05, as determined by a Mann-Whitney test, in comparison to the level for wt mice at the same time after inoculation. The limit of detection was 102 PFU/organ.
FIG. 4.

Weights of mice following peroral inoculation with T3SA+. C57BL/6 wt and cathepsin-deficient mice, 2 to 4 (d) days old, were inoculated perorally with 102 PFU T3SA+. Mice were weighed only on the day of harvest, as indicated. Results are displayed as the mean weights of 6 to 20 animals. *, P values of <0.05, as determined by Student's t test, in comparison to the level for wt mice at the same time after inoculation.
The trend displayed by Ctsl−/− mice differs from that of Ctsb−/− mice with respect to both peak titer and kinetics of disease progression. In contrast to what was found for Ctsb−/− mice, peak titers in all organs of Ctsl−/− mice were decreased in comparison to those for wt mice, reaching statistical significance in the heart at day 8 (Fig. 3B). However, viral clearance was delayed, as titers in Ctsl−/− mice were greater than those in wt mice at days 12, 16, and 20 postinoculation, reaching statistical significance in the intestine and liver at day 12. As expected from these results, the average weight of Ctsl−/− mice was significantly less than that of wt mice at day 20 (Fig. 4). Despite the lower dose of T3SA+ used in these experiments, several Ctsl−/− animals displayed overt signs of illness, including weakness and lethargy, and some died.
The kinetics of viral growth in Ctss−/− mice were similar to those in Ctsl−/− mice. Peak titers in all organs of Ctss−/− animals were lower than those in wt mice, reaching statistical significance in the liver at day 4, in the heart at days 4 and 8, and in the brain at day 8 (Fig. 3C). As in Ctsl−/− mice, viral titers did not decrease in Ctss−/− mice between days 8 and 12, and titers in the intestines of Ctss−/− mice were actually greater than those in wt mice at days 12 and 16. These data suggest that viral clearance was delayed in Ctss−/− mice. Also, like Ctsl−/− mice, Ctss−/− mice infected with T3SA+ displayed signs of illness, including weakness and lethargy, diminished weight gain (Fig. 4), and mortality. Thus, peak reovirus titers were decreased in all strains of cathepsin-deficient mice, but mice deficient in cathepsin L or cathepsin S had higher viral titers at late times after inoculation and increased disease severity.
Inflammation in the liver is more severe in cathepsin L- and cathepsin S-deficient mice.
To better understand why Ctsl−/− and Ctss−/− mice had greater susceptibility to reovirus infection than wt and Ctsb−/− mice, we compared histological sections of heart and liver from mice inoculated perorally with 106 PFU T3SA+. Animals chosen for histological analysis were matched for viral titer in the liver. Viral antigen staining in the hearts of all genotypes of mice localized primarily to the subepicardial myocardium, with little to moderate involvement of the deeper myocardium (data not shown). The extents of inflammatory cell infiltrates associated with infectious foci were consistent across all strains of mice.
In all mouse strains, infection in the liver centered on the bile duct epithelium (Fig. 5), as has been previously reported (5). Viral antigen and inflammation were more pronounced along large portal tracts; however, smaller portal tracts also were involved. Hepatic lobular involvement was present in all strains of mice in regions of increased reovirus antigen staining and inflammation. Although there was variability within each genotype of mice and some overlap between them, inflammation centered at the portal triads was more severe in Ctsl−/− and Ctss−/− than in wt and Ctsb−/− mice (Fig. 5, low-magnification panels showing increased numbers of leukocytes in panels C and D versus A and B).
FIG. 5.

Histological analysis of reovirus growth in the liver. C57BL/6 wt and cathepsin-deficient mice, 2 to 4 days old, were inoculated perorally with 106 PFU T3SA+. Livers were resected at day 8 postinfection, and a small wedge of liver was removed for titer determination by a plaque assay. The remaining liver was processed for histopathology, and consecutive sections were stained with hematoxylin and eosin or polyclonal reovirus antiserum. Representative samples from titer-matched livers are shown. Boxes indicate areas of enlargement shown in the panels on the right. Arrows indicate areas of inflammation. Original magnifications, ×10 and ×40. A, wt; B, Ctsb−/−; C, Ctsl−/−; D, Ctss−/−.
Liver enzyme levels are increased in cathepsin L- and cathepsin S-deficient mice.
To obtain biochemical evidence of biliary or hepatic injury in reovirus-infected mice, we assessed levels of total bilirubin (TBIL), alkaline phosphatase (ALK), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) in mice that were either mock infected or infected with 106 PFU T3SA+. No biomarker of liver injury was elevated in mock-infected mice at any time point or in any strain of reovirus-infected mice at day 8 postinfection (data not shown). In contrast, levels of TBIL and ALK were increased in Ctsl−/− mice and levels of all four markers were increased in Ctss−/− mice at day 12 postinfection (Table 2). These results indicate more-severe hepatobiliary damage in reovirus-infected Ctsl−/− and Ctss−/− mice than in wt and Ctsb−/− animals.
TABLE 2.
Bilirubin and liver enzyme levels in sera following reovirus infectiona
| Genotype | Concn in sera of indicated mouse group
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Mock infected
|
Reovirus infected
|
|||||||
| TBIL (mg/dl) | ALK (U/liter) | ALT (U/liter) | AST (U/liter) | TBIL (mg/dl) | ALK (U/liter) | ALT (U/liter) | AST (U/liter) | |
| wt | 1.5 | 634 | 127 | 461 | 0.9 (0.3) | 768 (140.8) | 295 (224.8) | 495 (231.2) |
| Ctsb−/− | 2.0 | 475 | 73 | 417 | 1.9 (0.2) | 783 (109.0) | 97.5 (31.1) | 460.5 (105.1) |
| Ctsl−/− | 1.5 | 567 | 57 | 409 | 9.2* (1.1) | 1282.4 (300.3) | 60 (20.8) | 439 (43.8) |
| Ctss−/− | 0.9 | 574 | 99 | 330 | 6.9* (2.0) | 1709* (258.9) | 858 (665.5) | 2083.75 (856.8) |
C57/Bl6 wt and cathepsin-deficient mice, 2 to 4 days old, were inoculated perorally with PBS alone or 106 PFU T3SA+. At day 12 postinoculation, the liver was resected and blood was collected. A small wedge of liver was removed for titer determination by a plaque assay. Mean values from two mock-infected mice or three to five reovirus-infected mice with approximately equivalent viral titers in the liver are shown. *, P values of <0.05, as determined by Student's t test, in comparison to the level for wt mice. The standard errors of the means are shown in parentheses.
Hematogenous dissemination of reovirus is unaffected by the absence of a single cathepsin protease following peroral inoculation.
We thought it possible that the capacity of reovirus to either disseminate to sites of secondary infection in cathepsin-deficient mice or establish infection once reaching those sites might be reduced. To distinguish between these possibilities, we quantified viral titers in blood samples following peroral inoculation. Newborn mice were inoculated perorally with 106 PFU of T3SA+, and blood samples were collected at various times after inoculation. Reovirus established viremia in all strains of mice, reaching peak titers that did not differ significantly (Fig. 6). Therefore, the lack of a single cathepsin protease does not appear to alter the efficiency of reovirus hematogenous dissemination in mice.
FIG. 6.

Viremia in wt and cathepsin-deficient mice following peroral inoculation. C57BL/6 wt and cathepsin-deficient mice, 2 to 4 days (d) old, were inoculated perorally with 106 PFU T3SA+. Blood samples were collected at the times shown and homogenized by freeze-thawing and sonication. Viral titers in blood samples were determined by a plaque assay. Results are presented as mean viral titers of 9 to 14 mice. The limit of detection was 50 PFU/ml.
Growth of reovirus in the brain is diminished in cathepsin L-deficient mice following intracranial inoculation.
To determine the effect of cathepsin deficiency on the capacity of reovirus to grow at a site of secondary replication, we quantified viral titers in the brain following intracranial inoculation of wt and cathepsin-deficient mice. This site was chosen for ease of direct inoculation. Titers of T3SA+ in the brains of wt, Ctsb−/−, and Ctss−/− mice did not differ statistically (Fig. 7), suggesting that cathepsins B and S are not required for reovirus growth in the brain following direct inoculation into that site. However, titers of T3SA+ in the brains of Ctsl−/− mice were significantly lower on day 9 postinoculation, suggesting that cathepsin L is required for efficient growth of reovirus in the brain.
FIG. 7.

Viral growth in the brains of wt and cathepsin-deficient mice following intracranial inoculation. C57BL/6 wt and cathepsin-deficient mice, 2 to 4 days (d) old, were inoculated intracranially with 102 PFU T3SA+. Brains were resected at the times shown and homogenized by freeze-thawing and sonication. Viral titers in brain homogenates were determined by a plaque assay. Results are expressed as mean viral titers in the brains of 9 to 13 mice. Error bars represent standard errors of the means. *, P values of <0.05, as determined by a Mann-Whitney test, in comparison to the level for wt mice at the same time after inoculation. The limit of detection was 102 PFU/brain.
Treatment with an inhibitor of cathepsin L promotes survival following reovirus infection.
Since titers of reovirus in organs of cathepsin-deficient mice are lower than those in wt mice, but mortality is increased in Ctsl−/− and Ctss−/− mice, we hypothesized that the immune defects accompanying genetic cathepsin L and cathepsin S deficiency might mask a cathepsin requirement for efficient viral growth. Therefore, we sought to determine whether reovirus disease could be ameliorated in wt mice by treatment with a cathepsin inhibitor. CLIK-148, a derivative of the pan-cysteine protease inhibitor E-64, inhibits cathepsin L in vivo (37, 38). Mice were treated with CLIK-148 at a dose of approximately 100 μg/g body weight via intraperitoneal injection 1 h prior to peroral inoculation with 107 PFU T3SA+ and then every 24 h for 7 days. In comparison to mice treated with vehicle alone, a significantly greater percentage of mice treated with CLIK-148 survived (Fig. 8A). At day 21, 40% of CLIK-148-treated mice succumbed, whereas 80% of vehicle-treated mice died. As a surrogate marker for disease, CLIK-148-treated mice exhibited only minimal weight loss, followed by substantial weight gain through the course of the experiment, whereas vehicle-treated mice exhibited more-severe weight loss and gained weight more slowly during the recovery phase (Fig. 8B). Mock-infected mice treated with CLIK-148 or vehicle alone showed no signs of drug toxicity and had survival rates of 100% (data not shown). These results suggest that treatment with a cathepsin L inhibitor dampens the severity of reovirus disease.
FIG. 8.

(A to C) Treatment with an inhibitor of cathepsin L decreases disease severity. C57BL/6 wt mice, 2 to 4 days (d) old, were inoculated intraperitoneally with CLIK-148 at a dose of 100 μg/g average litter body weight or vehicle control 1 h prior to peroral inoculation with 107 PFU (A, B) or 10 PFU (C) T3SA+. Mice were treated intraperitoneally with CLIK-148 thereafter for 7 days. Mice (n = 15) were monitored for survival (A) and weight gain (B). (A) *, P values of <0.05, as determined by a log rank test, in comparison to the level for vehicle-treated mice. (B) All living mice were weighed each day. (C) Mice (n = 9 or 10) were euthanized at day 8 postinoculation, and viral titers in organs were determined by a plaque assay. Error bars represent standard errors of the means. (D) CLIK-148 inhibits infection by virions but not by ISVPs. Monolayers of L cells were preincubated for 4 h in medium with or without CLIK-148 or E64 at the concentrations shown. The medium was removed, and cells were adsorbed with T3SA+ virions or ISVPs at a multiplicity of infection of 2 PFU per cell. After 1 h, the inoculum was removed, fresh medium with or without CLIK-148 or E64 was added, and cells were incubated for 0 or 24 h. Viral titers in cell lysates were determined by a plaque assay. The results are presented as mean viral yields, calculated by dividing the titer at 24 h by the titer at 0 h for each concentration of CLIK-148 or E64 for duplicate wells.
Treatment with an inhibitor of cathepsin L diminishes reovirus growth at sites of secondary replication.
To determine whether the cause of diminished mortality following treatment with CLIK-148 is related to viral growth, we quantified viral titers in various organs of wt mice treated with CLIK-148. Mice were treated with CLIK-148 at a dose of approximately 100 μg/g body weight via intraperitoneal injection 1 h prior to peroral inoculation with 10 PFU T3SA+ and then every 24 h for 7 days. On day 8 postinfection, organs were harvested and titers determined by a plaque assay. Although titers in vehicle-treated and CLIK-148-treated mice were equivalent in the intestine, titers were reduced at sites of secondary replication in the CLIK-148-treated mice in comparison to those in vehicle-treated mice (Fig. 8C). More striking, perhaps, was that of the five CLIK-148-treated animals with detectable titers in the intestine, only two exhibited virus dissemination to other organs. However, of the seven vehicle-treated mice with detectable titers in the intestine, six had disseminated virus. Thus, pharmacologic blockade of cathepsin L activity diminishes reovirus dissemination to sites of secondary replication.
Treatment with CLIK-148 inhibits reovirus entry into cells.
To define the block to infection imposed by CLIK-148, murine L cells were treated with various concentrations of CLIK-148 prior to infection with T3SA+ virions and in vitro-generated ISVPs. ISVPs are capable of penetrating cells at the plasma membrane and are resistant to inhibitors of proteolytic disassembly (2, 17, 19, 76, 86). In comparison to viral yields in untreated cells, yields in cells treated with 100 μM CLIK-148 were diminished 10-fold (Fig. 8D). Treatment with 200 μM E64, which blocks the activity of both cathepsin B and cathepsin L (3), diminished viral yields 100-fold. Yields produced following infection by ISVPs were not decreased in cells treated with either inhibitor, demonstrating that the block imposed by CLIK-148 occurs at a stage in viral replication prior to generation of ISVPs.
DISCUSSION
In this study, we found that cathepsins B, L, and S are individually required for development of peak viral titers at sites of secondary replication and thus influence reovirus disease (Table 3). Survival is enhanced in mice lacking cathepsin B but diminished in mice lacking cathepsin L or cathepsin S, likely reflecting the differential importance of these cathepsins in adaptive immunity. Importantly, treatment with an inhibitor of cathepsin L activity, which uncouples cathepsin functions in reovirus disassembly and immunity, enhances survival. These findings indicate a key role for cathepsin proteases in viral pathogenesis.
TABLE 3.
Reovirus pathogenesis in cathepsin-deficient mice
| Genotype | Survival rate | AST level | Level in mice inoculated perorally
|
Level of replication in brain in mice inoculated intracranially | ||||
|---|---|---|---|---|---|---|---|---|
| Replication in intestine | Replication in liver | Replication in heart | Replication in brain | Viremia | ||||
| Ctsb−/−b | ↑ | ↑ | ⇆ | ⇆ | ↓ | ↓ | ⇆ | ⇆ |
| Ctsl−/−b | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ⇆ | ↓ |
| Ctss−/−b | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ⇆ | ⇆ |
| wt plus CLIK-148c | ↑ | ND | ⇆ | ↓ | ↓ | ↓ | ND | ND |
↑, increase; ↓, decrease; ⇆, no change; ND, not determined.
Results shown are in comparison to the level for wt mice.
Results shown are in comparison to the level for wt mice treated with DMSO.
There are 11 cysteine proteases in the papain superfamily encoded by the human genome (8, 40, 82). Of these, several have been linked to disease in humans or animals. In addition to roles in Alzheimer's disease (31), atherosclerosis (42), cancer (49), and osteoporosis (81), cathepsins B, L, and S are important mediators of cell entry by several viruses (13, 23, 28, 36, 58). Although members of the cathepsin family show some redundancy of function, there exist specific roles for each protease. Therefore, we hypothesized that the proteases capable of mediating reovirus disassembly would serve nonredundant functions in reovirus pathogenesis by virtue of their specialized host functions.
Reovirus virions are uncoated in late endosomes or lysosomes by cathepsins B (23), L (23), or S (28). We reasoned that deficiency in the proteases that catalyze uncoating might lead to decreased viral growth and diminished disease severity. Only mice deficient in expression of cathepsin B fit this profile. Ctsb−/− mice had an increased survival rate in comparison to wt mice when infected with a high dose of reovirus, whereas Ctsl−/− and Ctss−/− mice displayed decreased survival rates. In concordance with this observation, reovirus produced lower titers at sites of secondary replication in Ctsb−/− mice than in wt mice, and Ctsb−/− mice displayed greater weight gain. These results suggest that cathepsin B is required for reovirus to establish high-titer infection and exert pathological effects.
The decreased survival rates of Ctsl−/− and Ctss−/− mice in comparison to that of wt mice raised the possibility that viral loads might be higher in these animals. However, reovirus produced lower peak titers at sites of secondary replication in both genotypes of mice in comparison to those in wt and Ctsb−/− mice. Diminished survival rates among Ctsl−/− and Ctss−/− mice are likely attributable to defects in immune function in these animals (15), with resultant failure to resolve viral infection. Indeed, viral titers in the intestine at day 12 were significantly higher in both Ctsl−/− and Ctss−/− mice than those in wt animals. As viral titers in wt mice decreased from day 8 to day 12, viral titers in Ctsl−/− and Ctss−/− mice did not. We think it most likely that the absence of functional CD4+ T cells leads to inefficient viral clearance. Consequently, tissue injury is sustained for a longer period of time. This conclusion is supported by the kinetics of survival following infection. Although Ctsl−/− and Ctss−/− mice die at higher frequency than wt mice, survival times of the cathepsin-deficient mice are prolonged. Thus, the immune deficiency displayed by mice lacking cathepsin L or cathepsin S is associated with enhanced reovirus virulence.
The paradox that Ctsl−/− and Ctss−/− mice display increased disease severity and mortality despite lower peak titers suggests that Ctsl−/− and Ctss−/− mice are more susceptible to the pathological effects of reovirus infection than are wt mice. The disease phenotype of these mice included lethargy, gallbladder distention, intestinal obstruction, and oily hair (5, 20, 87), a syndrome associated with viral replication in intrahepatic bile duct epithelium, biliary obstruction, and fat malabsorption (5, 59, 60, 62, 87). However, the disease symptoms did not include spastic movements of the extremities, overt seizures, or paralysis, indicative of encephalitis. We think that death of reovirus-infected animals lacking cathepsin L or cathepsin S resulted from damage to the liver and heart. Histological analysis of the heart did not reveal striking differences in pathological injury in the different strains of mice, perhaps due to the high viral loads in the hearts of all mice. However, the increase in inflammatory infiltrate surrounding portal triads, coupled with the results of liver enzyme profiling, supports the conclusion that damage to the liver contributed to the poor outcome of reovirus-infected Ctsl−/− and Ctss−/− mice.
The observation that cathepsin-deficient mice inoculated with reovirus had lower titers at sites of secondary replication prompted us to investigate how individual cathepsins promote viral pathogenesis. We envision two possibilities. First, cathepsin expression might allow the virus to disseminate systemically in the host. Second, cathepsin expression might be required for growth at sites of secondary replication once those sites are reached. All strains of mice had detectable virus in the blood, suggesting that cathepsin deficiency does not block reovirus spread from the intestine into the bloodstream. However, these proteases could be required for reovirus exit from the bloodstream into the surrounding tissues, for example, by promoting viral growth in endothelium or extravasation of infected lymphocytes from blood vessels into host tissues.
While cathepsins B and S are dispensable for reovirus growth in the brain, cathepsin L is not. Following intracranial inoculation, titers in Ctsl−/− mice were decreased in comparison to those in the other mouse strains tested. Cathepsin S is required for maximal growth of reovirus in the intestine, as titers of reovirus in the intestines of Ctss−/− mice are lower than those in all other strains. It is possible that decreased titers in the intestines of Ctss−/− mice are insufficient to allow efficient viral dissemination to sites of secondary replication. However, peak reovirus titers in the brains of Ctss−/− mice following peroral inoculation are greater than those in Ctsb−/− mice, suggesting that viral spread is independent of titer in the intestine. Since spread to the brain is less affected in Ctss−/− mice, and virus is present in the blood of Ctss−/− mice, cathepsin S may be important for reovirus growth at other sites of secondary replication, including the heart and liver, at which sites peak titers are less than those in wt mice. Cathepsin B also may be important for growth in the heart, as titers at that site in Ctsb−/− mice are decreased in comparison to those in wt mice, while titers in the intestine and liver are not. It is noteworthy that in mouse fibroblasts, although both cathepsin B and cathepsin L can mediate reovirus uncoating, cathepsin L is more efficient (23). Our data following both peroral and intracranial inoculation support this conclusion in that peak titers in all organs tested are lower in Ctsl−/− mice than in Ctsb−/− mice. These findings suggest that cathepsin L is required for efficient reovirus growth in tissues other than the intestine.
To eliminate the confounding variables present in the gene deletion experiments, we sought to determine the effect of a cathepsin inhibitor on reovirus pathogenesis. Mice treated with CLIK-148, which specifically inhibits cathepsin L (37, 38), had increased resistance to reovirus infection in comparison to vehicle-treated controls, as assessed by both survival and viral titers at sites of secondary replication. Because survival was increased in CLIK-148-treated mice but decreased in Ctsl−/− mice, we think that the inhibition of viral replication is specific and that immune functions are preserved. Furthermore, since the mice treated with CLIK-148 did not display defects in fur growth characteristic of Ctsl−/− mice (68), we conclude that pharmacologic inhibition of cathepsin L is not as complete as genetic ablation. Successful pharmacologic attenuation of reovirus disease with a cathepsin inhibitor raises the possibility that cathepsin inhibitor therapy could be effective for other viruses that require cathepsin proteolysis for cell entry. Ebola virus (13), Nipah virus (22, 57), Hendra virus (58), and severe acute respiratory syndrome coronavirus (36) utilize cathepsin proteases to enter cells. Treatment of cells with chloroquine inhibits Hendra and Nipah virus infection (64), most likely via interference with viral fusion glycoprotein processing by cathepsin L. The absence of inhibitor-associated toxicity in this study and others (37, 38), along with the efficacy of CLIK-148 treatment in the amelioration of disease (this study), suggests that cathepsin inhibitors should be evaluated for therapeutic efficacy against these viruses.
We have shown that cathepsin proteases are required for efficient reovirus infection in mice. Cathepsins promote optimal growth at specific sites of reovirus replication in the host and influence survival following reovirus infection. Organ-specific differences in infection of cathepsin-deficient mice highlight distinct roles for these proteases in vivo. A better understanding of the function of cathepsin proteases in the pathogenesis of viral infections should lead to novel therapeutics for a variety of important human pathogens.
Acknowledgments
We thank Mark Boothby, Graham Carpenter, Anne Kenworthy, and Borden Lacy for helpful suggestions. We thank Hal Chapman, Doug Hanahan, and Johanna Joyce for providing the mice used in these experiments. We thank Jessica Summers and the Vanderbilt University Division of Animal Care for expert animal husbandry. We thank Pam Wirth, Melissa Downing, and Frances Shook from the Vanderbilt Immunohistochemistry Core for sample preparation. We thank Guo-Ping Shi for help with CLIK-148 administration, Charles Cobb for fluorometry assistance, John Mort for provision of cathepsin L, and Greg Wilson for assistance with cathepsin digestion of virions.
This work was supported by Public Health Service awards T32 AI07611 (E.M.J.), T32 GM08554 (J.D.D.), and R01 AI32539 and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by Public Health Service awards P30 CA68485 for the Vanderbilt-Ingram Cancer Center and P60 DK20593 for the Vanderbilt Diabetes Research and Training Center.
Footnotes
Published ahead of print on 29 July 2009.
REFERENCES
- 1.Antar, A. A. R., J. L. Konopka, J. A. Campbell, R. A. Henry, A. L. Perdigoto, B. D. Carter, A. Pozzi, T. W. Abel, and T. S. Dermody. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baer, G. S., and T. S. Dermody. 1997. Mutations in reovirus outer-capsid protein σ3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J. Virol. 71:4921-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett, A. J., A. A. Kembhavi, M. A. Brown, H. Kirschke, C. G. Knight, M. Tamai, and K. Hanada. 1982. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 201:189-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton, E. S., J. L. Connolly, J. C. Forrest, J. D. Chappell, and T. S. Dermody. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276:2200-2211. [DOI] [PubMed] [Google Scholar]
- 5.Barton, E. S., B. E. Youree, D. H. Ebert, J. C. Forrest, J. L. Connolly, T. Valyi-Nagy, K. Washington, J. D. Wetzel, and T. S. Dermody. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Investig. 111:1823-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borsa, J., B. D. Morash, M. D. Sargent, T. P. Copps, P. A. Lievaart, and J. G. Szekely. 1979. Two modes of entry of reovirus particles into L cells. J. Gen. Virol. 45:161-170. [DOI] [PubMed] [Google Scholar]
- 7.Borsa, J., M. D. Sargent, P. A. Lievaart, and T. P. Copps. 1981. Reovirus: evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology 111:191-200. [DOI] [PubMed] [Google Scholar]
- 8.Bromme, D., and J. Kaleta. 2002. Thiol-dependent cathepsins: pathophysiological implications and recent advances in inhibitor design. Curr. Pharm. Des. 8:1639-1658. [DOI] [PubMed] [Google Scholar]
- 9.Bromme, D., A. Steinert, S. Friebe, S. Fittkau, B. Wiederanders, and H. Kirschke. 1989. The specificity of bovine spleen cathepsin S. A comparison with rat liver cathepsins L and B. Biochem. J. 264:475-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carmona, E., É. Dufour, C. Plouffe, S. Takebe, P. Mason, J. S. Mort, and R. Ménard. 1996. Potency and selectivity of the cathepsin L propeptide as an inhibitor of cysteine proteases. Biochemistry 35:8149-8157. [DOI] [PubMed] [Google Scholar]
- 11.Chandran, K., D. L. Farsetta, and M. L. Nibert. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J. Virol. 76:9920-9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandran, K., J. S. Parker, M. Ehrlich, T. Kirchhausen, and M. L. Nibert. 2003. The δ region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J. Virol. 77:13361-13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandran, K., N. J. Sullivan, U. Felbor, S. P. Whelan, and J. M. Cunningham. 2005. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang, C. T., and H. J. Zweerink. 1971. Fate of parental reovirus in infected cell. Virology 46:544-555. [DOI] [PubMed] [Google Scholar]
- 15.Chapman, H. A. 2006. Endosomal proteases in antigen presentation. Curr. Opin. Immunol. 18:78-84. [DOI] [PubMed] [Google Scholar]
- 16.Chapman, H. A., R. J. Riese, and G. P. Shi. 1997. Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 59:63-88. [DOI] [PubMed] [Google Scholar]
- 17.Clark, K. M., J. D. Wetzel, J. Bayley, D. H. Ebert, S. A. McAbee, E. K. Stoneman, G. S. Baer, Y. Zhu, G. J. Wilson, B. V. V. Prasad, and T. S. Dermody. 2006. Reovirus variants selected for resistance to ammonium chloride have mutations in viral outer-capsid protein σ3. J. Virol. 80:671-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danthi, P., C. M. Coffey, J. S. Parker, T. W. Abel, and T. S. Dermody. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the μ1 φ domain. PLoS Pathog. 4:e1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dermody, T. S., M. L. Nibert, J. D. Wetzel, X. Tong, and B. N. Fields. 1993. Cells and viruses with mutations affecting viral entry are selected during persistent infections of L cells with mammalian reoviruses. J. Virol. 67:2055-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derrien, M., J. W. Hooper, and B. N. Fields. 2003. The M2 gene segment is involved in the capacity of reovirus type 3Abney to induce the oily fur syndrome in neonatal mice, a S1 gene segment-associated phenotype. Virology 305:25-30. [DOI] [PubMed] [Google Scholar]
- 21.Deussing, J., W. Roth, P. Saftig, C. Peters, H. L. Ploegh, and J. A. Villadangos. 1998. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc. Natl. Acad. Sci. USA 95:4516-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diederich, S., L. Thiel, and A. Maisner. 2008. Role of endocytosis and cathepsin-mediated activation in Nipah virus entry. Virology 375:391-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebert, D. H., J. Deussing, C. Peters, and T. S. Dermody. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609-24617. [DOI] [PubMed] [Google Scholar]
- 24.Ehrlich, M., W. Boll, A. Van Oijen, R. Hariharan, K. Chandran, M. L. Nibert, and T. Kirchhausen. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591-605. [DOI] [PubMed] [Google Scholar]
- 25.Fleeton, M., N. Contractor, F. Leon, J. D. Wetzel, T. S. Dermody, and B. Kelsall. 2004. Peyer's patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J. Exp. Med. 200:235-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Funkelstein, L., T. Toneff, C. Mosier, S. R. Hwang, F. Beuschlein, U. D. Lichtenauer, T. Reinheckel, C. Peters, and V. Hook. 2008. Major role of cathepsin L for producing the peptide hormones ACTH, beta-endorphin, and alpha-MSH, illustrated by protease gene knockout and expression. J. Biol. Chem. 283:35652-35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furlong, D. B., M. L. Nibert, and B. N. Fields. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 62:246-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golden, J. W., J. A. Bahe, W. T. Lucas, M. L. Nibert, and L. A. Schiff. 2004. Cathepsin S supports acid-independent infection by some reoviruses. J. Biol. Chem. 279:8547-8557. [DOI] [PubMed] [Google Scholar]
- 29.Guicciardii, M. E., J. Deussing, H. Miyoshi, S. F. Bronk, P. A. Svingen, C. Peters, S. H. Kaufmann, and G. J. Gores. 2000. Cathepsin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 106:1127-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halangk, W., M. Lerch, B. Brandt-Nedelev, W. Roth, M. Ruthenbuerger, T. Reinheckel, W. Domschke, H. Lippert, C. Peters, and J. Deussing. 2000. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Investig. 106:773-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haque, A., N. L. Banik, and S. K. Ray. 2008. New insights into the roles of endolysosomal cathepsins in the pathogenesis of Alzheimer's disease: cathepsin inhibitors as potential therapeutics. CNS Neurol. Disord. Drug Targets 7:270-277. [DOI] [PubMed] [Google Scholar]
- 32.Honey, K., K. Benlagha, C. Beers, K. Forbush, L. Teyton, M. J. Kleijmeer, A. Y. Rudensky, and A. Bendelac. 2002. Thymocyte expression of cathepsin L is essential for NKT cell development. Nat. Immunol. 3:1069-1074. [DOI] [PubMed] [Google Scholar]
- 33.Honey, K., T. Nakagawa, C. Peters, and A. Rudensky. 2002. Cathepsin L regulates CD4+ T cell selection independently of its effect on invariant chain: a role in the generation of positively selecting peptide ligands. J. Exp. Med. 195:1349-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh, C. S., P. deRoos, K. Honey, C. Beers, and A. Y. Rudensky. 2002. A role for cathepsin L and cathepsin S in peptide generation for MHC class II presentation. J. Immunol. 168:2618-2625. [DOI] [PubMed] [Google Scholar]
- 35.Hsing, L. C., and A. Y. Rudensky. 2005. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 207:229-241. [DOI] [PubMed] [Google Scholar]
- 36.Huang, I.-C., B. J. Bosch, F. Li, W. Li, K. H. Lee, S. Ghiran, N. Vasilieva, T. S. Dermody, S. C. Harrison, P. R. Dormitzer, M. Farzan, P. J. Rottier, and H. Choe. 2006. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 281:3198-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katunuma, N., E. Murata, H. Kakegawa, A. Matsui, H. Tsuzuki, H. Tsuge, D. Turk, V. Turk, M. Fukushima, Y. Tada, and T. Asao. 1999. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 458:6-10. [DOI] [PubMed] [Google Scholar]
- 38.Katunuma, N., H. Tsuge, M. Nukatsuka, T. Asao, and M. Fukushima. 2002. Structure-based design of specific cathepsin inhibitors and their application to protection of bone metastases of cancer cells. Arch. Biochem. Biophys. 397:305-311. [DOI] [PubMed] [Google Scholar]
- 39.Kirschke, H., B. Wiederanders, D. Bromme, and A. Rinne. 1989. Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. Biochem. J. 264:467-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, N. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860-921. [DOI] [PubMed] [Google Scholar]
- 41.Lee, P. W. K., E. C. Hayes, and W. K. Joklik. 1981. Protein σ1 is the reovirus cell attachment protein. Virology 108:156-163. [DOI] [PubMed] [Google Scholar]
- 42.Lutgens, S. P., K. B. Cleutjens, M. J. Daemen, and S. Heeneman. 2007. Cathepsin cysteine proteases in cardiovascular disease. FASEB J. 21:3029-3041. [DOI] [PubMed] [Google Scholar]
- 43.Maehr, R., J. D. Mintern, A. E. Herman, A. M. Lennon-Dumenil, D. Mathis, C. Benoist, and H. L. Ploegh. 2005. Cathepsin L is essential for onset of autoimmune diabetes in NOD mice. J. Clin. Investig. 115:2934-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mann, M. A., D. M. Knipe, G. D. Fischbach, and B. N. Fields. 2002. Type 3 reovirus neuroinvasion after intramuscular inoculation: direct invasion of nerve terminals and age-dependent pathogenesis. Virology 303:222-231. [DOI] [PubMed] [Google Scholar]
- 45.Marsh, M., and A. Helenius. 2006. Virus entry: open sesame. Cell 124:729-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsunaga, Y., T. Saibara, H. Kido, and N. Katunuma. 1993. Participation of cathepsin B in processing of antigen presentation to MHC class II. FEBS Lett. 324:325-330. [DOI] [PubMed] [Google Scholar]
- 47.Meulendyke, K. A., M. A. Wurth, R. O. McCann, and R. E. Dutch. 2005. Endocytosis plays a critical role in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12643-12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mizuochi, T., S. T. Yee, M. Kasai, T. Kakiuchi, D. Muno, and E. Kominami. 1994. Both cathepsin B and cathepsin D are necessary for processing of ovalbumin as well as for degradation of class II MHC invariant chain. Immunol. Lett. 43:189-193. [DOI] [PubMed] [Google Scholar]
- 49.Mohamed, M. M., and B. F. Sloane. 2006. Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer 6:764-775. [DOI] [PubMed] [Google Scholar]
- 50.Moriyama, T., M. Wada, R. Urade, M. Kito, N. Katunuma, T. Ogawa, and R. D. Simoni. 2001. 3-hydroxy-3-methylglutaryl coenzyme A reductase is sterol-dependently cleaved by cathepsin L-type cysteine protease in the isolated endoplasmic reticulum. Arch. Biochem. Biophys. 386:205-212. [DOI] [PubMed] [Google Scholar]
- 51.Morrison, L. A., R. L. Sidman, and B. N. Fields. 1991. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. USA 88:3852-3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakagawa, T., W. Roth, P. Wong, A. Nelson, A. Farr, J. Deussing, J. A. Villadangos, H. Ploegh, C. Peters, and A. Y. Rudensky. 1998. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science 280:450-453. [DOI] [PubMed] [Google Scholar]
- 53.Nakagawa, T. Y., W. H. Brissette, P. D. Lira, R. J. Griffiths, N. Petrushova, J. Stock, J. D. McNeish, S. E. Eastman, E. D. Howard, S. R. Clarke, E. F. Rosloniec, E. A. Elliott, and A. Y. Rudensky. 1999. Impaired invariant chain degradation and antigen presentation and diminished collagen-induced arthritis in cathepsin S null mice. Immunity 10:207-217. [DOI] [PubMed] [Google Scholar]
- 54.Nibert, M. L., A. L. Odegard, M. A. Agosto, K. Chandran, and L. A. Schiff. 2005. Putative autocleavage of reovirus μ1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 345:461-474. [DOI] [PubMed] [Google Scholar]
- 55.Odegard, A. L., K. Chandran, X. Zhang, J. S. Parker, T. S. Baker, and M. L. Nibert. 2004. Putative autocleavage of outer capsid protein μ1, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J. Virol. 78:8732-8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Donnell, S. M., M. W. Hansberger, J. L. Connolly, J. D. Chappell, M. J. Watson, J. M. Pierce, J. D. Wetzel, W. Han, E. S. Barton, J. C. Forrest, T. Valyi-Nagy, F. E. Yull, T. S. Blackwell, J. N. Rottman, B. Sherry, and T. S. Dermody. 2005. Organ-specific roles for transcription factor NF-κB in reovirus-induced apoptosis and disease. J. Clin. Investig. 115:2341-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pager, C. T., W. W. Craft, Jr., J. Patch, and R. E. Dutch. 2006. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 346:251-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pager, C. T., and R. E. Dutch. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 79:12714-12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papadimitriou, J. M. 1968. The biliary tract in acute murine reovirus 3 infection. Am. J. Pathol. 52:595-601. [PMC free article] [PubMed] [Google Scholar]
- 60.Parashar, K., M. J. Tarlow, and M. A. McCrae. 1992. Experimental reovirus type 3-induced murine biliary tract disease. J. Pediatr. Surg. 27:843-847. [DOI] [PubMed] [Google Scholar]
- 61.Petanceska, S., P. Canoll, and L. A. Devi. 1996. Expression of rat cathepsin S in phagocytic cells. J. Biol. Chem. 271:4403-4409. [DOI] [PubMed] [Google Scholar]
- 62.Phillips, P. A., D. Keast, J. M. Papadimitriou, M. N. Walters, and N. F. Stanley. 1969. Chronic obstructive jaundice induced by reovirus type 3 in weanling mice. Pathology 1:193-203. [DOI] [PubMed] [Google Scholar]
- 63.Pluger, E. B., M. Boes, C. Alfonso, C. J. Schroter, H. Kalbacher, H. L. Ploegh, and C. Driessen. 2002. Specific role for cathepsin S in the generation of antigenic peptides in vivo. Eur. J. Immunol. 32:467-476. [DOI] [PubMed] [Google Scholar]
- 64.Porotto, M., G. Orefice, C. Yokoyama, B. Mungall, R. Realubit, M. Sganga, M. Aljofan, M. Whitt, F. Glickman, and A. Moscona. 2009. Simulating henipavirus multicycle replication in a screening assay leads to identification of a promising candidate for therapy. J. Virol. 83:5148-5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reinheckel, T., J. Deussing, W. Roth, and C. Peters. 2001. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol. Chem. 382:735-741. [DOI] [PubMed] [Google Scholar]
- 66.Richardson, B. A., and J. Overbaugh. 2005. Basic statistical considerations in virological experiments. J. Virol. 79:669-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Riese, R. J., G. P. Shi, J. Villadangos, D. Stetson, C. Driessen, A. M. Lennon-Dumenil, C. L. Chu, Y. Naumov, S. M. Behar, H. Ploegh, R. Locksley, and H. A. Chapman. 2001. Regulation of CD1 function and NK1.1(+) T cell selection and maturation by cathepsin S. Immunity 15:909-919. [DOI] [PubMed] [Google Scholar]
- 68.Roth, W., J. Deussing, V. Botchkarev, M. Pauly-Evers, P. Saftig, A. Hafner, P.Schmidt, W. Schmahl, J. Scherer, I. Anton-Lamprechet, K. Von Figura, R. Paus, and C. Peters. 2000. Cathepsin L deficiency as molecular defect of furless: hyperproliferation of keratinocytes and perturbation of hair follicle cycling. FASEB J. 14:2075-2086. [DOI] [PubMed] [Google Scholar]
- 69.Rubin, D. H., and B. N. Fields. 1980. Molecular basis of reovirus virulence: role of the M2 gene. J. Exp. Med. 152:853-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schiff, L. A., M. L. Nibert, and K. L. Tyler. 2007. Orthoreoviruses and their replication, p. 1853-1915. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 71.Shen, L., L. J. Sigal, M. Boes, and K. L. Rock. 2004. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 21:155-165. [DOI] [PubMed] [Google Scholar]
- 72.Shi, G. P., J. S. Munger, J. P. Meara, D. H. Rich, and H. A. Chapman. 1992. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J. Biol. Chem. 267:7258-7262. [PubMed] [Google Scholar]
- 73.Shi, G. P., A. C. Webb, K. E. Foster, J. H. Knoll, C. A. Lemere, J. S. Munger, and H. A. Chapman. 1994. Human cathepsin S: chromosomal localization, gene structure, and tissue distribution. J. Biol. Chem. 269:11530-11536. [PubMed] [Google Scholar]
- 74.Silverstein, S. C., C. Astell, D. H. Levin, M. Schonberg, and G. Acs. 1972. The mechanism of reovirus uncoating and gene activation in vivo. Virology 47:797-806. [DOI] [PubMed] [Google Scholar]
- 75.Simmons, G., D. N. Gosalia, A. J. Rennekamp, J. D. Reeves, S. L. Diamond, and P. Bates. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 102:11876-11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sturzenbecker, L. J., M. L. Nibert, D. B. Furlong, and B. N. Fields. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J. Virol. 61:2351-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tardieu, M., M. L. Powers, and H. L. Weiner. 1983. Age-dependent susceptibility to reovirus type 3 encephalitis: role of viral and host factors. Ann. Neurol. 13:602-607. [DOI] [PubMed] [Google Scholar]
- 78.Turk, V., B. Turk, and D. Turk. 2001. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 20:4629-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tyler, K. L., E. S. Barton, M. L. Ibach, C. Robinson, T. Valyi-Nagy, J. A. Campbell, P. Clarke, S. M. O'Donnell, J. D. Wetzel, and T. S. Dermody. 2004. Isolation and molecular characterization of a novel type 3 reovirus from a child with meningitis. J. Infect. Dis. 189:1664-1675. [DOI] [PubMed] [Google Scholar]
- 80.Tyler, K. L., R. T. Bronson, K. B. Byers, and B. N. Fields. 1985. Molecular basis of viral neurotropism: experimental reovirus infection. Neurology 35:88-92. [DOI] [PubMed] [Google Scholar]
- 81.Vasiljeva, O., T. Reinheckel, C. Peters, D. Turk, V. Turk, and B. Turk. 2007. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 13:387-403. [DOI] [PubMed] [Google Scholar]
- 82.Venter, J. C., M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Wang, A. Wang, X. Wang, J. Wang, M. Wei, R. Wides, C. Xiao, C. Yan, et al. 2001. The sequence of the human genome. Science 291:1304-1351. [DOI] [PubMed] [Google Scholar]
- 83.Virgin, H. W., IV, R. Bassel-Duby, B. N. Fields, and K. L. Tyler. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 62:4594-4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Virgin, H. W., K. L. Tyler, and T. S. Dermody. 1997. Reovirus, p. 669-699. In N. Nathanson (ed.), Viral pathogenesis. Lippincott-Raven, New York, NY.
- 85.Weiner, H. L., K. A. Ault, and B. N. Fields. 1980. Interaction of reovirus with cell surface receptors. I. Murine and human lymphocytes have a receptor for the hemagglutinin of reovirus type 3. J. Immunol. 124:2143-2148. [PubMed] [Google Scholar]
- 86.Wetzel, J. D., G. J. Wilson, G. S. Baer, L. R. Dunnigan, J. P. Wright, D. S. H. Tang, and T. S. Dermody. 1997. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J. Virol. 71:1362-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilson, G. A., L. A. Morrison, and B. N. Fields. 1994. Association of the reovirus S1 gene with serotype 3-induced biliary atresia in mice. J. Virol. 68:6458-6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang, M., Y. Zhang, J. Pan, J. Sun, J. Liu, P. Libby, G. K. Sukhova, A. Doria, N. Katunuma, O. D. Peroni, M. Guerre-Millo, B. B. Kahn, K. Clement, and G. P. Shi. 2007. Cathepsin L activity controls adipogenesis and glucose tolerance. Nat. Cell Biol. 9:970-977. [DOI] [PubMed] [Google Scholar]
- 89.Yasuda, Y., J. Kaleta, and D. Bromme. 2005. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv. Drug Deliv. Rev. 57:973-993. [DOI] [PubMed] [Google Scholar]