

Abstract
A three-step procedure for the synthesis of 2-arylpropionic acids (profens) from vinyl arenes in nearly enantiomerically pure form has been developed. Excellent yields (>97%), regioselectivities (>99%), and enantioselectivities (>97% ee) for the desired branched products were obtained in the asymmetric hydrovinylation reactions of vinyl arenes, and the products from these reactions were transformed into 2-arylpropionic acids via oxidative degradation. Subsequent Curtius or Schmidt rearrangements of these acids provided highly valued 1-arylethyl amines, including a prototypical primary amine with an α-chiral tertiary N-alkyl group, in very good yields.
Introduction
Non-steroidal antiinflammatory drugs (NSAIDs) are the most frequently prescribed analgesics with annual worldwide sales estimated to exceed $20 billion.1 Among these, 2-arylpropionic acids are the most important class of compounds. Biological activity of the 2-arylpropionic acids is largely, if not entirely, stereospecific with the (S)-enantiomer being more active2 in the inhibition of prostaglandin biosynthesis through non-selective inhibition of cyclooxygenase (both COX-1 and COX-2 enzymes).3 The enzymatic bioinversion of (R)-(−)-ibuprofen and other chiral 2-arylpropionic acids to the biologically active (S)-enantiomer has been proven to be a unidirectional process4 producing compounds with the desired analgesic effects. Flurbiprofen among other NSAIDs, has unique properties, as the (S)-enantiomer is an antiinflammatory agent and the (R)-enantiomer selectively targets γ-secretase lowering Aβ42,5 which may retard amyloid plaque formation in Alzheimer’s disease, without inducing COX inhibition. Currently, (R)-flurbiprofen is in Phase III clinical trials as a Selective Aβ42 Lowering Agent (SALA)6 under the trade name Flurizan® (generic name: tarenflurbil).
Our recent work in fine-tuning of hemi-labile phosphoramidites in the asymmetric hydrovinylation (HV)7a eliminated the isomerization and polymerization problems that in the past had plagued this important carbon-carbon bond forming reaction.7b As we begin to seek applications of asymmetric hydrovinylation reaction for natural product synthesis, transformations of the vinyl group are becoming important in these synthetic efforts. Oxidative cleavage of the 3-arylbutenes for the synthesis of NSAIDs is thus a natural progression of this work. Because of the diverse nature of the aryl units, this oxidative degradation was found to be more difficult than anticipated. For example, the most direct and practical ozonolysis route that works well for ibuprofen and flurbiprofen, was found not to be applicable for the electron-rich aromatic precursors of naproxen and fenoprofen. Thus we examined the most optimal oxidative methods for the syntheses of five of the most common 2-arylpropionic acids.8
Many amines bearing an α-chiral benzylic group are known to possess a wide range of therapeutic activities.9 Several successful drugs such as Zoloft® and Sensipar® belong to this class of compounds. Typically, chiral 1-arylethyl amines are produced via classical resolution of salts,10 asymmetric reduction (i.e. hydrogenation) of C=N bonds,11 and biotechnological transformations, such as transamination12 and enzymatic kinetic resolutions.13 As each of these methods for the production of chiral α-alkylbenzylamines has advantages, each also suffers from distinct disadvantages. New enabling technologies are needed when substrates are structurally complex, when the parent compounds have strongly electron-withdrawing groups attached, for amines carrying N-tertiary alkyl groups, and in cases where enzymes cannot produce the desired enantiomer of the amine with high selectivity. Access to enantiomerically pure 2-arylpropionic acids opens a facile route to these compounds via Curtius,14 Schmidt,15 or related reactions.
In this paper we provide the details of the syntheses of prototypical enantiopure 2-arylpropionic acids from 3-arylbutenes, and the subsequent transformations of these acids to 1-arylethyl amines.
Results and Discussion
Synthesis of 2-Arylpropionic Acids
During our initial investigations, mild, highly tolerant reaction conditions for the asymmetric hydrovinylation of vinylarenes were developed (Eq. 1).7,16 Using ligand L1 under the optimized conditions described Eq 1, the 3-arylbutenes were prepared. Racemic compounds were prepared in a reactions of ethylene and the vinylarenes of interest in the presence of a catalytic amount of [(allyl)NiBr]2, ligand L2 [an equimolar mixture of simplified ligand (RCRC) and (SCSC) enantiomers] and sodium tetrakis[(3,5-trifluoromethyl)phenyl]borate [NaBARF].
 |
(1) |
Initial experiments were conducted with 3-(4-isobutylphenyl)-1-butene (2), the precursor for ibuprofen (Scheme 1). In the original preparation of this alkene, complete conversion of 4-isobutylstyrene 1 to 2 was achieved in 98% yield and 96% ee after reaction with ethylene in the presence of 0.014 equiv. of a pre-catalyst produced from (S)-2,2’-binaphthoyl-benzyl-(R)-[l-(l-naphthylethyl)]aminoylphosphine (L1), [(allyl)NiBr]2 and NaBARF at −78°C (2 h).7a,16 Under these conditions, no (E) or (Z) olefins arising via isomerization of the primary product or by-products from oligomerization were observed. Hydrovinylation of a number of vinyl arenes, giving 3-arylbutenes 2–7, all precursors to 2arylpropionic acids, were carried out under optimal conditions and the results are shown in Table 1. In all cases, the conversions, yields, and selectivities were highly reproducible, with best values realized at low temperatures. In a typical example, selectivities of >96% ee and 92% ee were observed for the formation of 2 at −78 °C and at 0 °C respectively under otherwise identical conditions.
Scheme 1.

Synthesis of (S)-Ibuprofen from 4-Isobutylstyrene
Table 1.
Asymmetric Hydrovinylation of Vinylarenesa
| entry | product | conv.b | yieldc | selec.b,d | ee(%)e/conf.f |
|---|---|---|---|---|---|
| 1. | 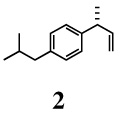 |
>99 | 97 | >99 | 96, R |
| 2. | 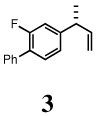 |
>99 | 97 | >99 | >97, R |
| 3. | 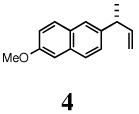 |
>99 | 98 | >99 | 99, R |
| 4. | 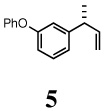 |
>99 | 98 | >99 | 97, R |
| 5. | 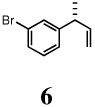 |
>99 | 99 | >99 | 91, R |
| 6. | 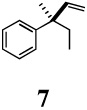 |
>99 | 99 | 99 | >97, R |
See experimental section for details.
Conversion and selectivities determined by GC analysis.
Yields determined by isolated mass after column chromatography.
Selectivity for the branched HV product.
Determined by GC except for 4, which was determined by HPLC.
Configuration assigned by comparison to GC and HPLC data with authentic samples (2 and 4). Others assigned by analogy.
To obtain the 2-arylpropionic acids from the branched HV products, we envisioned oxidative cleavage of the olefin followed by further oxidation of the intermediate aldehyde to the corresponding carboxylic acid (Scheme 1). A variety of methods are available for such oxidations.17 The enantiomeric excess of (S)-ibuprofen was verified by three independent methods (chiral stationary phase GC of 2 and (L)-(−)-menthyl esters 10, [α]D of 9) to be within experimental error at two different stages in the three step synthesis from 4-isobutylstyrene. Ozonolysis of 2 in a solution of CH2Cl2 and pyridine followed by reductive workup afforded the desired aldehyde (8), which was treated with NaClO2 to afford (S)-ibuprofen (9) in 92% yield (97% ee) over three steps (Scheme 1). The configuration and enantiomeric excess were established by conversion to the known (L)-menthyl esters.18 Gas chromatographic analysis of the (L)-menthyl esters (10) of ibuprofen using a Cyclodex-β column revealed baseline separation, with a diastereomeric excess of 96% for the (S)-ibuprofen ester. This confirms the overall selectivity and absolute configuration of the hydrovinylation product. Oxidative degradation using RuCl3/NalO4 gave 96% of the acid from 2.
Table 2 lists the conditions for the oxidative cleavage of all the hydrovinylation products. Only compounds 2, 3 and 6 underwent clean ozonation and subsequent oxidation under modified Pinnick conditions to afford the acids without epimerization (Table 2, entries 1,2 and 5). Substrates containing electron-rich aryl units, 4 and 5 proved to be incompatible with ozonolysis, presumably due to over oxidation of the aromatic ring, even when controlled ozonolysis was attempted.19 Hiyama has reported that olefins in proximity to electron-rich aromatic systems could be oxidatively cleaved and subsequently oxidized to the desired carboxylic acid at low temperatures with KMnO4.20 This method proved most successful in the oxidation of 4 to form naproxen, 12. Other common oxidants led to substrate decomposition. Synthesis of (S)-fenoprofen from 5 (entry 4) was best achieved under Sharpless’ improved conditions for ruthenium tetroxide-catalyzed oxidations of organic compounds,17c which afforded 13 in 91% overall yield from the 3-phenoxystyrene. Ibuprofen and flurbiprofen can also be made by using RuCl3, even though the ozonolysis route has clear advantages if large scale synthesis is to be attempted. For 5, the fenoprofen precursor, KMnO4/NaIO4 oxidation gave only 40% yield. The acid 15 is best prepared by the RuCl3-mediated oxidation of the corresponding alkene.
Table 2.
Oxidation of Hydrovinylation Products. Synthesis of 2-Arylpropionic Acidsa
| entry | alkene | product | yieldb | ee(%)c/oxidation method | ||
|---|---|---|---|---|---|---|
| RuCl3, NalO4 | O3, NaClO2 | KMnO4, NalO4 | ||||
| 1. | 2 | 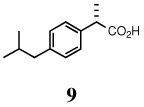 |
96 | 98 | 78 | 96/ozone |
| 2. | 3 | 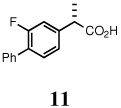 |
91 | 95 | 68 | 97/ozone |
| 3. | 4 | 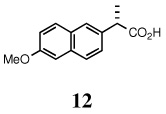 |
59 | --d | 67 | 99e/RuCl3 |
| 4. | 5 | 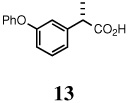 |
91 | --d | 40 | 97/RuCl3 |
| 5. | 6 | 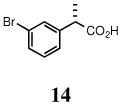 |
--d | 79 | 93 | 91/KMnO4 |
| 6. | 7 | 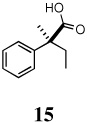 |
84 | -- | -- | 97/RuCl3 |
See experimental section for details.
Yields determined by isolated mass after column chromatography.
Determined by GC.
decomposition of starting alkene.
Enantiomeric excess assigned by comparison to literature rotation (ref. 21)
We have reported that 3-(bromoaryl)-butenes can serve as precursors for 2-arylpropionic acids with substituents on the aromatic ring. For example, 3-(4-bromophenyl)-1-butene has been converted into ibuprofen by Ni-catalyzed cross-coupling (Kumada Coupling) with i-butylmagnesium bromide followed by oxidation.18 Likewise, 3-(3-bromophenyl)-1-butene (6), prepared in >99% yield by asymmetric hydrovinylation of 3-bromostyrene can serve as a precursor for another important 2-arylpropionic acid, (S)-ketoprofen. A viable sequence for the synthesis of ketoprofen from the 3-bromoaryl-butene in the racemic series is shown in Scheme 2. Lithium halogen exchange reaction, followed by the addition of benzaldehyde gives the diastereomeric alcohol(s) 16. Subsequent global oxidation of the alcohols without further purification led to ketoprofen (17) in 72% overall yield in three steps from commercially available materials.
Scheme 2.

Synthesis of Ketoprofen from 3-Bromostyrene
Synthesis of 1-Arylethyl Amines
Curtius14 and Schmidt15 reactions are widely used methods for the conversion of acids to amines with one less carbon atom. These methods were chosen as each allows for the synthesis of either the free amine, or a number of other synthetically and pharmaceutically useful derivatives including carbamates, ureas, and amides. Further, the configuration of the α-carbon of the amine derivative is reliably predictable since the migration to the electron-deficient nitrogen proceeds with retention of configuration. Thus, (S)-2-methyl-2-phenylbutanoic acid (15), prepared from the corresponding hydrovinylation adduct 7 (Table 2, entry 6), upon reaction with diphenylphosphoryl azide22 in benzene, followed by capture of the isocyanate with benzyl alcohol, yields the CBZ-derivative 18 in 81% yield (Eq 2). Schmidt reaction of the acid 15 with hydrazoic acid gives the corresponding free amine upon aqueous work up. This product was trapped and identified as the corresponding propionamide 19. Products 20–22 were also prepared by similar routes. Careful HPLC analysis of 22 was conducted to guarantee that the configuration of the asymmetric carbon of 3 as derived from the hydrovinylation remained unaltered both in the oxidative cleavage to 11 and the subsequent rearrangement to the amide. The true advantage of this procedure lies in the ability to generate quaternary heteroatom-substituted chiral centers and in the ability to create chiral 1-artylethyl amines with a wide variety of structural and electronic features for the generation of analog libraries for pharmaceutical evaluation.
 |
(2) |
Conclusions
Enantiopure 2-arylpropionic acids ibuprofen, naproxen, flurbiprofen, and fenoprofen were prepared from the corresponding 3-arylbutenes by oxidative degradation of the vinyl group. This approach represent the highest overall selectivity obtained in the enantioselective syntheses of the title compounds that does not rely upon classical resolution or dynamic kinetic resolution.23 Curtius and Schmidt rearrangements of the carboxylic acids also produce the corresponding 1-aryletheyl amine derivatives in high enantiomeric purity.
EXPERIMENTAL
General methods
Typical procedure for asymmetric hydrovinylation7a,16
The pre-catalyst was prepared as follows in a glovebox: To [di(µ-bromo)bis(η-allyl)nickel(II)24 (2.6 mg, 0.007 mmol for substrate 1) in CH2Cl2 (1 mL) was added a solution of ligand (L1, 0.014 mmol for substrate 1) in CH2Cl2 (1 mL) at ambient temperature. The resulting solution was added to a suspension of NaBARF25,16 (12.4 mg, 0.014 mmol for 4-isobutylstyrene) dissolved in dichloromethane (1 mL) and the mixture was stirred at ambient temperature in a 10 mL pear shaped flask for 2 h affording a dark brown solution containing a small amount of fine particulate (NaBr).
In a fumehood, a 25 mL three-necked round bottomed flask equipped with a rubber septum, flow-controlled nitrogen inlet, a temperature probe, and a magnetic stirring bar was flame-dried and purged with nitrogen. The flask was then charged with 5 mL of dry dichloromethane. The catalyst solution prepared above now removed from the drybox, was introduced to the vessel via cannula. The flask containing the catalyst solution was further rinsed with 2 mL of CH2Cl2, and this solution was also transferred to the reaction mixture. Upon completion of pre-catalyst transfer, the system was closed at the flow-controlled stopcock and cooled to −78 °C, creating a vacuum. Dry ethylene (passed through a 0.5” × 4” column of Drierite®) was introduced via needle through the serum stopper and the vessel atmosphere was slowly evacuated 3 times with a 20 mL syringe. After cooling the solution to −78 °C, a solution of p-isobutylstyrene (160 mg, 1.00 mmol) in 2 mL dry CH2Cl2 was introduced dropwise into the solution of pre-catalyst over a one minute period via syringe followed by a 1 mL rinse with CH2Cl2. The vessel was then maintained at −78 °C for a period of 1 h. At the end of this period, the ethylene line is removed and the system exposed to nitrogen. Deionized H2O (1 mL) was introduced into the flask, the septum was pierced with a needle, and nitrogen was blown through the flask to remove any remaining ethylene. The biphasic solution was then poured into a 100-mL separatory funnel containing 20 mL H2O and the CH2Cl2 was collected. The aqueous layer was then extracted with three 10 mL portions of diethyl ether. The organic layers were combined, dried, filtered through a sintered glass funnel and the volume was reduced by rotary evaporation, yielding a free-flowing yellow oil. The resulting oil was filtered by flash column chromatography (eluted with isocratic pentane) to afford the desired crude hydrovinylation product as a colorless oil, which was then used to acquire all analytical data without further purification.
1-Isobutyl-4-[(R)-1-methylallyl]benzene (2)
clear liquid, 1H NMR (400 MHz, CDCl3): δ 7.13-7.07 (m, 4H), 6.05-5.97 (m, 1H), 5.07-5.00 (m, 2H), 3.48-3.41 (m, 1H), 2.44 (d, J = 6.80 Hz, 2H), 1.85 (septet, J = 6.80 Hz, 1H), 1.35 (d, J = 7.20 Hz, 3H), 0.90 (d, J = 6.80 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 143.5, 142.7, 139.4, 129.1, 126.9, 112.8, 45.0, 42.8, 30.2, 22.4, 20.7. [α]D22 = −9.1 ± 0.02 (c 0.50, CHCl3). HRMS 188.1562 (M+; calcd for C14H2O 188.1565). GC [poly(dimethylsiloxane] conditions: 5 min at 100°C, 5°C/min, 5 min at 200°C; retention time (min): 15.23. GC (chiral) conditions: (Cyclodex-β) 45 min at 100°C (isothermal); retention time (min): 37.72 (R), 38.37 (S).
2-Fluoro-1-phenyl-4-[(R)-1-methylallyl]benzene (3)
clear liquid, 1H NMR (400 MHz, CDCl3):δ 7.56-7.54 (m, 2H), 7.46-7.42 (m, 2H), 7.38-7.34 (m, 2H), 7.08 (dd, J = 7.60, 1.60 Hz, 1H), 7.02 (dd, J = 8.00, 1.60 Hz, 1H), 6.06-5.97 (m, 1H), 5.14-5.08 (m, 2H), 3.53-3.50 (m, 1H), 1.40 (d, J = 6.80 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 161.0, 147.3, 142.4, 135.8, 130.6, 130.5, 129.0, 128.9, 128.4, 127.4, 123.2, 114.9, 114.7, 113.8, 42.7, 20.6. [α]D22 = −18.7 ± 0.02 (c 1.00, CHCl3). HRMS 249.1051 (M+’; calcd for C16H15F + Na 249.1055). GC [poly(dimethylsiloxane] conditions: 5 min at 100°C, 5°C/min, 5 min at 200°C; retention time (min): 25.14. GC (chiral) conditions: (Cyclodex-β) 120 min at 130°C (isothermal); retention time (min): 91.98 (R), 93.24 (S).
2-Methoxy-6-[(R)-1-methylallyl]naphthalene (4)
Crystalline solid, mp <10 °C; 1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 9.60 Hz, 2H), 7.58 (s, 1H), 7.33 (dd, J = 8.40, 1.60 Hz, 1H), 7.15-7.12 (m, 2H), 6.13-6.04 (m, 1H), 5.13-5.06 (m, 2H), 3.92 (s, 3H), 3.62-3.59 (m, 1H), 1.45 (d, J = 7.20 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 157.3, 143.3, 140.7, 133.2, 129.1, 129.0, 126.8, 126.7, 125.0, 118.7, 113.2, 105.6, 55.3, 43.0, 20.7. [α]D22 = −18.9 ± 0.02 (c 1.00, CHCl3). HRMS 212.1207 (M+; calcd for C15H160 212.1201). GC [poly(dimethylsiloxane] conditions: 5 min at 100°C, 10°C/min, 10 min at 250°C; retention time (min): 18.90. HPLC (chiral) conditions: (Chiracel OJ-H) hexanes:isopropanol = 95:5, 0.50 mL/min; retention time (min): 25.52 (R), 26.80 (s).
l-Phenoxy-3-[(R)-1-methylallyl]benzene (5)
Clear liquid, 1H NMR (400 MHz, CDCl3): δ 7.34 (t, J = 7.80 Hz, 2H), 7.26 (t, J = 7.80 Hz, 1H), 7.10 (t, J = 7.40 Hz, 1H), 7.01 (d, J = 7.60 Hz, 2H), 6.97 (d, J = 8.00 Hz, 1H), 6.92 (s, 1H), 6.83 (dd, J= 8.00, 2.40 Hz, 1H), 6.03-5.95 (m, 1H), 5.08-5.03 (m, 2H), 3.47-3.44 (m, 1H), 1.35 (d, J = 6.80 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 157.3, 157.2, 147.8, 142.8, 129.7, 129.6, 123.0, 122.2, 118.7, 118.0, 116.5, 113.4, 43.1, 20.7. [α]D22 = −7.8 ± 0.02 (c 1.00, CHCl3). HRMS 224.1197 (M+; calcd for C16H160 224.1201). GC [poly(dimethylsiloxane] conditions: 5 min at 100°C, 5°C/min, 5 min at 250°C; retention time (min): 24.81. GC (chiral) conditions: (Cyclodex-β) 100 min at 100°C, 0.3°C/min, 91.67 min at 125°C; retention time (min): 215.68 (R), 216.85 (S).
l-Bromo-3-[(R)-1-methylallyl]benzene (6)
Clear liquid, 1H NMR (400 MHz, CDCl3): δ 7.36-7.32 (m, 2H), 7.19-7.14 (m, 2H), 6.00-5.92 (m, 1H), 5.09-7.07 (m, 1H), 5.06-5.04 (m, 1H), 3.47-3.41 (m, 1H), 1.35 (d, J = 6.80 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 147.9, 142.3, 130.4, 130.0, 129.2, 126.0, 122.5, 113.8, 42.9, 20.6. [α]D22 = −23.3 ± 0.02 (c 1.00, CHCl3). HRMS 210.0040 (M+; calcd for C10H11Br 210.0044). GC [poly(dimethylsiloxane] conditions: 5 min at 100°C, 5°C/min, 5 min at 200°C; retention time (min): 20.38. GC (chiral) conditions: (Cyclodex-β) 70 min at 85°C (isothermal); retention time (min): 54.01 (R), 55.81 (S).
(R)-3-Methyl-3-phenyl-1-pentene (7)
Clear liquid, 1H NMR (500 MHz, CDCl3): δ 7.34-7.28 (m, 4 H), 7.21-7.16 (m, 1 H), 6.03 (dd, J = 17.40, 10.40 Hz, 1 H), 5.10 (dd, J = 10.70, 1.20 Hz, 1 H), 5.04 (dd, J = 17.60, 1.20 Hz, 1 H), 1.89-1.72 (ABX3; νAB = 0.035, νA = 1.84, vB = 1.77; JAB = 13.70 Hz, JAX = 7.40 Hz, JBx = 7.40 Hz; 2 H), 1.35 (s, 3H), 0.77 (t, J = 7.40 Hz, 3 H). 13C NMR (125 MHz, CDCl3): δ 147.4, 146.9, 128.0, 126.7, 125.7, 111.7, 44.5, 33.4, 24.4, 8.9. [α]D22 = −22.3 (c 1.05, CHCl3). HRMS 160.1250 (M+; calcd for Cl2Hl6 160.1252). GC [poly(dimethylsiloxane] conditions: 5 min at 100°C, 5°C/min, 5 min at 200°C, retention time (min): 10.61. GC (chiral) conditions: (Cyclodex-β) 40 min at 70°C, 5°C/min, 10 min at 90°C, retention time (min): 53.90 (R), 55.65 (S).
Typical procedure for the oxidation of 2-arylpropionic acid precursors
Procedure 126
A solution of 1-isobutyl-4-[(R)-1-methylallyl]benzene (100 mg, 0.53 mmol) in dichloromethane-pyridine (9:1, 30 mL) was cooled to −78°C, and ozone was passed through the solution until the blue color persisted. It was stirred for 20 min at −78°C, while nitrogen was passed through the solution to remove excess ozone, then dimethylsulfide (0.5 mL) was added to the mixture. The resulting mixture was permitted to warm to 0°C, and stirring was continued for 1 h at 0°C and for another hour at ambient temperature. It was concentrated under reduced pressure, diluted with water, and extracted with diethyl ether (3 × 15 mL). The organic extract was washed with saturated aqueous sodium bicarbonate, dried over magnesium sulfate, and concentrated to dryness in vacuo to afford 106 mg of the intermediate aldehyde as a colorless oil. The crude product was used for the next step without further purification.
To a solution of crude aldehyde in diethyl ether (3 mL) was added 2,3-dimethyl-2-butene (2 mL) and was cooled to 0°C. Sodium chlorite (90mg, 1.00 mmol) which had been finely powdered, was added and the resulting mixture was stirred vigorously. The reaction mixture was allowed to warm to ambient temperature and was stirred for a further 15 minutes, at which time water (5 mL) was added and stirred for three minutes. Sulfuric acid (3 mL of a 10% v:v solution) was added and the solution was stirred for an additional ten minutes before the slurry was extracted into diethyl ether (3×10 mL). The combined organic extracts were dried over anhydrous MgSO4, filtered through a sintered glass funnel, and concentrated to afford 107 mg (98%) of (S)-(+)-ibuprofen.
Procedure 217c
A solution of 1-Phenoxy-3-[(R)-1-methylallyl]benzene (90 mg, 0.40 mmol) in 1:2 CCl4:H2O (lOmL) was added NalO4 (0.427 g, 2.0 mmol). A solution of RuCl3-H2O (0.80 mg, 0.004 mmol) in CH3CN (4 mL) was added in a dropwise fashion to the biphasic solution containing the olefin. The reaction was stirred at ambient temperature for 3 h at which time it was poured into a 100 mL separatory funnel containing CH2Cl2 (20 mL) and 10% H2SO4 (20 mL). The DCM was drained off and the remaining aqueous solution was extracted with DCM (2 × 15 mL). The resulting organic layers were combined, dried over anhydrous MgSO4, filtered, and concentration by rotary evaporation. The crude material was purified by column chromatography (6:1 hexanes/ethyl acetate) to afford (S)-fenoprofen (88 mg, 91%).
Procedure 327
To a solution of 2-(3-bromophenyl)-1-butene (100 mg, 0.46 mmol) in 1:2 t-BuOH/H2O (36 mL), was added KMnO4 (216 mg, 1.38 mmol), NalO4 (1.76 g, 8.2 mmol) and K2CO3 (446 mg, 3.2 mmol). The pH of the reaction solution was adjusted to 8 with 2 M KOH aqueous solution. The reaction was stirred for 3 h at ambient temperature. Concentrated HCl was added to adjust the pH of the solution to 1, and NaHSO3 was added to reduce KMnO4 until the reaction mixture turned yellow. The mixture was extracted with dichloromethane and the CH2Cl2 layer was extracted with 2 M KOH aqueous solution (5 mL). The aqueous layer was acidified with 10% H2SO4 and then was extracted with CH2Cl2 (3 × 15 mL). The dried organic layer was evaporated and the product was subjected to flash column chromatography (10:1 CH2Cl2/MeOH) to afford 2-(3- bromophenyl)-propionic acid (100 mg, 93%).
(S)-(+)-ibuprofen (9)
White solid, mp 76–77 °C; 1H NMR (400 MHz, CDCl3): δ 11.13 (br s, 1H), 7.22 (d, J = 8.00 Hz, 2H), 7.10 (d, J = 8.00 Hz, 2H), 3.71 (q, J = 7.20 Hz, 1H), 2.44 (d, J = 7.20 Hz, 2H), 1.85 (septet, J = 6.80 Hz, 1H), 1.50 (d, J = 7.20 Hz, 3H), 0.89 (d, J= 6.80 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 181.3, 140.8, 137.0, 129.4, 127.3, 45.0 (2), 30.1, 22.4, 18.1. [α]D22 = 55.1 ± 0.02 (c 1.00, CHCl3). HRMS 229.1200 (M+; calcd for C13H18O2 + Na 229.1204). To ascertain the absolute configuration and the enantioselectivity, the crude product (0.5 mg in 100 µL of dichloromethane) was converted to the corresponding ibuprofen (−)-menthyl esters by mixing with 100 µL of an esterification solution, prepared as described below, and then stirring the resulting mixture for 30 min. The esterification solution was prepared by dissolving 3.5 g of (−)-menthol, 0.12 g of dicyclohexylcarbodiimide, 6 mg of 4-dimethylaminopyridine, and 25 µL of 1M HCl in 1 mL of dichloromethane. GC (chiral stationary phase, Cyclodex-P) conditions for the (L)-menthyl esters, 60 min at 160°C (isothermal): retention time (min): 39.55 (S), 42.55 (R). The major acid isomer was identified as R-ibuprofen (96% ee) by comparison of retention times with that of authentic samples. The diastereomeric menthyl esters were prepared from commercially available racemic ibuprofen using the esterification solution. A slight preference for the formation of one of the diastereomeric esters was noticed in this preparation.
(S)-(+)-flurbiprofen (11)
White solid, mp 115–117 °C; 1H NMR (400 MHz, CDCl3): δ 11.48 (br s, 1H), 7.55-7.52 (m, 2H), 7.46-7.35 (m, 4H), 7.19-7.14 (m, 2H), 3.79 (q, J = 7.20 Hz, 1H), 1.57 (d, J = 7.20 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 179.9, 140.9, 135.4, 130.9, 128.9, 128.4, 127.7, 123.7, 115.5, 115.3, 44.8, 18.0. [α]D22 = 41.2 ± 0.02 (c 1.00, CHCl3). HRMS 244.0904 (M+; calcd for C15H13O2F 244.0900). GC (chiral) conditions, (L)-menthyl esters: (Chirasil-S-Val) 120 min at 200°C (isothermal); retention time (min): 82.24 (R), 87.37 (S).
(S)-(+)-naproxen (12)
White solid, mp 154–155 °C; 1H NMR (400 MHz, CDCl3):δ 7.72 (s, 1H), 7.69 (s, 2H), 7.42 (dd, J = 8.40, 1.60 Hz, 1H), 7.15 (dd, J = 8.80, 1.60 Hz, 1H), 7.11 (d, J = 2.00 Hz, 1 H), 3.92 (s, 3H), 3.88 (q, J = 7.20 Hz, 1H), 1.60 (d, J = 7.20 Hz). 13C NMR (100 MHz, CDCl3): δ 180.8, 157.7, 134.8, 133.8, 129.3, 128.9, 127.2, 126.2, 126.1, 119.0, 105.6, 55.3, 45.3, 18.1. [α]D22 = 66.5 ± 0.02 (c 1.00, CHCl3). HRMS 248.1045 (M+; calcd for C14H14O3 + H2O 248.1049).
(S)-(+)-fenoprofen (13)
Beige solid, mp 169–170 °C; 1H NMR (400 MHz, CDCl3): δ 11.48 (br s, 1H), 7.33-7.28 (m, 2H), 7.25-7.20 (m, 2H), 7.09 (t, J = 7.20 Hz, 1H), 7.04 (d, J = 7.60 Hz, 1H), 7.01-6.97 (m, 3H), 6.87 (dd, J = 8.00, 2.40 Hz, 1H), 3.69 (q, J = 7.20 Hz, 1H), 1.49 (d, J = 7.20 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 180.5, 157.5, 156.9, 141.6, 129.9, 129.7, 123.4, 122.3, 119.0, 118.2, 117.5, 45.2, 18.0. [α]D22 = 22.3 ± 0.02 (c 0.50, CHCl3). HRMS 265.0836 (M+; calcd for C15H14O3 + Na 265.0841). GC (chiral) conditions, (L)-menthyl esters: (Chirasil-S-Val) 120 min at 195°C (isothermal); retention time (min): 94.33 (R), 95.81 (S).
(S)-2-(3-bromophenyl)-propionic acid (14)
Light yellow oil, 1H NMR (400 MHz, CDCl3): δ 10.17 (br s, 1H), 7.48 (s, 1H), 7.40 (d, J = 8.00 Hz, 1H), 7.25 (d, J = 8.80 Hz, 1H), 7.20 (d, J = 8.00 Hz, 1H), 3.70 (q, J = 7.20 Hz, 1H), 1.50 (d, J = 7.20 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 179.9, 141.8, 130.8, 130.6, 130.2, 126.3, 122.7, 45.0, 18.0. [α]D22 = 33.8 ± 0.02 (c 1.00, CHCl3). HRMS 249.9600 (M+; calcd for C9H8O2BrNa 249.9605). GC (chiral) conditions, (L)-menthyl esters: (Chirasil-S-Val) 45 min at 190°C (isothermal); retention time (min): 25.77 (R), 26.70 (S).
Synthesis of (±)-ketoprofen from racemic 6
A 25 mL three-necked round bottomed flask equipped with a rubber septum, flow-controlled nitrogen inlet, a temperature probe, and a magnetic stirring bar was flame-dried and purged with nitrogen. The flask was then charged with 6 (0.10 g, 0.46 mmol) and freshly distilled hexanes (5 mL) and the vessel was subsequently cooled to −78°C, at which time, t-BuLi (0.92 mmol, 0.55 mL of 1.7 M solution in hexanes) was added to the stirred mixture in a dropwise fashion, maintaining the temperature below −75°C. After addition was complete, the solution was allowed to stir for 1 h before addition of benzaldehyde (0.06 mL, 0.55 mmol). Upon completion of addition, the vessel was warmed to ambient temperature and allowed to react for 18 hr. Upon completion of reaction, the mixture was poured into water (20 mL) and washed with diethyl ether (3 × 20 mL). The ethereal layers were combined and washed with an ethanolic solution of NaHSO3 (20 mL). The ethereal portion was dried with Na2SO4 (1.5 g), filtered, and reduced. The crude alcohol (0.100 g, 0.45 mmol) was used without further purification. The olefin was suspended in a solution of 2:1 H2O:t-BuOH (36 mL) with vigorous stirring in a 100 mL single-necked round bottomed flask. When the mixture became homogenous, the solution was cooled to 0°C and the olefin was treated with KMnO4(0.213 g, 1.35 mmol), NalO4 (1.73 g, 8.10 mmol), and K2CO3 (0.44 g, 3.20 mmol), which were added as a single portion. The pH was adjusted to 8 with a solution of 2 M KOH. The reaction was stirred for 2 h at 0°C. Concentrated HCl was added to adjust the pH of the solution to 1, and NaHSO3 was added to reduce KMnO4 until the reaction mixture turned yellow. The mixture was extracted with dichloromethane and the CH2Cl2 layer was extracted with 2 M KOH aqueous solution. The aqueous layer was acidified with concentrated 10% H2SO4 and then was extracted with CH2Cl2 (3 × 15 mL). The dried organic layer was evaporated and the product was subjected to chromatography (10:1 CH2Cl2/MeOH) to afford racemic ketoprofen (83 mg, 72%) as a white crystalline solid.
(±)-ketoprofen (17)
White solid, mp 94–96 °C; 1H NMR (400 MHz, CDCl3): δ 7.81-7.78 (m, 3H), 7.69 (d, J = 7.60 Hz, 1H), 7.61-7.56 (m, 2H), 7.49-7.43 (m, 3H), 3.83 (q, J = 7.20 Hz, 1H), 1.56 (d, J = 7.20 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 196.4, 179.9, 140.1, 137.9, 137.4, 132.5, 131.6, 130.1, 129.3, 129.2, 128.6, 128.3, 45.2, 18.1. HRMS 276.0761 (M+; calcd for C16H13O3Na 276.0762).
(S)-2-methyl-2-phenylbutanoic acid (15)
Beige solid, mp 67–69 °C; 1H NMR (400 MHz, CDCl3):δ 11.68 (br s, 1H), 7.40-7.32 (m, 4H), 7.27-7.24 (m, 1H), 2.16-1.95 (m, 2H), 1.57 (s, 3H), 0.86 (t, J = 7.60 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 182.5, 142.8, 128.4, 126.9, 126.3, 50.4, 31.6, 21.7, 9.0. HRMS 196.1095 (M+; calcd for C11H16O3 196.1099).
Typical procedure for the Curtius rearrangement of 2-arylpropionic acids14
A 10 mL two-necked round bottomed flask equipped with a magnetic stirring bar, rubber septum, reflux condenser, and a gas inlet was evacuated, flame-dried, and purged with nitrogen. The flask was then charged with (S)-2-methyl-2-phenylbutanoic acid (0.l00g, 0.56 mmol), diphenylphosphoryl azide (0.24 mL, 1.12 mmol), triethylamine (0.12 mL, 0.84 mmol), and benzene (4 mL). The solution was then heated to reflux and maintained for 12 h, at which time benzyl alcohol (0.29 mL, 2.80 mmol) was added in a single portion and the reaction was maintained at reflux for an additional 6 h. Upon completion, the reaction was cooled to ambient temperature, treated with 1 M NaOH (4 mL), and the slurry was poured into a separatory funnel containing deionized H2O (15 mL). The solution was then extracted with dichloromethane (3×10 mL). The organic layers were then combined and washed with 10% H2SO4 (20 mL). The organic layer was then dried with MgSO4, filtered, and reduced. The product was then subjected to chromatography (6:1 hexanes/ethyl acetate) to afford (S)-benzyl-2-phenylbutan-2-ylcarbamate (18, 128 mg, 81%) as a clear oil.
(S)-benzyl-2-phenylbutan-2-ylcarbamate (18)
Clear oil, 1H NMR (500 MHz, CDCl3): δ 7.37-7.31 (m, 9H), 7.24-7.22 (m, 1H), 5.11 (br s, 1H), 5.04 (s, 2H), 2.04-1.99 (m, 1H), 1.92-1.88 (m, 1H), 1.70 (s, 3H), 0.80 (t, J = 7.50 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 154.4, 145.7, 136.7, 129.8, 128.4, 128.2, 128.1, 127.7, 126.5, 125.2, 120.1, 66.2, 58.3, 34.8, 25.1, 8.2. IR (neat) cm−1: 3432, 3344, 2974, 1718, 1497, 737. [α]D22 = 1.4 ± 0.02 (c 1.00, CHCl3). HRMS 283.1570 (M+; calcd for C18H21NO2 283.1572).
(S)-ethyl-2-(4-isobutylphenyl)butan-2-ylcarbamate (20)
Clear oil, 1H NMR (500 MHz, CDCl3): δ 7.21 (d, J = 8.00 Hz, 2H), 7.10 (d, J = 8.00 Hz, 2H), 4.99 (br s, 1H), 4.83 (br s, 1H), 4.14-4.07 (m, 2H), 2.45 (d, J= 7.50 Hz, 2H), 1.85 (septet, J = 7.00 Hz, 1H), 1.47 (d, J= 6.50 Hz, 3H), 1.22 (t, J = 7.00 Hz, 3H), 0.90 (d, J = 6.50 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ 155.8, 140.8, 140.6, 129.3, 125.7, 60.6, 50.5, 50.2, 45.0, 30.1, 22.3, 14.5. IR (neat) cm−1: 3437, 3334, 2957, 1713, 1510, 740. [α]D22 = 12.1 ± 0.02 (c 1.00, CHCl3). HRMS 277.2045 (M+; calcd for C17H27NO2 277.2042).
(S)-ethyl-2-(3-phenoxyphenyl)butan-2-ylcarbamate (21)
Light yellow oil, 1H NMR (500 MHz, CDCl3): δ 7.33 (dt, J = 8.50, 1.50 Hz, 2H), 7.28 (t, J = 8.00 Hz, 1H), 7.10 (t, J = 7.50 Hz, 1H), 7.04 (d, J = 8.00 Hz, 1H), 7.01-6.98 (m, 3H), 6.86 (dd, J = 8.00, 2.00 Hz, 1H), 4.94 (d, J = 7.00 Hz, 1H), 4.82 (br s, 1H), 4.13-4.07 (m, 2H), 1.45 (d, J = 6.50 Hz, 3H), 1.22 (t, J = 7.00 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 157.5, 157.1, 155.8, 146.0, 129.9, 129.8, 123.3, 120.8, 118.9, 117.4, 116.4, 60.9, 50.4, 22.6, 14.6. IR (neat) cm−1: 3442, 3329, 3015, 2981, 1704, 1584, 1245, 757. [α]D22 = −42.8 ± 0.02 (c 0.50, CHCl3). HRMS 313.1674 (M+; calcd for C19H23NO3 313.1678).
Typical procedure for the Schmidt rearrangement of 2-arylpropionic acids15
A 10 mL two-necked round bottomed flask equipped with a magnetic stirring bar, rubber septum, reflux condenser, and a gas inlet was evacuated, flame-dried, and purged with nitrogen. The flask was then charged with (S)-2-methyl-2-phenylbutanoic acid (15, 0.l00g, 0.56 mmol), and chloroform (4 mL) distilled from calcium hydride and the reaction was cooled to 0°C. Methanesulfonic acid (0.36 mL, 5.60 mmol) was then introduced over a period of 10 min. Once addition was complete, the reaction was stirred for 10 min before sodium azide (0.18 g, 2.80 mmol) was added as a single portion, maintaining the temperature at 0°C. The reaction mixture was then warmed to reflux for a period of 12 h. Upon completion, the reaction was cooled to ambient temperature and the solution was neutralized via addition of 1 M NaOH (3 mL). The mixture was then poured into deionized H2O (10 mL) and extracted into DCM (3 × 10 mL). The organic layers were then combined, dried over MgSO4, filtered, and reduced to a syrup which was then dissolved in DCM (3 mL). Propionyl chloride (0.5 mL) was then added to the solution in a dropwise fashion and stirred for 1 h. The reaction was then neutralized with saturated NaHCO3 (10 mL). The product was extracted into DCM (3 × 10 mL). The combined organic layers were washed with 1 M NAOH (10 mL), and then brine (10 mL). The organic layer was then dried over MgSO4, filtered, and reduced in vacuo. The product was then purified by passage through a silica gel column (4:1 hexanes/ethyl acetate) to afford 74 mg (64 %) of the amide as a clear oil.
(S)-N-(2-phenylbutan-2-yl)propionamide (19)
Clear oil, 1H NMR (500 MHz, CDCl3): δ 7.34-7.31 (m, 4H), 7.25-7.21 (m, 1H), 4.14 (q, J = 7.00 Hz, 1H), 2.15-2.08 (m, 1H), 1.99-1.92 (m, 1H), 1.53 (s, 3H), 1.19 (t, J = 7.00 Hz, 3H), 0.85 (t, J = 7.00 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 176.2, 144.0, 128.2, 126.5, 126.0, 60.6, 50.6, 31.8, 22.2, 14.1, 9.1. IR (neat) cm−1: 3447, 2879, 1272, 1265, 738. [α]D22 = 2.0 ± 0.02 (c 0.50, CHCl3). HRMS 205.1463 (M+; calcd for C13H19NO 205.1467).
(S)-N-(l-(2-fluorobiphenyl-4-yl)ethyl)propionamide (22)
Clear oil, 1H NMR (500 MHz, CDCl3): δ 7.55 (d, J = 8.00 Hz, 2H), 7.45 (t, J = 7.50 Hz, 2H), 7.42-7.35 (m, 2H), 7.16 (t, J = 8.00 Hz, 2H), 4.23-4.12 ([(ABq)q]; νAB = 0.036, νA = 4.21, νB = 4.14; JAB = 11.50, JAX = 7.00, JBX = 7.00 Hz; 2H), 3.75 (q, J = 7.00 Hz, 1H), 1.54 (d, J = 7.00 Hz, 3H), 1.25 (d, J = 7.00 Hz, 3H). 13C NMR (125 MHz, CDCl3):δ 174.0, 159.7 (d, JC–F = 246.0 Hz), 142.0, 135.5, 130.7, 128.9, 128.4, 127.8, 127.6, 123.5, 115.3, 115.1, 61.0, 45.1, 18.4, 14.1. IR (neat) cm−1: 3445, 2981, 1730, 1484, 1183, 737. [α]D22 = 22.7 ± 0.02 (c 1.00, CHCl3). HRMS 271.1370 (M+; calcd for C17H18NOF 271.1372). HPLC (chiral) conditions: (Chiralpak AD-H) isocratic hexanes, 0.50 mL/min; retention time (min): 42.82 (R), 47.43 (S).
Supplementary Material
Spectroscopic and chromatographic data for characterization of compounds. This material is available free of charge via the Internet at a. http://pubs.acs.org.
Figure 1.

Curtius (20 and 21) and Schmidt (22) reaction products
Acknowledgement
Financial assistance for this research by NSF (CHE-0610349) and NIH (General Medical Sciences, R01 GM075107) is gratefully acknowledged. We also thank the ACS Organic Division for a graduate fellowship sponsored by The Schering Plough Corporation to Craig Smith.
REFERENCES
- 1.(a) Jüni P, Dieppe P. Age and Ageing. 2004;33:100. doi: 10.1093/ageing/afh035. [DOI] [PubMed] [Google Scholar]; (b) Dieppe PA, Ebrahim S, Juni P. British Med. J. 2004;329:867. doi: 10.1136/bmj.329.7471.867. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Knijff-Dutmer EAJ, Kalsbeek-Batenburg EM, Koerts J, van de Laar MAFJ. Rheumatology. 2002;41:458. doi: 10.1093/rheumatology/41.4.458. [DOI] [PubMed] [Google Scholar]; (d) Hungin APS, Kean WF. Am. J. Med. 2001;110:8S. doi: 10.1016/s0002-9343(00)00628-8. [DOI] [PubMed] [Google Scholar]
- 2.(a) Adams SS, Bresloff P, Mason CG. J. Pharm. Pharmacol. 1976;28:256. doi: 10.1111/j.2042-7158.1976.tb04144.x. [DOI] [PubMed] [Google Scholar]; (b) Hutt AJ, Caldwell J. J. Pharm. Pharmacol. 1983;35:693. doi: 10.1111/j.2042-7158.1983.tb02874.x. [DOI] [PubMed] [Google Scholar]; (c) Caldwell J, Hutt AJ, Fournel-Gigleux S. Biochem. Pharmacol. 1988;37:105. doi: 10.1016/0006-2952(88)90762-9. [DOI] [PubMed] [Google Scholar]; (d) Hutt AJ, Caldwell J. Clin. Pharmacokinet. 1984;9:371. doi: 10.2165/00003088-198409040-00007. [DOI] [PubMed] [Google Scholar]
- 3.(a.) Hawkey CJ. Lancet. 1999;353:307. doi: 10.1016/s0140-6736(98)12154-2. [DOI] [PubMed] [Google Scholar]; (b.) Golden BD, Abramson SB. Rheum. Dis. Clin. North Am. 1999;25:359. doi: 10.1016/s0889-857x(05)70073-9. [DOI] [PubMed] [Google Scholar]; (c.) Garavito RM, DeWitt DL. Biochim. Biophys. Acta. 1999;1441:278. doi: 10.1016/s1388-1981(99)00147-x. [DOI] [PubMed] [Google Scholar]; (d.) Pepper GA. Rheumatology. 2000;35:223. [Google Scholar]
- 4.(a) Nakamura Y, Yamaguchi T, Hashimoto S, Iwatani S, Nakagawa Y. Pharmacobiol. Dynamics. 1981;4:S1. [Google Scholar]; (b) Williams K, Day R, Knihiricki R, Duffield A. Biochem. Pharmacol. 1986;35:3403. doi: 10.1016/0006-2952(86)90443-0. [DOI] [PubMed] [Google Scholar]; (c) Knihinicki R, Williams KM, Day RO. Biochem. Pharmacol. 1989;38:4389. doi: 10.1016/0006-2952(89)90647-3. [DOI] [PubMed] [Google Scholar]; (d) Cox SR. Clin. Pharmacol. Ther. 1988;43:146. [Google Scholar]; (e) Jarnali F, Mehvar R, Russell AS, Sattari S, Yakimets WW, Koo J. J. Pharm. Sci. 1992;81:221. doi: 10.1002/jps.2600810306. [DOI] [PubMed] [Google Scholar]; (f) Hall SD, Rudy AC, Knight PM, Brater DC. Clin. Pharmacol. Ther. 1993;53:393. doi: 10.1038/clpt.1993.42. [DOI] [PubMed] [Google Scholar]
- 5.(a) Morihara T, Chu T, Ubeda O, Beech W, Cole GM. J. Neurochem. 2002;83:1009. doi: 10.1046/j.1471-4159.2002.01195.x. [DOI] [PubMed] [Google Scholar]; (b) Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. J. Biol. Chem. 2003;278:31837. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]; (c) Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. J. Biol. Chem. 2003;278:30748. doi: 10.1074/jbc.M304824200. [DOI] [PubMed] [Google Scholar]; (d) Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE. J. Clin. Invest. 2003;112:440. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lleö A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT. Nat. Med. 2004;10:1065. doi: 10.1038/nm1112. [DOI] [PubMed] [Google Scholar]
- 6.(a.) Koo EH, Golde TE, Galasko DR, Weggen S. 6911466. U.S. Patent. 2005; (b.) Koo EH, Golde TE, Weggen S. 7097998. U.S. Patent. 2006
- 7.Smith CR, RajanBabu TV. Org. Lett. 2008;10:1657. doi: 10.1021/ol800395m.(b) For a review, see: RajanBabu TV. Chem. Rev. 2003;103:2845. doi: 10.1021/cr020040g.Ligand synthesis: Smith CR, Mans D, RajanBabu TV. Org. Synth. 2008;85:238.
- 8.For a review of synthesis of 2-arylpropionic acids, see: Stahly GP, Starrett RM. In: Chirality in Industry II. Collins AN, Sheldrake GN, Crosby J, editors. Chichester: John Wiley; 1997.
- 9.Importance of chiral amines: Lawrence SA. Amines, Synthesis Properties and Applications. Cambridge: Cambridge University Press; 2004. Breuer M, Ditrich K, Habicher T, Hauer B, Keβeler M, Stürmer R, Zelinski T. Angew. Chem. Int. Ed. 2004;43 doi: 10.1002/anie.200300599.788 and references cited therein.
- 10.(a) Jacques J, Collett A, Wilen S. Enantiomers, Racemates, and Resolutions. New York: Wiley & Sons; 1980. [Google Scholar]; (b) Ingersoll AW. Org. Synth. Coll. 1950;Vol. II:506. [Google Scholar]; (c) Ault A. Org. Synth. Coll. 1973;Vol. V:932. [Google Scholar]; (d) Bryker W, Avila LA. 4983771. U. S. Patent. 1989 (Chem. Abstr.. 1991, 114, 206739).; (e) Yamakawa Yakuhin Ko 56026848. Japanese Patent. 1979 (Chem. Abstr.1981, 95, 115012).; (f) Brown E, Voit F. Tetrahedron Lett. 1985;26:4451. [Google Scholar]; (g) Vries T, Wynberg H, van Echten E, Koek J, ten Hoeve W, Kellogg RM, Broxterman QB, Minnaard A, Kaptein B, van der Sluis S, Hulshof L, Kooistra J. Angew. Chem. Int. Ed. 1998;37:2349. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2349::AID-ANIE2349>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lensink C, de Vries JG. Tetrahedron: Asymmetry. 1992;3:325. [Google Scholar]; (b) Lensink C, de Vries JG. Tetrahedron: Asymmetry. 1993;4:215. [Google Scholar]; (c) Burk MJ, Casy G, Johnson NB. J. Org. Chem. 1998;63:6084. doi: 10.1021/jo9809332. [DOI] [PubMed] [Google Scholar]; (d) Spindler F, Blaser H-U. Adv. Synth. Catal. 2001;343:68. [Google Scholar]; (e) Krzyzanowska B, Stec WJ. Synthesis. 1978:521. [Google Scholar]; (f) Krzyzanowska B, Stec WJ. Synthesis. 1982:270. [Google Scholar]; (g) Togni A, Breutel C, Schnyder A, Spindler F, Landert H, Tijani A. J. Am. Chem. Soc. 1994;116:4061. [Google Scholar]
- 12.(a) Sterling DI, Zeitlin AL, Matcham GW. 4950606. U. S. Patent. 1989 (Chem. Abstr.1991, 114, 41044).; (b) Matcham GW, Rozzell JD, Sterling DI, Zeitlin AL. 5169780. U. S. Patent. 1990 (Chem. Abstr.1993, 118, 190144).; (c) Iwasaki A, Yamada Y, Kizaki N, Ikenaka Y, Hasegawa J. Appl. Microbiol. Biotechnol. 2006;69:499. doi: 10.1007/s00253-005-0002-1. [DOI] [PubMed] [Google Scholar]
- 13.(a) Phillips GT, Shears JH. 399589. European Patent. 1989 (Chem. Abstr.. 1991, 115, 69951).; (b) Schmidt H, Fischer A, Fischer P, Schmidt RD, Stelzer U. 812363. European Patent. 1995 (Chem. Abstr.1996, 125, 219744).; (c) Balkenhohl F, Ditrich K, Hauer B, Ladner W. J. Prakt. Chem. 1997;339:381. [Google Scholar]; (d) Ditrich K, Ladner W, Melder J-P. 19913256. German Patent. 1999 (Chem. Abstr.2000, 133, 252151).; (e) Ditrich K. Synthesis. 2008:2283. [Google Scholar]
- 14.(a) Buchner E, Curtius T. Chem. Ber. 1885;18:2371. [Google Scholar]; (b) Curtius T. Ber. 1890;23:3023. [Google Scholar]; (c) Smith PAS. Org. React. 1946;3:337. [Google Scholar]; (d) Nagumo S, Nishida A, Yamazaki C, Matoba A, Murashige K, Kawahara N. Tetrahedron. 2002;58:4917. [Google Scholar]; (e) Boger DL, Cassidy KC, Nakahara S. J. Am. Chem. Soc. 1993;115:10733. [Google Scholar]; (f) Kim S, Ko H, Kim E, Kim D. Org. Lett. 2002;4:1343. doi: 10.1021/ol0256419. [DOI] [PubMed] [Google Scholar]
- 15.(a) Schmidt KFZ. Angew. Chem. 1923;36:511. [Google Scholar]; (b) Wolff H. Org. React. 1946;3:307. [Google Scholar]; (c) Wrobleski A, Sahasrabudhe K, Aube J. J. Am. Chem. Soc. 2002;124:9974. doi: 10.1021/ja027113y. [DOI] [PubMed] [Google Scholar]; (d) Tanaka M, Oba M, Tamai K, Suemune H. J. Org. Chem. 2001;66:2667. doi: 10.1021/jo001423m. [DOI] [PubMed] [Google Scholar]
- 16.Smith CR, Zhang A, Mans D, RajanBabu TV. Org. Synth. 2008;85:248. doi: 10.15227/orgsyn.085.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Bailey PS. Ozonation in Organic Chemistry. Vol. 2. San Diego: Academic; 1982. [Google Scholar]; (b) Pryor WA, Giamalva D, Church DF. J. Am. Chem. Soc. 1983;105:6858. doi: 10.1021/ja00284a035. [DOI] [PubMed] [Google Scholar]; (c) Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J. Org. Chem. 1981;46:3936. [Google Scholar]; (d) Fraunhoffer KJ, White MC. J. Am. Chem. Soc. 2007;129:7274. doi: 10.1021/ja071905g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ramminger C, Zim D, Lando VR, Fassina V, Monteiro AL. J. Braz. Chem. Soc. 2000;11:105. [Google Scholar]; (f) Lee DG, Lamb SE, Chang VS. Org. Synth. 1981;60:11. [Google Scholar]
- 18.Park H, Kumareswaran R, RajanBabu TV. Tetrahedron. 2005;61:6352. [Google Scholar]
- 19.Veysoglu T, Mitschler LA, Swayze JK. Synthesis. 1980:807. [Google Scholar]
- 20.Hiyama T, Wakasa N. Tetrahedron Lett. 1985;26:3259. [Google Scholar]
- 21.Koul S, Parshad R, Taneja SC, Qazi GN. Tetrahedron: Asymmetry. 2003;14:2459. [Google Scholar]
- 22.Shioiri T, Ninomiya K, Yamada S. J. Am. Chem. Soc. 1972;94:6203. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]; (a) Shioiri T, Yamada S. Org. Synth. Coll. Vol. VII. 1990;7:206. [Google Scholar]
- 23.For the best asymmetric routes to date, Naproxen via Ru-catalyzed asymmetric hydrogenation of 2-arylacrylic acids: Ohta T, Takaya H, Kitamura M, Nagai K, Noyori R. J. Org. Chem. 1987;52:3174.(98% ee). Ni-catalyzed asymmetric hydrocyanation: RajanBabu TV, Casalnuovo AL. J. Am. Chem. Soc. 1996;1186325 and references cited therein (95% ee). Ibuprofen via Ru-catalyzed hydrogenation: Uemura T, Zhang X, Matsumura K, Sayo N, Kumobayashi H, Ohta T, Nozaki K, Takaya H. J. Org. Chem. 1996;61:5510. doi: 10.1021/jo961689m.(97% ee). Rh-catalyzed asymmetric hydroformylation: Nozaki K, Sakai N, Nanno T, Higashijima T, Mano S, Horiuchi T, Takaya H. J. Am. Chem. Soc. 1997;119:4413.(92% ee). Hydrovinylation:Zhang A, RajanBabu TV. Org. Lett. 2004;6:1515. doi: 10.1021/ol0495063.(91%). Zn-Cu catalyzed allylic substitution: Harrington-Frost N, Leuser H, Calaza ML, Kneisel FF, Knochel P. Org. Lett. 2003;5:2111. doi: 10.1021/ol034525i.(97% ee). Flurbiprofen via DKR Norinder J, Bogär K, Kanupp L, Bäckvall J-E. Org. Lett. 2007;9:5095. doi: 10.1021/ol702261t.(>97% ee). No useful catalytic asymmetric methods are known for other (S)-2-arylpropionic acid precursors.
- 24.Herrmann WA, Böhm VPW, Reisinger C-P. J. Organomet. Chem. 1999;576:23. [Google Scholar]
- 25.(a) Kobayashi H, Sonoda A, Iwamoto H, Yoshimura M. Chem. Lett. 1981;10:579. [Google Scholar]; (b) Brookhart M, Grant B, Volpe AF., Jr Organometallics. 1992;11:3920–3922. [Google Scholar]
- 26.(a) Criegee R. Angew. Chem. Int. Ed. Engl. 1975;87:745. [Google Scholar]; (b) Tietze LF, Bratz M. Org. Synth. 1993;71:214. [Google Scholar]
- 27.(a) Lemieux RU, von Rudloff E. Can. J. Chem. 1955;33:1701. [Google Scholar]; (b) von Rudloff E. Can. J. Chem. 1965;43:1784. [Google Scholar]; (c) von Rudloff E. Can. J. Chem. 1956;34:1413. [Google Scholar]; (d) von Rudloff E. Can. J. Chem. 1965;43:2660. [Google Scholar]; (e) Suga T, von Rudloff E. Can. J. Chem. 1969;47:3682. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectroscopic and chromatographic data for characterization of compounds. This material is available free of charge via the Internet at a. http://pubs.acs.org.