

Abstract
Hereditary Inclusion Body Myopathy (HIBM) is an autosomal recessive, quadriceps sparing type commonly referred to as HIBM but also termed h-IBM or Inclusion Body Myopathy 2 (IBM2). The clinical manifestations begin with muscle weakness progressing over the next 10–20 years uniquely sparing the quadriceps until the most advanced stage of the disease. Histopathology of an HIBM muscle biopsy shows rimmed vacuoles on Gomori’s trichrome stain, small fibers in groups and tubulofilaments without evidence of inflammation. In affected individuals distinct mutations have been identified in the GNE gene, which encodes the bifunctional enzyme uridine diphospho-N-acetylglucosamine (UDP-GlcNAc) 2-epimerase/N-acetyl-mannosamine (ManNAc) kinase (GNE/MNK). GNE/MNK catalyzes the first two committed steps in the biosynthesis of acetylneuraminic acid (Neu5Ac), an abundant and functionally important sugar. The generation of HIBM animal models has led to novel insights into both the disease and the role of GNE/MNK in pathophysiology. Recent advances in therapeutic approaches for HIBM, including administration of N-acetyl mannosamine (ManNAc), a precursor of Neu5Ac will be discussed.
Keywords: muscular dystrophy, sialic acid synthesis, UDP-GlcNAc 2-epimerase/ManNAc kinase, hyposialylation, HIBM mouse model, ManNAc therapy, GNE mutations
1. Introduction
Inclusion Body Myositis (IBM) was described by Yunis and Samaha on the basis of distinctive inclusions containing tubulofilaments in a subset of patients with polymyositis [1]. IBM, defined by the pathologic presence of rimmed vacuoles and tubulofilaments on muscle histology is further classified into “sporadic inclusion body myositis” (s-IBM; OMIM#137421), which invariably has inflammation, and “hereditary inclusion body myopathy” which shows familial inheritance and no inflammation [2, 3]. This review will focus on the molecular basis, pathophysiology and clinical features of a specific type of Hereditary Inclusion Body Myopathy, the autosomal recessive, quadriceps sparing type commonly referred to as HIBM but also termed h-IBM, or Inclusion Body Myopathy 2 (IBM2) (OMIM#600737), which is allelic to the Japanese disorder Distal Myopathy with Rimmed Vacuoles (DMRV) or Nonaka Myopathy (OMIM#605820) [4, 5]. We henceforth refer to this disorder as HIBM.
2. Clinical features and pathology
2.1. Clinical features
Argov and Yarom [6] first described the disorder HIBM in Jews of Persian descent characterized clinically by progressive proximal and distal muscle weakness and wasting of the upper and lower limbs usually beginning after age 20. Apart from the Persian-Jewish population, affected individuals have now been described worldwide, including patients of Caucasian, Indian, Thai, Japanese and African descent [5, 7–10].
The clinical course of HIBM is relentless. Progression of muscle weakness after onset continues over the next 10 to 20 years. Typically, however, there is sparing of the quadriceps muscles, partially or completely, even in the advanced stages of the disease, a unique feature of this disorder [6]. Weakness and atrophy of the foot extensors manifests as impaired foot dorsiflexion at an early stage of the disease presenting as gait difficulties. Subsequently, forearm flexors, girdle and axial muscles become more involved. The progressive course is gradual without involvement of the ocular, pharyngeal, and respiratory muscles. Cognition, cranial nerves, sensation and coordination remain normal. In more advanced stages of this disorder the muscles of the shoulder girdle are severely affected, with relative sparing of the deltoid, biceps, and triceps. As lower extremity weakness becomes widespread the most characteristic clinical finding, sparing of the quadriceps, becomes obvious. Even as muscle weakness progresses in other groups, the quadriceps remains strong so that affected individuals are able to stand and walk until the clinical pathology is quite advanced [5, 8]. By two to three decades after diagnosis affected individuals require a wheelchair for mobility. HIBM has also been associated with cardiac involvement in a small number of affected patients with severe muscle disease.
Creatine kinase levels are normal or only mildly elevated and nerve conduction velocity is typically normal. MRI T1 weighted images of the thighs showed fatty or fibrous replacement of the hamstring muscles with sparing of the quadriceps (Fig. 1). The diagnosis of HIBM is based on both clinical symptoms as well as the histopathology of a muscle biopsy.
Fig. 1.

T1 weighted magnetic resonance images of the thigh of an individual affected with HIBM. (A) Axial image showing fibrotic muscles of the posterior compartment or “hamstring” muscles (H) with comparatively less involvement of the quadriceps femoris (Q). (B) Coronal image showing similar findings.
2.2. Histopathology
Histopathology of a muscle biopsy from an HIBM affected individual typically demonstrates red rimmed vacuoles with Gomori’s trichrome stain, small fibers in groups, occasionally amyloid deposit, seen with Congo-red staining visualized with rhodamine filters, and 15 to18nm tubulofilaments [2, 6, 11]. These “non-storage vacuoles” have granular staining, basophilic on H&E and red on Gomori trichrome stains. It was suggested that these vacuoles are autophagic [12]. Presumptive evidence of an autophagocytic process in the rimmed vacuole areas is supported by high acid phosphatase activity, reactivity with lysosomal markers, and the presence of multilammelar bodies on electron microscopy [13].
HIBM muscle immunohistochemistry shows normal cytoskeletal and membrane protein staining patterns. Many degenerating, vacuolated muscle cells show immunoreactivity to neural cell adhesion molecule, NCAM1, which is a fetal muscle antigen. NCAM1 is almost undetectable in normal control muscles, however, it is detectable in regenerating fibers [14]. There is no apparent autoimmune basis for the myopathy as only macrophages around necrotic fibers were noted without the presence of lymphocytes [4]. Electron microscopy on muscle biopsy reveals cytoplasmic and nuclear inclusion bodies containing membrane degradation products with some proliferation of mitochondria with irregular size and shape as well as cytoplasmic tubulofilaments.
3. Molecular genetics
3.1. GNE gene identification
Initial genome-wide linkage analyses in nine Persian Jewish families with HIBM revealed evidence for both autosomal recessive inheritance as well as linkage to 9p1-q1 [15]. Linkage results to the same region in Japanese families with autosomal recessive distal myopathy suggested that this disorder, Nonaka myopathy or DMRV, and HIBM were allelic [16]. The gene was further localized to a 700kb region within 9p13-p12 in Middle Eastern Jews and haplotype analysis of the chromosomal region in 104 affected people from 47 Middle Eastern families revealed one unique ancestral founder chromosome [17, 18]. Single non-Jewish families from India, U.S., and the Bahamas with linkage to the same region, had three distinct haplotypes. Using a candidate gene approach in Middle Eastern patients, Eisenberg et al. identified a shared single homozygous missense mutations in the GNE gene, p.M712T; while affected individuals of other ethnic origins were compound heterozygotes for other distinct mutations [19].
The GNE gene (GenBank NM_005476) spans ~ 44kb of genomic DNA and its major transcript consists of 13 exons, exons 1 and 13 are non-coding. GNE is ubiquitously expressed, with the highest levels in liver.
3.1. GNE mutation analysis
Over the last 8 years, the genetic heterogeneity of HIBM has expanded. There are now over 60 GNE mutations described worldwide associated with HIBM/DMRV in patients of different ethnic backgrounds (listed in Table 1). These mutations are predominantly missense (82%) and scattered throughout the GNE gene.
Table 1.
GNE mutations associated with HIBM
| # | Nucleotide substitution | Amino acid substitution | GNE exon | Ethnicity | Protein domain | Refs |
|---|---|---|---|---|---|---|
| 1 | c.C31T | p.R11W | 2 | India | Epimerase | [33] |
| 2 | c.G38C | p.C13S | 2 | Japan, Korea | Epimerase | [24, 27] |
| 3 | c.79C>T | p.P27S | 2 | Italy | Epimerase | [28] |
| 4 | c.86T>C | p.M29T | 2 | Korea | Epimerase | [27] |
| 5 | c.107C>T | p.P36L | 2 | Italy | Epimerase | [21] |
| 6 | ins10 bp | Frameshift* | 2 | Japan | Epimerase | [5] |
| 7 | c.265G>C | p.G89R | 3 | Thailand | Epimerase | [10] |
| 8 | c.386G>A | p.R129Q | 3 | Japan, Korea | Epimerase | [24, 27] |
| 9 | c.396C>G | p.H132Q | 3 | Japan | Epimerase | [5] |
| 10 | c.404G>T | p.G135V | 3 | USA, Irish, English | Epimerase | [30] |
| 11 | c.484C>T | p.R162C | 3 | Italy | Epimerase | [71] |
| 12 | c.511A>G | p.M171V | 3 | Italy | Epimerase | [23] |
| 13 | c.578A>T | p.D176V | 3 | Japan | Epimerase | [5, 24] |
| 14 | c.529C>T | p.R177C | 3 | Japan | Epimerase | [5] |
| 15 | c.598A>T | p.I200F | 3 | USA | Epimerase | [21] |
| 16 | c.605G>T | p.R202L | 3 | Greece | Epimerase | [33] |
| 17 | c.616G>A | p.G206S | 3 | Italy | Epimerase | [28] |
| 18 | c.617delG | p.G206fsX4 | 3 | Italy | Epimerase | [28] |
| 19 | c.647T>C | p.V216A | 4 | USA | Epimerase | [33, 72] |
| 20 | c.673G>A | p.D225N | 4 | Bahamas | Epimerase | [19] |
| 21 | c.722T>G | p.I241S | 4 | Taiwan | Epimerase | [29, 73] |
| 22 | c.736C>T | p.R246W | 4 | USA | Epimerase | [22, 30] |
| 23 | c.737G>A | p.R246Q | 4 | Bahamas, Italy, Taiwan | Epimerase | [19, 28, 29] |
| 24 | IVS4+4A>G | Exon 4 skipping | Intron 4 | Japan | Epimerase | [5] |
| 25 | c.829C>T | p.R277C | 5 | France | Epimerase | [32] |
| 26 | c.847C>T | p.P283S | 5 | Japan | Epimerase | [24] |
| 27 | c.907–908TG>GT | p.C303V | 5 | Japan | Epimerase | [25] |
| 28 | c.909T>A | p.C303X | 5 | India | Epimerase | [19] |
| 29 | c.917G>A | p.R306Q | 5 | Japan | Epimerase | [5] |
| 30 | c.992T>C | p.V331A | 6 | Japan | Epimerase | [5] |
| 31 | c.1003C>T | p.R335W | 6 | USA | Epimerase | [74] |
| 32 | c.1039_1042 delCTGCinsA | p.L347del; H348N | 6 | USA | Epimerase | [74] |
| 33 | IVS6+2dupT (c.1070+2dupT) | p.352fsX15; p.Y355_C357del | 6 | Italian | Epimerase | [28] |
| 34 | c.1099G>A | p.V367I | 7 | Iran | Epimerase | [75] |
| 35 | c.1130delT | p.I377fsX16 | 7 | Italian | Epimerase | [28] |
| 36 | c.1183G>T | p.D378Y | 7 | USA, Japan, Ireland | Epimerase** | [5, 21] |
| 37 | c.1136T>A | p.L379H | 7 | Tunesia | Epimerase?** | [76] |
| 38 | c.1258C>T | p.R420X | 7 | Japan | Kinase | [24] |
| 36 | c.1262T>C | p.V421A | 7 | Japan | Kinase | [24] |
| 37 | c.1295delA | p.K432fsX17 | 8 | India | Kinase | [77] |
| 38 | c.1379C>T | p.A460V | 8 | Japan | Kinase | [26] |
| 39 | c.1415T>C | p.I472T | 9 | Japan | Kinase | [5, 78] |
| 40 | c.1532C>A | p.P511H | 9 | Japan | Kinase | [31] |
| 41 | c.1532C>T | p.P511L | 9 | Thailand | Kinase | [10] |
| 42 | c.1539G>A | p.W513X | 9 | Taiwan | Kinase | [73] |
| 43 | c.1556A>G | p.N519S | 9 | Italy | Kinase | [28] |
| 44 | c.1571C>T | p.A524V | 9 | Thailand, Mexico, South America, France | Kinase | [10, 22, 32] |
| 45 | c.1583T>G | p.F528C | 9 | Germany | Kinase | [21] |
| 46 | c.1670T>C | p.I557T | 10 | Italy, Japan | Kinase | [21, 24] |
| 47 | c.1675G>C | p.G559R | 10 | Greece | Kinase | [33] |
| 48 | c.1714G>C | p.V572L | 10 | Japan, Korea, Asian | Kinase | [25–27] |
| 49 | c.1727G>A | p.G576E | 10 | USA | Kinase | [19] |
| 50 | c.1760T>C | p.I587T | 10 | USA, Italy, Algeria | Kinase | [21, 32] |
| 51 | c.1771G>A | p.A591T | 10 | Korea | Kinase | [27] |
| 52 | c.1798G>A | p.A600T | 10 | Italy | Kinase | [28] |
| 53 | c.1888G>A | p.A630T | 11 | Japan | Kinase | [5] |
| 54 | c.1891G>A | p.A631T | 11 | USA, Senegal | Kinase | [19, 32] |
| 55 | c.1892C>T | p.A631V | 11 | Germany, Ireland, Japan, USA | Kinase | [5, 21, 25, 72] |
| 56 | c.1967T>A | p.I656N | 11 | Thailand | Kinase | [10] |
| 57 | c.2023T>C | p.Y675H | 12 | Mexico, South America | Kinase | [22] |
| 58 | c.2036T>G | p.V679G | 12 | France | Kinase | [32] |
| 59 | c.2086G>A | p.V696M | 12 | Thailand, India, Algeria | Kinase | [10, 19, 32, 33] |
| 60 | c.2122G>A | p.G708S | 12 | Japan | Kinase | [24] |
| 61 | c.2135T>C | p.M712T | 12 | Persian Jewish | Kinase | [19, 22–24, 76] |
| 62 | del ex3-ex9 (>35.7kb) | Large deletion | 3–9 | Italy | Epimerase + Kinase | [71] |
Gray background; severe mutations, likely resulting in nonsense mediated RNA decay and limited GNE/MNK protein expression.
GNE/MNK amino acid residues 1–378 are suggested to regulate epimerase enzymatic activity, and residues 410–722 regulate kinase enzymatic activity [38].
Of all 62 reported GNE mutations associated with HIBM so far, only 11 (18%) are ‘null’ mutations, nonsense or frame shift mutations, highlighted in gray in Table 1, which likely result in nonsense mediated RNA decay and limited or no protein expression. GNE null mutations have never been identified on both alleles in a patient; this would most likely be lethal, also suggested by a Gne knock-out mouse model, which did not survive past the embryonic stage [20].
Among the GNE missense mutations there appear to exist at least 3 founder mutations. p.M712T is the most predominant, identified in patients of Persian-Jewish decent [19, 21, 22], however, surprisingly this mutation has also been described in an Italian patient [23], a Japanese patient [24], and two unrelated Middle Eastern Moslem families [21]. The second most common GNE mutation is p.V572L, predominantly identified in patients of Japanese descent, but also found in some other Asian patients [25–27]. A third GNE founder mutation is p.D176V, occurring in the Japanese population [5, 25].
Other interesting findings among GNE mutations suggest a presence of genetic “hotspots” for mutation. Several distinct amino acids are mutated in different ways: p.G206S or p.G206fsX4 [28], p.R246W or p.R246Q [19, 22, 28–30], p.303V or p.303X [19, 25], p.P511H or p.P511L [10, 31], and p.A631T and p.A631V [5, 19, 21, 25, 32]. Also some specific GNE mutations arose presumably independently in multiple ethnicities: p.R246Q in Italy, Bahamas and Taiwan [19, 28, 29], p.D378Y in Japan and Ireland [5, 21], p.A524V in Thailand, Mexico and France [10, 22, 32], p.I557T in Italy and Japan [21, 24], p.A631V in Germany, Ireland and Japan [5, 21, 25], and p.V696M in Thailand, India and Algeria [10, 19, 32, 33].
4. Protein function and biochemistry
GNE mRNA is translated into the 722 amino acid bifunctional enzyme uridine diphospho-N-acetylglucosamine (UDP-GlcNAc) 2-epimerase/N-acetyl-mannosamine (ManNAc) kinase (GNE/MNK) [34–36]. GNE/MNK is ubiquitously expressed and catalyzes the first 2 committed, rate-limiting steps in the biosynthesis of 5-N-acetylneuraminic acid (Neu5Ac), also known as sialic acid. Neu5Ac, further referred to as ‘sialic acid’, is the most abundant mammalian sialic acid and is typically found as the terminal sugar on glycoconjugates, where it plays a role in a variety of cellular signaling functions [37].
The N-terminal portion of GNE/MNK (amino acids 1–378) has UDP-GlcNAc 2-epimerase catalytic activity (EC 5.1.3.14) [38], which catalyzes the epimerization of UDP-GlcNAc to ManNAc with release of UDP. The C-terminal portion (amino acids 410–722) has ManNAc kinase catalytic activity (EC 2.7.1.60), which phosphorylates ManNAc to ManNAc-6-P and phosphoenolpyruvate. ManNAc-6-P is then further condensed to sialic acid (Fig. 2). The exact locations of the active sites within these domains remain to be determined. The activated form of sialic acid, cytidine monophosphate (CMP)-sialic acid, is utilized as substrate for sialyltransferases by the Golgi-complex in the sialylation of glycoconjugates [39]. Cytosolic CMP-sialic acid regulates GNE/MNK epimerase catalytic activity through a negative feedback mechanism at its allosteric site (amino acids 263–266) [40–42]. Interestingly, heterozygous missense mutations within the GNE/MNK allosteric site lead to another human disorder, sialuria (OMIM#269921). Sialuria cells have lost GNE/MNK feedback inhibition by CMP-sialic acid, resulting in cytoplasmic accumulation and urinary excretion of large quantities of free sialic acid [40–42].
Fig. 2.
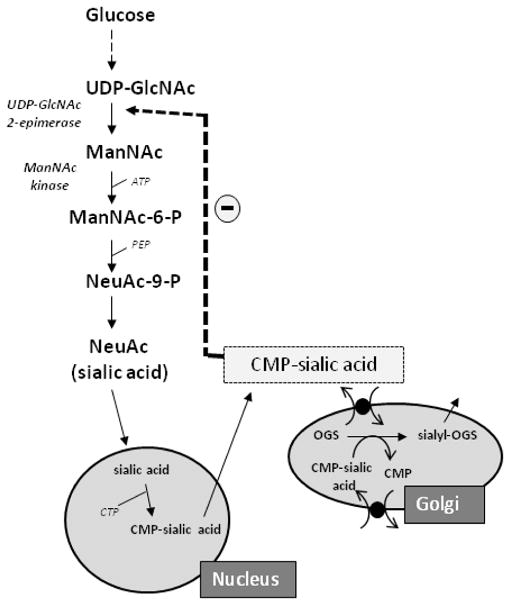
Sialic acid synthesis pathway. The biosynthesis of sialic acid (Neu5Ac) occurs in the cytosol, where glucose undergoes several modifications to become UDP-GlcNAc. The UDP-GlcNAC 2-epimerase activity of GNE/MNK then epimerises UDP-GlcNAc into ManNAc, after which its ManNAc kinase activity further converts this to ManNAc-6-P, which is then converted in several steps to the downstream product CMP-sialic acid. CMP-sialic acid is utilized by the Golgi complex to sialylate glycoconjugates. CMP-sialic acid can feedback-inhibit the UDP-GlcNAc 2-epimerase enzymatic activity in its allosteric site. For more details, see text.
GNE/MNK is a soluble protein, localizing to the cytoplasm, the Golgi-region and the cell nucleus [43]. The role of GNE/MNK in the nucleus remains elusive. GNE/MNK is not predicted to undergo glycosylation, but it has several potential phosphorylation sites [44]. In addition, the GNE/MNK enzyme forms a homohexamer by oligomerization. As a monomer, GNE/MNK has no enzymatic activity; its dimer exhibits only MNK activity, and the hexameric state displays both GNE and MNK activities [34, 38, 45].
5. HIBM Pathology
Since mutations in the GNE gene are associated with HIBM, the presumed mechanism of pathophysiology would be: GNE mutations (mostly missense, Table 1) lead to decreased GNE/MNK enzymatic activities resulting in decreased production of sialic acid. The decrease in intracellular sialic acid content would then lead to the muscle degeneration in HIBM. Although excellent experimental work has been done in pursuit of supportive evidence of this hypothesis, the exact cellular mechanisms behind the development of the myopathy in HIBM has remained elusive.
The effects of GNE mutations on the enzymatic properties of GNE/MNK were assessed by assays of both GNE-epimerase and MNK-kinase activities, which were reduced, but not absent, in HIBM muscle biopsies, as well as in cultured HIBM fibroblasts, lymphoblasts, and myoblasts [5, 30, 45, 46]. In vitro studies, in which specific human GNE mutations were expressed in Sf6 insect cells [38, 47], in COS-7 cells [45], or in a cell-free in vitro transcription-translation system [30], revealed that the reduction in GNE and MNK enzymatic activity is mutation-dependent. Moreover, mutations in one enzymatic domain affect not only that domain’s enzyme activity but also the activity of the other domain. Compared with enzyme activities in a cell-free system, fibroblasts exhibited higher residual activities of both GNE and MNK, suggesting the presence in fibroblasts of additional sugar epimerases and kinases with overlapping substrate specificity [30, 48].
These experiments revealed that the mechanism of pathology is not that of a typical autosomal recessive disorder with low enzyme activity in the gene product. Rather the total functional activity of GNE/MNK activity in a cell may be dependent both on the location or domain of the mutation in the GNE/MNK protein as well as the activity of other enzymes in metabolically interconnected pathways.
Equally enigmatic are the results of several investigators analyzing sialic acid levels in tissue from individuals affected with HIBM. Hinderlich et al. demonstrated normal membrane bound sialic acid levels in lymphoblastoid lines with the p.M712T mutation [49]. Yet, cultured muscle cells from patients with a variation of GNE mutations, showed variable sialylation, ranging from the normal range to significantly decreased [45, 46, 50]. These studies suggested that muscle cells with a strong reduction in epimerase activity, below 35% of normal, resulting from at least one GNE mutation in the epimerase domain, consistently showed measurable decreased sialyation [45, 46, 50]. However, isoelectric focusing studies of serum transferrin, which contains only N-GlcNAc linked glycans, and serum apolipoprotein CIII, which contains only O-GalNAc linked glycans, appeared normal in all HIBM patients tested so far. This suggests that unaffected serum N-GlcNAc linked and O-GalNAc linked glycosylation in hepatically derived serum glycoproteins in individuals with HIBM [51, 52]. Hyposialylation of specific glycosylated proteins in HIBM muscle was reported for PSA, polysialic acid, on NCAM [14] and for α-dystroglycan [33, 53], but was reported to be unaffected in other studies [46, 54]. Hyposialylation of O-linked glycans in HIBM muscle cells was also demonstrated by use of specific lectins [45, 53]. Importantly, Noguchi et al. showed that the hyposialylation of these cells can be reversed by the addition of ManNAc, a substrate in sialic acid synthesis, or sialic acid itself to the media of the cells [45].
Since HIBM is an adult onset disease, and patients have residual GNE/MNK enzymatic activity, the effects of sialic acid deficiency may appear gradually. Some glycoconjugates, for example N-linked, might be more readily sialylated than others, for example O-linked or PSA linked. Thus, when a shortage of sialic acid occurs, specific proteins may be inadequately glycosylated, such as PSA-NCAM or alpha-dystroglycan, contributing to the pathology of HIBM.
Apart from hyposialylation, other hypotheses have arisen for the role of mutated GNE/MNK in the pathology of HIBM. These include the unusual compartmentalization of GNE/MNK in cells [43], leading to speculation of additional GNE/MNK enzymatic activities in cells. Exploration of this phenomenon showed no difference in the compartmentalization of GNE/MNK in either skeletal muscle or primary myoblasts from individuals affected with HIBM [55]. In addition, two novel isoforms of GNE/MNK (GNE1) were identified, which have extended (GNE2) or partially deleted N-termini (GNE3), and display tissue-specific expression [56], which may contribute to the pathology of HIBM. Furthermore, impaired apoptotic signaling in HIBM cells was reported, implicating involvement of apoptotic pathways in HIBM pathophysiology [57]. Another intriguing finding is that GNE/MNK may control sialyltransferase expression, ganglioside production and modulation of proliferation and apoptosis, independent of sialic acid production [58]. In another study, microarray RNA expression and muscle morphology analysis indicated that mitochondrial processes may be affected in HIBM muscle [59]. And recently, co-immunoprecipitation assays identified alpha-actinin 1, an actin binding and crosslinking protein, as a ligand of GNE/MNK [60]. The relevance of α-actinin 1 function in skeletal muscle and its role in HIBM pathophysiology remains elusive.
The above findings underscore that there is more to be learned about the cellularsite of pathology and the mechanism of muscle cell degeneration in HIBM. To further analyze these pathways on a whole animal, as well as explore potential treatment methods, efforts to study HIBM mouse models are ongoing.
6. HIBM mouse models
Generating a mouse model for HIBM has been complicated because initial trials showed that a complete knock-out of the Gne gene led to embryonic lethality [20]. Despite this hurdle, two models were generated by different groups.
Malicdan et al. developed an animal model for HIBM showing the pathologic muscle phenotype over time. They generated a transgenic mouse which expressed the human GNE cDNA with the p.D176V epimerase domain mutation, common among Japanese patients, on a mouse background with a disrupted mouse Gne gene; Gne(−/−)hGNED176V-Tg [61, 62]. The mutant offspring of this cross appeared normal at birth but had decreased levels of sialic acid in serum and different organs. These mice developed poor motor performance and increasing serum creatine kinase levels mimicking some aspects of the muscle phenotype seen in the human disease. By 40 weeks of age the mice showed significant changes in muscle pathology with intracytoplasmic rimmed vacuoles which were immunoreactive to lysosomal markers, amyloid and phosphorylated tau and neurofilaments. Ultrastructural and immunohistochemical studies confirmed the presence of autophagosomes in affected mouse muscle. Of interest, the cardiac muscle of these mice also showed rimmed vacuoles suggesting the possible involvement of other muscles types besides skeletal muscle. The phenotype of this p.D176V transgenic mouse model appeared reminiscent of the clinical, pathologic and biochemical features of HIBM/DMRV in humans [61, 62].
A second HIBM mouse model created by our group produced an intriguing, unexpected outcome. This Gne knock-in mouse was created by homologous recombination which introduced the p.M712T kinase domain mutation, common among Persian-Jews, into the endogenous mouse Gne gene; GneM712T/M712T [63]. Surprisingly, mutant mice died within 72 hours of birth with severe glomerular disease including proteinuria, podocytopathy, segmental splitting of the glomerular basement membrane and effacement of the podocyte foot processes. Biochemical analysis of the mutant mice kidneys revealed decreased Gne/Mnk expression and activity and deficient sialylation of the major podocyte sialoprotein, podocalyxin, suggesting that decreased production of sialic acid may lead to lethality in these mice.
We then proceeded with oral administration of the sialic acid precursor N-acetylmannosamine (ManNAc) to the pregnant and nursing mothers, which resulted in survival of 43% of the mutant GneM712T/M712T pups beyond 72 hours. Mutant survivors exhibited improved renal histology, increased sialylation of podocalyxin, and increased Gne/Mnk protein expression and Gne-epimerase activities. These findings established this GneM712T/M712T knock-in mouse as the first genetic model of podocyte injury due to hyposialylation [63, 64].
In HIBM patients, no indications of renal abnormalities have been reported. Humans and mice may differ in the relative importance of sialic acid to the kidney, and protein glycosylation patterns also vary; it is known that podoxalyxin differs among species in the contingent of O-and N-linked glycosylation sites [65]. The type of sialic acid present also differs, most mammalian species utilize the sialic acid N-glycolylneuraminic acid (Neu5Gc), but humans have lost the ability to synthesize Neu5Gc [66], and mainly utilize N-acetyl neuraminic acid (Neu5Ac).
Our recent findings of abnormal histology in muscle tissue starting at 6 months of age in surviving mutant GneM712T/M712T mice (not receiving ManNAc) are encouraging in that this may mimic the human disorder (authors unpublished data). Further research is required to elucidate phenotypic differences between the transgenic Gne(−/−)hGNED176V-Tg model and the knock-in GneM712T/M712T model.
In spite of these still obscure phenotypic differences between HIBM mouse models, the encouraging results of the ManNAc supplementation of the murine knock-in model [63] support evaluation of ManNAc, a well-tolerated intervention, not only as a treatment for HIBM, but also as a treatment for renal disorders involving proteinuria and hematuria due to podocytopathy and/or segmental splitting of the glomerular basement membrane.
7. Treatment
No therapies are currently available for HIBM. Dietary modifications were proposed, including avoidance of excess selenium, copper and zinc (inhibitors of GNE/MNK activity), reduced consumption of ethanol, ethanol promotes hydrolysis of sialoconjugates, and dietary promotion of magnesium, an essential co-factor of GNE/MNK [67].
Other suggested strategies of therapy for HIBM invoke the concept that hyposialylation is the basis of the pathophysiology in affected individuals. The hypothesis that increasing total body sialic acid through exogenous means will lead to clinical benefit, was recently tested in our center through a pilot study on four affected patients (http://clinicaltrials.gov: Identifier NCT00195637) [68]. The HIBM patients were loaded with 1g/kg intravenous immunoglobulin G (IVIG) on two consecutive days followed by 3 doses of 400mg/kd at weekly intervals. It was hypothesized that the large sialic acid content on IgG (~ 8 μmol of sialic acid/g) could be utilized to sialylate other glycoproteins. This study showed improvement in mean quadriceps strength both after loading (+22%) and at the end of the study (+35%). Mean shoulder strength showed similar findings at (+44%) and (+46%) respectively. A composite of 8 other muscle groups showed improvements as well after loading (+8%) and (+19) at the end of the study [68].
Although immunohistochemical staining and immunoblotting of muscle biopsies for alpha-dystroglycan and NCAM did not show indisputable evidence of increased sialylation after IVIG treatment, patients did report subjective improvement in their ability to perform routine daily activities. Though follow-up studies are needed, this protocol reveals issues surrounding therapeutic strategies for HIBM and opens the door to other possible therapeutic options. These include the administration of other forms of sialic acid, in particular the sialic acid precursor ManNAc.
ManNAc is an uncharged, natural compound and feeds into the sialic acid biosynthesis pathway distal to the rate-limiting GNE-epimerase step (Fig. 2). Residual MNK activity in HIBM patients, or ancillary kinases (e.g., GlcNAc kinase) [48], might convert ManNAc into ManNAc-6P and aid synthesis of free sialic acid. Hyposialylated, Gne-deficient mouse embryonic stem cells [20] and human HIBM/DMRV cultured myotubes [45] became resialylated after the growth medium was supplemented with ManNAc. Furthermore, incubation of cultured cells with ‘unnatural’ ManNAc derivatives (ManLev, N-levulinoylmannosamine or ManNAz, N-azidoacetylmannosamine) resulted in incorporation of the downstream sialic acid analogs, SiaLev (N-levulinoyl sialic acid) or SiaNAz (N-azidoacetyl sialic acid), into cell surface glycoconjugates [69, 70]. Of greatest significance, the salutary effect of oral ManNAc supplementation on survival and sialylation status of our HIBM knock-in mouse model [63] holds promise for potential benefit in a future human clinical treatment protocol. Our center is pursuing the design of a formal trial of ManNAc in humans, which has yet to be approved by regulatory authorities.
Apart from manipulating products and/or substrates in the GNE/MNK pathway, another future treatment option could be the delivery of a healthy GNE gene, gene therapy, or a healthy GNE/MNK enzyme via stem cells, to patients’ cells and tissues, in particular to the muscle. Continued work in this field will elucidate insights both into the pathophysiology of this devastating disorder as well as other human diseases caused by the perturbation of glycobiologic pathways.
Acknowledgments
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA.
References
- 1.Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest. 1971;25:240–248. [PubMed] [Google Scholar]
- 2.Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, Rowland LP. Inclusion body myositis and myopathies. Ann Neurol. 1995;38:705–713. doi: 10.1002/ana.410380504. [DOI] [PubMed] [Google Scholar]
- 3.Askanas V, Engel WK. New advances in the understanding of sporadic inclusion-body myositis and hereditary inclusion-body myopathies. Curr Opin Rheumatol. 1995;7:486–496. doi: 10.1097/00002281-199511000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. 1981;51:141–155. doi: 10.1016/0022-510x(81)90067-8. [DOI] [PubMed] [Google Scholar]
- 5.Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, Nagata T, Chida K, Takahashi T, Takusa Y, Ohi T, Nishimiya J, Sunohara N, Ciafaloni E, Kawai M, Aoki M, Nonaka I. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689–1693. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- 6.Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. 1984;64:33–43. doi: 10.1016/0022-510x(84)90053-4. [DOI] [PubMed] [Google Scholar]
- 7.Zlotogora J. Hereditary disorders among Iranian Jews. Am J Med Genet. 1995;58:32–37. doi: 10.1002/ajmg.1320580108. [DOI] [PubMed] [Google Scholar]
- 8.Sivakumar K, Dalakas MC. The spectrum of familial inclusion body myopathies in 13 families and a description of a quadriceps-sparing phenotype in non-Iranian Jews. Neurology. 1996;47:977–984. doi: 10.1212/wnl.47.4.977. [DOI] [PubMed] [Google Scholar]
- 9.Sadeh M, Gadoth N, Hadar H, Ben-David E. Vacuolar myopathy sparing the quadriceps. Brain. 1993;116(Pt 1):217–232. doi: 10.1093/brain/116.1.217. [DOI] [PubMed] [Google Scholar]
- 10.Liewluck T, Pho-Iam T, Limwongse C, Thongnoppakhun W, Boonyapisit K, Raksadawan N, Murayama K, Hayashi YK, Nishino I, Sangruchi T. Mutation analysis of the GNE gene in distal myopathy with rimmed vacuoles (DMRV) patients in Thailand. Muscle Nerve. 2006;34:775–778. doi: 10.1002/mus.20583. [DOI] [PubMed] [Google Scholar]
- 11.Massa R, Weller B, Karpati G, Shoubridge E, Carpenter S. Familial inclusion body myositis among Kurdish-Iranian Jews. Arch Neurol. 1991;48:519–522. doi: 10.1001/archneur.1991.00530170083024. [DOI] [PubMed] [Google Scholar]
- 12.Nishino I, Malicdan MC, Murayama K, Nonaka I, Hayashi YK, Noguchi S. Molecular pathomechanism of distal myopathy with rimmed vacuoles. Acta Myol. 2005;24:80–83. [PubMed] [Google Scholar]
- 13.Nonaka I, Noguchi S, Nishino I. Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep. 2005;5:61–65. doi: 10.1007/s11910-005-0025-0. [DOI] [PubMed] [Google Scholar]
- 14.Ricci E, Broccolini A, Gidaro T, Morosetti R, Gliubizzi C, Frusciante R, Di Lella GM, Tonali PA, Mirabella M. NCAM is hyposialylated in hereditary inclusion body myopathy due to GNE mutations. Neurology. 2006;66:755–758. doi: 10.1212/01.wnl.0000200956.76449.3f. [DOI] [PubMed] [Google Scholar]
- 15.Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet. 1996;5:159–163. doi: 10.1093/hmg/5.1.159. [DOI] [PubMed] [Google Scholar]
- 16.Ikeuchi T, Asaka T, Saito M, Tanaka H, Higuchi S, Tanaka K, Saida K, Uyama E, Mizusawa H, Fukuhara N, Nonaka I, Takamori M, Tsuji S. Gene locus for autosomal recessive distal myopathy with rimmed vacuoles maps to chromosome 9. Ann Neurol. 1997;41:432–437. doi: 10.1002/ana.410410405. [DOI] [PubMed] [Google Scholar]
- 17.Eisenberg I, Thiel C, Levi T, Tiram E, Argov Z, Sadeh M, Jackson CL, Thierfelder L, Mitrani-Rosenbaum S. Fine-structure mapping of the hereditary inclusion body myopathy locus. Genomics. 1999;55:43–48. doi: 10.1006/geno.1998.5630. [DOI] [PubMed] [Google Scholar]
- 18.Eisenberg I, Hochner H, Shemesh M, Levi T, Potikha T, Sadeh M, Argov Z, Jackson CL, Mitrani-Rosenbaum S. Physical and transcriptional map of the hereditary inclusion body myopathy locus on chromosome 9p12–p13. Eur J Hum Genet. 2001;9:501–509. doi: 10.1038/sj.ejhg.5200665. [DOI] [PubMed] [Google Scholar]
- 19.Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–87. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 20.Schwarzkopf M, Knobeloch KP, Rohde E, Hinderlich S, Wiechens N, Lucka L, Horak I, Reutter W, Horstkorte R. Sialylation is essential for early development in mice. Proc Natl Acad Sci USA. 2002;99:5267–5270. doi: 10.1073/pnas.072066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenberg I, Grabov-Nardini G, Hochner H, Korner M, Sadeh M, Bertorini T, Bushby K, Castellan C, Felice K, Mendell J, Merlini L, Shilling C, Wirguin I, Argov Z, Mitrani-Rosenbaum S. Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat. 2003;21:99. doi: 10.1002/humu.9100. [DOI] [PubMed] [Google Scholar]
- 22.Darvish D, Vahedifar P, Huo Y. Four novel mutations associated with autosomal recessive inclusion body myopathy (MIM: 600737) Mol Genet Metab. 2002;77:252–256. doi: 10.1016/s1096-7192(02)00141-5. [DOI] [PubMed] [Google Scholar]
- 23.Broccolini A, Pescatori M, D’Amico A, Sabino A, Silvestri G, Ricci E, Servidei S, Tonali PA, Mirabella M. An Italian family with autosomal recessive inclusion-body myopathy and mutations in the GNE gene. Neurology. 2002;59:1808–1809. doi: 10.1212/01.wnl.0000031808.04545.e0. [DOI] [PubMed] [Google Scholar]
- 24.Tomimitsu H, Shimizu J, Ishikawa K, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles (DMRV): new GNE mutations and splice variant. Neurology. 2004;62:1607–1610. doi: 10.1212/01.wnl.0000123115.23652.6c. [DOI] [PubMed] [Google Scholar]
- 25.Tomimitsu H, Ishikawa K, Shimizu J, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles: novel mutations in the GNE gene. Neurology. 2002;59:451–454. doi: 10.1212/wnl.59.3.451. [DOI] [PubMed] [Google Scholar]
- 26.Kayashima T, Matsuo H, Satoh A, Ohta T, Yoshiura K, Matsumoto N, Nakane Y, Niikawa N, Kishino T. Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene (GNE) J Hum Genet. 2002;47:77–79. doi: 10.1007/s100380200004. [DOI] [PubMed] [Google Scholar]
- 27.Kim BJ, Ki CS, Kim JW, Sung DH, Choi YC, Kim SH. Mutation analysis of the GNE gene in Korean patients with distal myopathy with rimmed vacuoles. J Hum Genet. 2006;51:137–140. doi: 10.1007/s10038-005-0338-5. [DOI] [PubMed] [Google Scholar]
- 28.Broccolini A, Ricci E, Cassandrini D, Gliubizzi C, Bruno C, Tonoli E, Silvestri G, Pescatori M, Rodolico C, Sinicropi S, Servidei S, Zara F, Minetti C, Tonali PA, Mirabella M. Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion-body myopathy. Hum Mutat. 2004;23:632. doi: 10.1002/humu.9252. [DOI] [PubMed] [Google Scholar]
- 29.Chu CC, Kuo HC, Yeh TH, Ro LS, Chen SR, Huang CC. Heterozygous mutations affecting the epimerase domain of the GNE gene causing distal myopathy with rimmed vacuoles in a Taiwanese family. Clin Neurol Neurosurg. 2007;109:250–256. doi: 10.1016/j.clineuro.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Sparks SE, Ciccone C, Lalor M, Orvisky E, Klootwijk R, Savelkoul PJ, Dalakas MC, Krasnewich DM, Gahl WA, Huizing M. Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human hereditary inclusion body myopathy. Glycobiology. 2005;15:1102–1110. doi: 10.1093/glycob/cwi100. [DOI] [PubMed] [Google Scholar]
- 31.Motozaki Y, Komai K, Hirohata M, Asaka T, Ono K, Yamada M. Hereditary inclusion body myopathy with a novel mutation in the GNE gene associated with proximal leg weakness and necrotizing myopathy. Eur J Neurol. 2007;14:e14–15. doi: 10.1111/j.1468-1331.2007.01905.x. [DOI] [PubMed] [Google Scholar]
- 32.Behin A, Dubourg O, Laforet P, Pecheux C, Bernard R, Levy N, Eymard B. Distal myopathy due to mutations of GNE gene: clinical spectrum and diagnosis. Rev Neurol (Paris) 2008;164:434–443. doi: 10.1016/j.neurol.2008.02.040. [DOI] [PubMed] [Google Scholar]
- 33.Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, Krasnewich D, Gahl WA, Dalakas MC. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. 2004;81:196–202. doi: 10.1016/j.ymgme.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 34.Hinderlich S, Stasche R, Zeitler R, Reutter W. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272:24313–24318. doi: 10.1074/jbc.272.39.24313. [DOI] [PubMed] [Google Scholar]
- 35.Keppler OT, Hinderlich S, Langner J, Schwartz-Albiez R, Reutter W, Pawlita M. UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science. 1999;284:1372–1376. doi: 10.1126/science.284.5418.1372. [DOI] [PubMed] [Google Scholar]
- 36.Huizing M. Disease mechanisms associated with mutations of the GNE gene. Drug Discovery Today Disease Mechanisms. 2005;2:519–527. [Google Scholar]
- 37.Varki A. Sialic acids as ligands in recognition phenomena. FASEB J. 1997;11:248–255. doi: 10.1096/fasebj.11.4.9068613. [DOI] [PubMed] [Google Scholar]
- 38.Effertz K, Hinderlich S, Reutter W. Selective loss of either the epimerase or kinase activity of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase due to site-directed mutagenesis based on sequence alignments. J Biol Chem. 1999;274:28771–28778. doi: 10.1074/jbc.274.40.28771. [DOI] [PubMed] [Google Scholar]
- 39.Harduin-Lepers A, Mollicone R, Delannoy P, Oriol R. The animal sialyltransferases and sialyltransferase-related genes: a phylogenetic approach. Glycobiology. 2005;15:805–817. doi: 10.1093/glycob/cwi063. [DOI] [PubMed] [Google Scholar]
- 40.Seppala R, Tietze F, Krasnewich D, Weiss P, Ashwell G, Barsh G, Thomas GH, Packman S, Gahl WA. Sialic acid metabolism in sialuria fibroblasts. J Biol Chem. 1991;266:7456–7461. [PubMed] [Google Scholar]
- 41.Seppala R, Lehto VP, Gahl WA. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am J Hum Genet. 1999;64:1563–1569. doi: 10.1086/302411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leroy JG, Seppala R, Huizing M, Dacremont G, De Simpel H, Van Coster RN, Orvisky E, Krasnewich DM, Gahl WA. Dominant inheritance of sialuria, an inborn error of feedback inhibition. Am J Hum Genet. 2001;68:1419–1427. doi: 10.1086/320598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krause S, Hinderlich S, Amsili S, Horstkorte R, Wiendl H, Argov Z, Mitrani-Rosenbaum S, Lochmuller H. Localization of UDP-GlcNAc 2-epimerase/ManAc kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res. 2005;304:365–379. doi: 10.1016/j.yexcr.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Horstkorte R, Nohring S, Danker K, Effertz K, Reutter W, Lucka L. Protein kinase C phosphorylates and regulates UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase. FEBS Lett. 2000;470:315–318. doi: 10.1016/s0014-5793(00)01331-4. [DOI] [PubMed] [Google Scholar]
- 45.Noguchi S, Keira Y, Murayama K, Ogawa M, Fujita M, Kawahara G, Oya Y, Imazawa M, Goto Y, Hayashi YK, Nonaka I, Nishino I. Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem. 2004;279:11402–11407. doi: 10.1074/jbc.M313171200. [DOI] [PubMed] [Google Scholar]
- 46.Salama I, Hinderlich S, Shlomai Z, Eisenberg I, Krause S, Yarema K, Argov Z, Lochmuller H, Reutter W, Dabby R, Sadeh M, Ben-Bassat H, Mitrani-Rosenbaum S. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem Biophys Res Commun. 2005;328:221–226. doi: 10.1016/j.bbrc.2004.12.157. [DOI] [PubMed] [Google Scholar]
- 47.Penner J, Mantey LR, Elgavish S, Ghaderi D, Cirak S, Berger M, Krause S, Lucka L, Voit T, Mitrani-Rosenbaum S, Hinderlich S. Influence of UDP-GlcNAc 2-epimerase/ManNAc kinase mutant proteins on hereditary inclusion body myopathy. Biochemistry. 2006;45:2968–2977. doi: 10.1021/bi0522504. [DOI] [PubMed] [Google Scholar]
- 48.Hinderlich S, Berger M, Schwarzkopf M, Effertz K, Reutter W. Molecular cloning and characterization of murine and human N-acetylglucosamine kinase. Eur J Biochem. 2000;267:3301–3308. doi: 10.1046/j.1432-1327.2000.01360.x. [DOI] [PubMed] [Google Scholar]
- 49.Hinderlich S, Salama I, Eisenberg I, Potikha T, Mantey LR, Yarema KJ, Horstkorte R, Argov Z, Sadeh M, Reutter W, Mitrani-Rosenbaum S. The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered overall cellular sialylation in hereditary inclusion body myopathy. FEBS Lett. 2004;566:105–109. doi: 10.1016/j.febslet.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 50.Saito F, Tomimitsu H, Arai K, Nakai S, Kanda T, Shimizu T, Mizusawa H, Matsumura K. A Japanese patient with distal myopathy with rimmed vacuoles: missense mutations in the epimerase domain of the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene accompanied by hyposialylation of skeletal muscle glycoproteins. Neuromuscul Disord. 2004;14:158–161. doi: 10.1016/j.nmd.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 51.Wopereis S, Grunewald S, Huijben KM, Morava E, Mollicone R, van Engelen BG, Lefeber DJ, Wevers RA. Transferrin and apolipoprotein C-III isofocusing are complementary in the diagnosis of N- and O-glycan biosynthesis defects. Clin Chem. 2007;53:180–187. doi: 10.1373/clinchem.2006.073940. [DOI] [PubMed] [Google Scholar]
- 52.Savelkoul PJ, Manoli I, Sparks SE, Ciccone C, Gahl WA, Krasnewich DM, Huizing M. Normal sialylation of serum N-linked and O-GalNAc-linked glycans in hereditary inclusion-body myopathy. Mol Genet Metab. 2006;88:389–390. doi: 10.1016/j.ymgme.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 53.Tajima Y, Uyama E, Go S, Sato C, Tao N, Kotani M, Hino H, Suzuki A, Sanai Y, Kitajima K, Sakuraba H. Distal myopathy with rimmed vacuoles: impaired O-glycan formation in muscular glycoproteins. Am J Pathol. 2005;166:1121–1130. doi: 10.1016/S0002-9440(10)62332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Broccolini A, Gliubizzi C, Pavoni E, Gidaro T, Morosetti R, Sciandra F, Giardina B, Tonali P, Ricci E, Brancaccio A, Mirabella M. alpha-Dystroglycan does not play a major pathogenic role in autosomal recessive hereditary inclusion-body myopathy. Neuromuscul Disord. 2005;15:177–184. doi: 10.1016/j.nmd.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 55.Krause S, Aleo A, Hinderlich S, Merlini L, Tournev I, Walter MC, Argov Z, Mitrani-Rosenbaum S, Lochmuller H. GNE protein expression and subcellular distribution are unaltered in HIBM. Neurology. 2007;69:655–659. doi: 10.1212/01.wnl.0000267426.97138.fd. [DOI] [PubMed] [Google Scholar]
- 56.Reinke SO, Hinderlich S. Prediction of three different isoforms of the human UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. FEBS Lett. 2007;581:3327–3331. doi: 10.1016/j.febslet.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 57.Amsili S, Shlomai Z, Levitzki R, Krause S, Lochmuller H, Ben-Bassat H, Mitrani-Rosenbaum S. Characterization of hereditary inclusion body myopathy myoblasts: possible primary impairment of apoptotic events. Cell Death Differ. 2007;14:1916–1924. doi: 10.1038/sj.cdd.4402208. [DOI] [PubMed] [Google Scholar]
- 58.Wang Z, Sun Z, Li AV, Yarema KJ. Roles for UDP-GlcNAc 2-epimerase/ManNAc 6-kinase outside of sialic acid biosynthesis: modulation of sialyltransferase and BiP expression, GM3 and GD3 biosynthesis, proliferation, and apoptosis, and ERK1/2 phosphorylation. J Biol Chem. 2006;281:27016–27028. doi: 10.1074/jbc.M604903200. [DOI] [PubMed] [Google Scholar]
- 59.Eisenberg I, Novershtern N, Itzhaki Z, Becker-Cohen M, Sadeh M, Willems PH, Friedman N, Koopman WJ, Mitrani-Rosenbaum S. Mitochondrial processes are impaired in hereditary inclusion body myopathy. Hum Mol Genet. 2008;17:3663–3674. doi: 10.1093/hmg/ddn261. [DOI] [PubMed] [Google Scholar]
- 60.Amsili S, Zer H, Hinderlich S, Krause S, Becker-Cohen M, MacArthur DG, North KN, Mitrani-Rosenbaum S. UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) binds to alpha-actinin 1: novel pathways in skeletal muscle? PLoS ONE. 2008;3:e2477. doi: 10.1371/journal.pone.0002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malicdan MC, Noguchi S, Nonaka I, Hayashi YK, Nishino I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. 2007;16:2669–2682. doi: 10.1093/hmg/ddm220. [DOI] [PubMed] [Google Scholar]
- 62.Malicdan MC, Noguchi S, Hayashi YK, Nishino I. Muscle weakness correlates with muscle atrophy and precedes the development of inclusion body or rimmed vacuoles in the mouse model of DMRV/hIBM. Physiol Genomics. 2008;35:106–115. doi: 10.1152/physiolgenomics.90219.2008. [DOI] [PubMed] [Google Scholar]
- 63.Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, Starost MF, Zerfas PM, Hoffmann VJ, Hoogstraten-Miller S, Krasnewich DM, Gahl WA, Huizing M. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quaggin SE. Sizing up sialic acid in glomerular disease. J Clin Invest. 2007;117:1480–1483. doi: 10.1172/JCI32482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kershaw DB, Beck SG, Wharram BL, Wiggins JE, Goyal M, Thomas PE, Wiggins RC. Molecular cloning and characterization of human podocalyxin-like protein. Orthologous relationship to rabbit PCLP1 and rat podocalyxin. J Biol Chem. 1997;272:15708–15714. doi: 10.1074/jbc.272.25.15708. [DOI] [PubMed] [Google Scholar]
- 66.Chou HH, Takematsu H, Diaz S, Iber J, Nickerson E, Wright KL, Muchmore EA, Nelson DL, Warren ST, Varki A. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci USA. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darvish D. Magnesium may help patients with recessive hereditary inclusion body myopathy, a pathological review. Med Hypotheses. 2003;60:94–101. doi: 10.1016/s0306-9877(02)00339-0. [DOI] [PubMed] [Google Scholar]
- 68.Sparks S, Rakocevic G, Joe G, Manoli I, Shrader J, Harris-Love M, Sonies B, Ciccone C, Dorward H, Krasnewich D, Huizing M, Dalakas MC, Gahl WA. Intravenous immune globulin in hereditary inclusion body myopathy: a pilot study. BMC Neurol. 2007;7:3. doi: 10.1186/1471-2377-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luchansky SJ, Yarema KJ, Takahashi S, Bertozzi CR. GlcNAc 2-epimerase can serve a catabolic role in sialic acid metabolism. J Biol Chem. 2003;278:8035–8042. doi: 10.1074/jbc.M212127200. [DOI] [PubMed] [Google Scholar]
- 70.Charter NW, Mahal LK, Koshland DE, Jr, Bertozzi CR. Biosynthetic incorporation of unnatural sialic acids into polysialic acid on neural cells. Glycobiology. 2000;10:1049–1056. doi: 10.1093/glycob/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 71.Del Bo R, Baron P, Prelle A, Serafini M, Moggio M, Fonzo AD, Castagni M, Bresolin N, Comi GP. Novel missense mutation and large deletion of GNE gene in autosomal-recessive inclusion-body myopathy. Muscle Nerve. 2003;28:113–117. doi: 10.1002/mus.10391. [DOI] [PubMed] [Google Scholar]
- 72.Vasconcelos OM, Raju R, Dalakas MC. GNE mutations in an American family with quadriceps-sparing IBM and lack of mutations in s-IBM. Neurology. 2002;59:1776–1779. doi: 10.1212/01.wnl.0000039780.13681.ad. [DOI] [PubMed] [Google Scholar]
- 73.Ro LS, Lee-Chen GJ, Wu YR, Lee M, Hsu PY, Chen CM. Phenotypic variability in a Chinese family with rimmed vacuolar distal myopathy. J Neurol Neurosurg Psychiatry. 2005;76:752–755. doi: 10.1136/jnnp.2004.048876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fisher J, Towfighi J, Darvish D, Simmons Z. A case of hereditary inclusion body myopathy: 1 patient, 2 novel mutations. J Clin Neuromuscul Dis. 2006;7:179–184. doi: 10.1097/01.cnd.0000211406.94445.f0. [DOI] [PubMed] [Google Scholar]
- 75.Krause S, Schlotter-Weigel B, Walter MC, Najmabadi H, Wiendl H, Muller-Hocker J, Muller-Felber W, Pongratz D, Lochmuller H. A novel homozygous missense mutation in the GNE gene of a patient with quadriceps-sparing hereditary inclusion body myopathy associated with muscle inflammation. Neuromuscul Disord. 2003;13:830–834. doi: 10.1016/s0960-8966(03)00140-8. [DOI] [PubMed] [Google Scholar]
- 76.Amouri R, Driss A, Murayama K, Kefi M, Nishino I, Hentati F. Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscul Disord. 2005;15:361–363. doi: 10.1016/j.nmd.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 77.Voermans NC, Guillard M, Doedee R, Lammens M, Huizing M, Padberg GW, Wevers RW, van Engelen BG, Lefeber DJ. Clinical features, lectin staining, and a novel GNE frameshift mutation in HIBM, Neuromuscul. Disord. 2009 under revision. [PMC free article] [PubMed] [Google Scholar]
- 78.Yabe I, Higashi T, Kikuchi S, Sasaki H, Fukazawa T, Yoshida K, Tashiro K. GNE mutations causing distal myopathy with rimmed vacuoles with inflammation. Neurology. 2003;61:384–386. doi: 10.1212/01.wnl.0000061520.63546.8f. [DOI] [PubMed] [Google Scholar]