

Abstract
The p53 protein responds to cellular stress and regulates genes involved in cell cycle, apoptosis, and DNA repair. Under normal conditions, p53 levels are kept low through MDM2-mediated ubiquitination and proteosomal degradation. In search for novel proteins that participate in this regulatory loop, we performed an MDM2 peptide pull-down assay and mass spectrometry to screen for potential interacting partners of MDM2. We identified ribosomal protein S3 (RPS3), whose interaction with MDM2, and notably p53, was further established by His and GST pull-down assays, fluorescence resonance energy transfer and an in situ proximity ligation assay. Additionally, in cells exposed to oxidative stress, p53 levels increased slightly over 24 hrs, whereas MDM2 levels declined after 6 hrs exposure, but rose over the next 18 hrs of exposure. Conversely, in cells exposed to oxidative stress and harboring siRNA to knockdown RPS3 expression, decreased p53 levels and loss of the E3 ubiquitin ligase domain possessed by MDM2 was observed. DNA pull-down assays using a 7, 8-dihydro-8-oxoguanine duplex oligonucleotide as a substrate found that RPS3 acted as a scaffold for the additional binding of MDM2 and p53, suggesting that RPS3 interacts with important proteins involved in maintaining genomic integrity.
Keywords: Ribosomal protein S3, p53, MDM2, oxidative stress, ubiquitination
1. Introduction
P53 is a transcription factor that can regulate numerous downstream targets to induce cell cycle arrest, apoptosis and DNA repair [1, 2]. It is activated by a variety of circumstances including DNA damage, oxidative stress and hypoxia. Cells that encounter such conditions either arrest cell division or undergo apoptosis if they have a functional p53 pathway.
In the absence of stress signals, the p53 protein is kept in check by MDM2 through different mechanisms, one of which includes acting as an E3 ubiquitin ligase to target p53 for proteosomal degradation [3, 4]. Also, low levels of MDM2 induce monoubiquitination and nuclear export of p53, whereas high levels promote its polyubiquitination and nuclear degradation [3–6]. This pathway also involves AKT1, as it phosphorylates MDM2 and results in the translocation of MDM2 to the nucleus where it binds to p53 and targets it for degradation by the proteosome [7, 8].
When cells are exposed to stress, DNA damage inducible kinases are rapidly activated to phosphorylate MDM2 at an N-terminal Serine [9], leading to destabilization of this protein through MDM2-mediated autoubiquitination. In regards to p53, DNA damage activates ATM kinase to phosphorylate S15 of p53, with a subsequent phosphorylation occurring at T18 and S20. These amino acids ordinarily interact with a hydrophobic pocket in the MDM2 N-terminus, but upon phosphorylation of T18 of p53, a destabilization results with subsequent dissociation of the p53/MDM2 complex [10]. Furthermore, MDM2 is a substrate for DNA-PK that can modify S17 which causes a structural change that prevents its association with p53 [11].
In an attempt to identify other novel proteins that interact with MDM2, we performed a co-precipitation of proteins bound to a biotinylated MDM2 peptide attached to streptavidin beads in which ribosomal protein S3 (RPS3) was one of the two proteins captured, and subsequently led to the discovery that RPS3 also interacts with p53 to protect it from MDM2-mediated ubiquitination.
2. Materials and methods
2.1. Cell culture and exposure to hydrogen peroxide and Nutlin-3
HEK 293 cells were cultured and exposed for up to 24 h to hydrogen peroxide (0.125 mM to 0.25 mM) as previously described [12]. Exposure of cells to hydrogen peroxide in the presence of Nutlin-3 (Sigma) was performed by pre-treating the cells with 10 μM inhibitor or DMSO vehicle for 1 h prior to the addition of hydrogen peroxide.
2.2. Cloning
Complete coding sequences of human RPS3 (wild type, T42D and T42A), p53 and MDM2 were cloned into pEYFP-C1, pEYFP-N1, pECFP-C1, and pECFP-N1 (Clontech) expression vectors to generate CFP tagged and YFP tagged constructs. For subcloning wild type RPS3 sequence from pcDNA-RPS3 into CFP and YFP vectors, EcoR1 and BamH1 restriction sites were used. T42D and T42A constructs were subcloned from previously described GFP constructs [12] by using EcoR1 and BamH1 sites. Wild type p53 sequence was amplified by PCR from p53-SN3 [13] construct using primers containing EcoR1 and BamH1 sites (forward primer: 5′-GCG GAT CCA TGG AGG AGC CAC AGT CAG A-3′; reverse primer: 5′-GCG AAT TCT TAG TCC GAG TCA GGC CCC-3′) and then used for inserting p53 sequence into pECFP and pEYFP vectors. HindIII and BamHI restriction sites were used to generate CFP and YFP constructs of MDM2. All restriction enzymes were purchased from New England Biolabs and cloning was performed according to the standard molecular biology protocols. Briefly, pECFP/pEYFP vectors and plasmids carrying the sequence of interest were digested with indicated restriction enzymes according to the manufacturer’s instructions. The digestion reactions were cleaned with QIAquick nucleotide removal kit (Qiagen) or QIAquick gel extraction kit (Qiagen) and subsequently used for ligating RPS3, p53 and MDM2 fragments into pECFP and pEYFP vectors in the presence of T4 DNA ligase. Chemically competent TOP10 E. coli (Invitrogen) was transformed using 5 μl of the ligation mixture, and bacterial colonies bearing the insert were selected on LB-agar plates containing 50 μg/ml of kanamycin. Plasmid DNAs were purified from individual clones using a QIAprep spin miniprep kit (Qiagen) and the presence of the expected insert was confirmed by sequencing.
2.3. Transient transfection
HEK 293 cells were transiently co-transfected with CFP and YFP tagged constructs using FuGENE HD transfection reagent (Roche Applied Science). Transient knockdown of cellular RPS3 was achieved by transfecting with RPS3 specific siRNA (Dharmacon) using Dharmafect 1 transfection reagent (Dharmacon) as previously described [14].
2.4. Fluorescence resonance energy transfer (FRET) analysis by laser scanning confocal microscopy
Cells co-transfected with CFP and YFP constructs were fixed in 10% neutral buffered formalin and washed in PBS before being mounted onto slides using Vectashield mounting media. Using acceptor photobleaching method [15], protein:protein interactions were analyzed by using a Zeiss laser scanning confocal microscope (LSM 510 Meta). FRET efficiency (E) was calculated using the equation E=1−(IDA/ID), where IDA and ID represent steady state CFP fluorescence in the presence and absence of the YFP, respectively. FRET efficiency was determined for a minimum of 50 cells of same fluorescence intensity and used for statistical manipulations.
2.5. Antibodies
Custom synthesized rabbit monoclonal RPS3 antibody (Proteintech) was used for immunoblotting and immunofluorescence. Anti-p53 antibody (DO-1) was purchased from Santa Cruz Biotechnology. The mouse monoclonal cocktail prepared from IF2, 4B11 and 2A10 antibodies from EMD Biosciences was used for detecting MDM2 by immunoblotting. MDM2 antibody, clone IF2 was used for immunofluorescence applications. Anti-glyceraldehyde 3-phosphate dehydrogenase antibody (GAPDH) was purchased from Chemicon.
2.6. Duolink in situ proximity ligation assay for protein: protein interactions
Duolink proximity ligation assay kit composed of anti-rabbit PLA probe plus, anti-mouse PLA probe minus and detection kit 613 was purchased from Olink Bioscience. Formalin fixed cells were permeabilized using 0.1% triton-X100 and blocked overnight at 4 °C in 1% BSA. Primary antibody mixtures were prepared in the blocking solution by adding RPS3 (1:200) to p53 (DO1, 1:100) or MDM2 (IF2, 1:200) antibodies and cells were incubated with the mixture for 1 h at room temperature. All subsequent incubations were performed in a humidifying chamber maintained at 37 °C. PLA probes were diluted in blocking solution and all other Duolink reagents were diluted according to the manufacturer’s instructions. After 90 min incubation with the PLA probes, cells were washed in PBS and incubated with the hybridization mixture for 15 min and ligation mixture for an additional 15 min with a TBS-T (10 mM Tris [pH 7.5], 150 mM Nacl and 0.1% Tween 20) wash in between. After washing in TBS-T, cells were incubated with the amplification mixture for 90 min followed by the detection mixture for 1 h. The cells were then washed in 2 × SSC, 1 × SSC, 0.2 × SSC, 0.02 × SSC followed by 70% ethanol wash. Samples were air dried and mounted with Olink mounting media containing DAPI nuclear stain. Detection of the interaction signals was carried out by fluorescence microscopy using Zeiss Axioplan 2 upright microscope equipped with Photometrics Coolsnap HQ CCD camera. The filter sets used for visualizing the fluorescent signals include DAPI (EX 360/40, EM 460/50) and Texas Red (EX 560/55, EM 645/75).
2.7. 8-oxodG oligonucleotide pull-down assay
5′ biotinylated 8-oxodG oligonucleotide, a 37mer containing a single 8-oxodG residue at position 21 and control oligonucleotide having the same sequence as the 8-oxodG oligonucleotide except for the unmodified guanine at position 21, were custom synthesized by Sigma Genosys. Both oligonucleotides were subjected to duplex synthesis in individual reactions by incubating with equal amount of complementary strand oligonucleotide in annealing buffer (10 mM Tris [pH 7.6], 10 mM MgCl2, 1 mM EDTA) for 10 min at 75 °C. Duplexes were then allowed to cool down at room temperature and DNA concentration measured. The duplexes (750 ng) were then immobilized onto anti-biotin antibody labeled agarose beads by incubating with 20 μl of the beads in binding buffer (20 mM Hepes [pH 7.9], 100 mM KCl, 2 mM MgCl2, 0.5 mM EDTA, 1 mM DTT, 0.4 mM ZnSO4, 40 μM ZnCl2, 10% Glycerol and 0.1% Triton X-100) for 1 h at 4 °C. DNA bound beads were collected, washed two times in the binding buffer and incubated with a 150 ng aliquot of purified RPS3, p53 and MDM2 proteins at 25 °C for 30 min. Protein complexes bound to beads were then collected, washed 4 times in binding buffer and eluted into 20 μl of Laemmli sample buffer. After separating on SDS-PAGE and electrotransfer, proteins were immunoblotted with antibodies to RPS3, p53 and MDM2.
2.8. Purification of recombinant proteins
Overexpression and subsequent purification of GST-RPS3 and His-tagged p53 and MDM2 were according to previously described procedures [8,11,16]
2.9. His6 pull-down assay
NiNTA beads re-suspended in binding buffer (PBS [pH 7.6], and 1 mM DTT) were mixed with purified His-p53 (500 ng) or His-MDM2 (2 μg) and incubated on a rotator at 4 °C for 1 h. The complex was then pelleted, washed 3 times in binding buffer and re-suspended in 1 ml of binding buffer containing 500 ng of GST-RPS3 and returned to the rotator for 1 h incubation. After washing the beads for four times in binding buffer, protein complexes were eluted into Laemmli sample buffer and denatured at 100 °C. After SDS-PAGE and electrotransfer to PVDF membranes, proteins were immunoblotted using anti-GST and anti-MDM2 primary antibodies and HRP-goat-anti-mouse secondary antibody.
2.10. GST pull-down assay
Whole cell lysates were prepared from HEK 293 cells in modified RIPA buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP40, 10% glycerol, 25 mM NaF) supplemented with protease and phosphatase inhibitors. Equal amount of protein (1 mg) from the clarified lysate was added to GST, GST-RPS3, or GST-RPS3 deleted for the KH domain (GST-RPS3-ΔKH; aa 41–111) immobilized onto glutathione- agarose beads and incubated on a shaker for 4 h at 4 °C. Protein complexes bound to agarose beads were then collected, washed 4 times in modified RIPA buffer, and eluted into Laemmli sample buffer. After separation of proteins by SDS-PAGE, gels were either Coomassie stained or electrotransferred for subsequent immunoblotting analysis using p53 and MDM2 antibodies.
2.11. In vitro p53 ubiquitination assay
The in vitro ubiquitination reaction contained UbE1 (activating enzyme), UBcH5c (conjugating enzyme), MDM2, ubiquitin, and p53. Wild type MDM2 or ubiquitin ligase defective mutant, MDM2-C464S, at 2 μg concentration in combination with E1 (4.5 μM), E2 (130 μM) and 1 mg of ubiquitin were added to 500 ng of p53 in reaction buffer. For the reactions carried out in the presence of RPS3, various concentrations of purified GST-RPS3 was added. After incubation at 37 °C for 2 h, p53 ubiquitination was examined by resolving the reactions on SDS-PAGE, transferring on to PVDF membrane and immunoblotting with p53 DO-1 antibody.
2.12. MDM2 peptide pull-down and mass-spectrometry identification of proteins
Whole cell lysates were prepared from approximately 18 × 106 H1299 cells in 4.5 ml NP40 lysis buffer (50 mM Tris-HCl [pH 7.5], 10% (v/v) glycerol, 0.1% NP-40, 150 mM NaCl), supplemented with complete protease inhibitor mixture. Biotin labeled MDM2 peptides (12 μg) were coupled to 30 μl of packed streptavidin-agarose beads in PBS for 1 h at 4 °C and then incubated for 1 h at 4 °C with 1 ml aliquots of clarified cell lysates. After washing the beads four times with NP40 lysis buffer, protein complexes were eluted and separated by SDS-PAGE for subsequent Coomassie gel staining. Bands appearing exclusively in the samples obtained with the wild type MDM2 peptide were excised, eluted, and identified by mass spectrometry. Mass spectrometry analysis was performed by the Fingerprints proteomics unit, Wellcome Trust Biocenter, Dundee, Scotland, UK. Tryptic peptides were analyzed on a Perspective Biosystems Elite STR matrix-assisted laser desorption time of flight-mass spectrometer (Framingham, MA), with saturated -cyanocinnamic acid as the matrix. The mass spectrum was acquired in the positive reflector mode and internally mass calibrated. The tryptic peptide ions obtained were scanned against the Swiss-Prot and Genpep data bases using the MASCOT program. SDS-PAGE separated samples were also analyzed by western blot using a rabbit polyclonal serum raised against a peptide encompassing the 20 C-terminal residues of human RPS3.
3. Results
3.1. RPS3 is trapped by a MDM2 peptide pull-down assay
In an effort to identify novel proteins that interact with MDM2, we used a biotinylated MDM2 acidic domain peptide (Fig. 1A) bound to streptavidin beads to capture proteins that interact with MDM2. Whole cell extracts from H1299 cells were prepared, and incubated with biotinylated peptides immobilized onto streptavidin beads. Two proteins were subsequently identified by Coomassie staining as major co-precipitants (Fig. 1C, left panel, lane 3). Mass spectrometry found sequence similarities to ribosomal proteins S3 (RPS3; Fig. 1D) and S20. Western blot analysis of the same samples showed that the 28 Kd band cross reacted with a rabbit polyclonal antibody to human RPS3 (Fig. 1C, right panel, lane 3). Notably, when a human MDM2 acidic domain mutant peptide (Fig. 1B) was used as bait, RPS3 and S20 were not co-precipitated (Fig. 1C, left panel, lane 4). However, when full length MDM2 possessing the acidic domain mutation was used as bait, binding to RPS3 was not affected (Fig. 1E), suggesting that the interaction domain between RPS3 and MDM2 is more complex than a total reliance on its acidic domain. The interaction of p53 and MDM2 is similar in this regard as it requires a site in the N-terminus of MDM2 as well as MDM2’s acidic domain region [17].
Fig. 1.

RPS3 interacts with MDM2 as determined by biotin tagged peptide pull -down assay using MDM2 peptides. (A) Biotin-labeled MDM2-acidic domain peptide comprising the amino acid segment 243–257, which lies within the central acidic domain of MDM2 (AA 230 – AA 300). (B) MDM2 acidic domain mutant peptide with three amino acids (EVE) within the wild type peptide mutated to alanine. (C) Whole cell lysates prepared from H1299 cells were incubated with Biotin-labeled MDM2-acidic domain peptide or MDM2 acidic domain mutant peptide that were immobilized onto streptavidin-agarose beads. After washing the beads, protein complexes were purified, separated on SDS-PAGE and Coomassie stained (left panel) Lane 1 is the total cell extract, lane 2 is the beads only control, lane 3 is the wild type MDM2 peptide and lane 4 is the acidic domain mutant MDM2 peptide. Protein bands appearing only in lane 3 were excised and subjected to tryptic digestion and mass spectrometry analysis. RPS3 and RPS20 were identified in this way. In the right panel, proteins were electro-transferred and analyzed by immunoblotting the membrane with an antibody specific to RPS3. The same result was obtained when using U2OS cells (not shown) (D) Amino acid sequence of the human RPS3, showing the tryptic peptides (red) used for protein identification. (E) U2OS cells were transfected with plasmids expressing human MDM2, an MDM2 mutant with the same mutation in the acidic domain as the peptide in (B), and/or an expression vector for HA-tagged RPS3. Cell extracts were immunoprecipitated with 2A10 mouse monoclonal antibody against MDM2. Proteins were detected by Western blot using an anti-HA antibody or the mouse monoclonal antibody 4B2 against MDM2.
3.2. RPS3 interacts with MDM2 and p53
Using a His-tagged pull-down assay, we show that a physical interaction exists between RPS3 and MDM2. Purified His-MDM2 immobilized on NiNTA beads was mixed with GST or GST-RPS3, incubated for 3 h, and protein complexes then eluted into Laemmli buffer and subjected to electrophoresis and immunoblotting with anti-GST or anti-MDM2 antibody. This experiment showed a physical interaction existed between RPS3 and MDM2 (Fig. 2A). No interaction was detected between GST and MDM2 (Fig. 2A).
Fig. 2.

Interaction of MDM2 and p53 with RPS3 by protein pull-down assays. (A) Purified His-MDM2 immobilized on to NiNTA beads were mixed with GST or GST-RPS3 contained in binding buffer and incubated for 1 h at room temperature. Beads were then collected, washed and protein complexes eluted into Laemmli buffer. After resolving the protein complexes on SDS-PAGE, the interaction of RPS3 with MDM2 was analyzed by immunoblotting by probing the membrane with GST and MDM2 antibodies. (B) Reaction mixtures containing His-p53 bound to NiNTA beads and purified GST or GST-RPS3 proteins were processed as in (A) and the interaction of RPS3 with p53 was demonstrated by using an antibody to GST. (C) GST pull-down was performed by incubating HEK 293 whole cell lysate with GST (lane 2), GST-RPS3 (lane 3) or GST-RPS3-ΔKH (lane 4) immobilized onto agarose beads. After elution and SDS-PAGE, proteins were observed by Coomassie staining (left panel) and identified by immunoblotting with p53 and MDM2 antibodies (right panel). The whole cell lysate used for the pull-down (lane 1) is shown as a control for total protein.
A similar experiment was performed to that shown in Fig. 2A, but in this case we tested whether RPS3 interacted with p53. Notably, RPS3 was found to have a robust association with p53 (Fig. 2B). In order to confirm the interaction with p53, we performed a GST pull-down assay using HEK 293 cell lysate and demonstrated by immunoblot that RPS3 binds to endogenous p53 (Fig. 2C, top right panel, lane 3). We also detected binding to MDM2 (Fig. 2C, bottom right panel, lane 3), thus establishing that a physical interaction existed between RPS3, p53, and MDM2.
To identify the interaction domain of RPS3 involved in its binding to p53 and MDM2 as well as to eliminate the possibility of non-specific protein-protein interactions that might be a reason for our in vitro results, we conducted additional GST pull-down assays with a GST-RPS3-ΔKH deletion (aa 41–111) that has previously been shown to be needed for the interaction between RPS3 and p65 [14]. We found that the same KH domain was essential for the interaction of RPS3 with p53 and with MDM2 (Fig. 2C, right panel, lane 4)
3.3. Fluorescence resonance energy transfer (FRET) and in situ proximity ligation assay (PLA) confirm RPS3 interacts with both MDM2 and p53
We used confocal microscopy to test that the interaction between RPS3, MDM2, and p53 takes place at a single cell level. Wild type constructs of p53 or MDM2 were tagged at the N-terminus with cyan fluorescent protein (CFP-p53 or CFP-MDM2). These constructs were co-expressed with wild type RPS3 tagged at the N-terminus with yellow fluorescent protein (YFP-RPS3). FRET analysis was then employed, which is a system based upon energy transfer from a donor fluorophore to an acceptor fluorophore with an efficiency that depends on the distance between the two fluorophores (< 100 Å). Under conditions where no interactions were detected for vectors alone (CFP and YFP, data not shown), interaction of RPS3 and p53 by FRET was detected in cells co-transfected with CFP-p53 and YFP-RPS3 (Figure 3A, left panel). The interaction between p53 and RPS3 appeared to be evenly distributed between the cytosol and nucleus. Tests to determine the interaction between RPS3 and MDM2 showed that a robust interaction occurred in the cytoplasm between CFP-MDM2 and YFP-RPS3, whereas the level of nuclear interaction was virtually undetectable (Fig. 3A, right panel).
Fig. 3.

FRET analysis to determine the interactions between RPS3/p53 and RPS3/MDM2. (A) HEK 293 cells co-transfected for 24 h with pECFP-N1-p53 and pEYFP-N1-RPS3 (left panel), pECFP-N1-MDM2 and pEYFP-N1-RPS3 (right panel) were left untreated or treated with 0.25 mM H2O2. At 24 h of treatment with H2O2, samples were fixed, washed and mounted onto slides and analyzed for FRET by confocal microscopy using acceptor photobleaching method. Cells showing equal prebleach intensities of YFP were photobleached with 514 nm laser using time bleach protocol and a total of 10 images recorded to calculate the FRET efficiency. After plotting the fluorescence intensities of 5 pre-bleach and 5 post-bleach images over time, the efficiency of FRET (E) was calculated using E=1−(IDA/ID) where IDA and ID represent steady state CFP fluorescence in the presence and absence of the YFP, respectively. (B) FRET analysis was performed on HEK 293 cells which were transfected similar to that described for corresponding left and right panels of (A) but pre-treated with 10 μM Nutlin-3 or the carrier DMSO before being treated with 0.25 mM H2O2 for 24 h. (C) HEK 293 cells were co-transfected for 24 h with phosphorylation mimic T42D or phosphorylation deficient T42A mutants of RPS3 cloned into pEYFP-N1 and pECFP-N1-p53 (left panel) or, pECFP-N1-MDM2 (right panel). After treatment with H2O2 for 24 h, FRET analysis was performed as described for (a).
Previous results have shown that there is a distinct pool of RPS3 that is cytosolic and ribosome-free. It is this population of RPS3 that is hypothesized to play a major role in NF-κB signaling [14] as well as its translocation to the nucleus upon oxidative stress [12]. In the later case, we have previously shown that roughly 15% of total RPS3 is nuclear but, under oxidative stress conditions, cytosolic RPS3 is translocated via an ERK1/2 mediated pathway that raises the nuclear pool to 40%. We therefore tested whether the interaction between RPS3 and p53 or MDM2 intensified under oxidative stress. In cells exposed to hydrogen peroxide (over 80% survival, [12]), the interaction between RPS3 and p53 increased over two-fold in both the cytosol and the nucleus (Fig. 3A, left panel). An increase due to oxidative stress was not apparent for the interaction between RPS3 and MDM2 in the cytosol, but became significant in the nucleus (Fig. 3A, right panel).
To understand at a greater depth the nature of the interaction between RPS3 and p53 or MDM2, we used the cis-imidazoline analog Nutlin-3, which is known to be a potent inhibitor of the MDM2 interaction with p53 [18]. The addition of Nutlin-3 and hydrogen peroxide to cells ectopically expressing CFP-p53 and YFP-RPS3 produced a decrease in the interaction between p53 and RPS3 (E=15) in the cytosol when compared to cells exposed to hydrogen peroxide but not Nutlin-3 (E= 25). Conversely, cells exposed to Nutlin-3 and hydrogen peroxide created over a two-fold decrease in the RPS3-p53 interaction in the nucleus (E=35) when compared to cells not exposed to Nutlin-3 (E=15; Fig. 3B, left panel). On the other hand, Nutlin-3 had little if any effect on the cytoplasmic or nuclear interaction between RPS3 and MDM2 (Fig. 3A and B, right panels). It therefore appears that for optimal RPS3 interaction levels with p53 in the nucleus, MDM2 would appear to be necessary to be present and interact with p53.
As an additional means of confirming that RPS3’s interaction with p53 and MDM2 is legitimate, we took advantage of RPS3 mutant-constructs that are abrogated at the ERK1/2 phosphorylation site (T42). One of the RPS3 mutants has previously been shown [12] to be present exclusively in the nucleus as a phosphorylation mimic (T42D), whereas the other (T42A) is totally found in the cytoplasm since it is refractive to ERK1/2 mediated phosphorylation. Using FRET analysis, we found that cells ectopically expressing YFP-T42D no longer was able to interact with cytoplasmic CFP-p53 (compare left panels of Fig. 3A and Fig. 3C), but did maintain the ability to interact with nuclear CFP-p53 (Fig. 3C, left panel). For the mutant refractive to ERK1/2 mediated phosphorylation, we found that YFP-T42A only interacted with cytoplasmic CFP-p53 (Fig. 3C, left panel). The same experiments were performed using ectopically expressed CFP-MDM2 (Fig. 3C, right panel), in which we failed to once again see interaction in the nucleus for the phosphorylation-minus mutant YFP-T42A, as well as a lack of interaction in the cytoplasm for the phosphorylation mimic YFP-T42D. It also appears that the intensity of interactions between RPS3 and MDM2 are unaffected by oxidative stress.
As a final means of establishing the interaction between RPS3 and p53-MDM2, we employed in situ PLA. This technique combines dual recognition of target proteins using secondary antibodies with attached DNA strands as proximity probes, forming templates for rolling circle amplification for detecting protein interactions. In untreated cells, only a modest amount of interaction was visualized between RPS3 and p53 or MDM2 (Fig. 4, panel ii and v). However, when cells were challenged with oxidative stress, interactions were clearly visible (Fig. 4, panels iii and vi). In the case of RPS3 and p53, the overlap detected by fluorescence microscopy showed that the interaction between these two proteins was evenly distributed between the nucleus and cytoplasm, as appeared to be the case for RPS3 and MDM2 as well. Taken together, the above experiments specifically connect RPS3 with both p53 and MDM2, in which the intensity of interaction in this case seems to improve under oxidative stress conditions.
Fig. 4.

In Situ proximity ligation assay (PLA) confirms the interactions of RPS3 with p53 and MDM2. Olink in situ PLA was performed on HEK 293 cells grown on chamber slides. Untreated or H2O2 (0.25mM) treated cells for 24 h were fixed and processed for in situ PLA by incubating with specific antibody mixtures of p53/RPS3 (panels ii and iii) and MDM2/RPS3 (panels v and vi). After the subsequent incubation steps with PLA probes, hybridization, ligation, polymerization and detection mixtures, samples were mounted with DAPI containing mounting medium and subjected to epifluorescence microscopy using texas red (red) and DAPI (blue) filters. For each sample, a representative image, which is a merge of the red and blue channels, is shown. In panels i (p53 antibody only) and iv (MDM2 antibody only), RPS3 antibody was omitted and used as negative controls to demonstrate the specificity of the protein:protein interactions.
3.4. DNA pull-down assays show that the interaction between RPS3 and p53/MDM2 can take place at sites of 7, 8-dihydro-8-oxoguanine (8-oxodG)
We have previously shown that the translocation of cytosolic RPS3 to the nucleus can be traced by immunofluorescence microscopy to its co-localization with the mutagenic DNA lesion 8-oxodG [12]. The use of surface plasmon resonance confirmed that RPS3 possesses an extremely robust apparent binding affinity (4.82 × 10 −11 M) for sensor chips containing 8-oxodG [19]. We therefore questioned whether the binding of RPS3 to 8-oxodG could act as scaffold for the subsequent recruitment of p53 and/or MDM2. To test this, we used a duplex 5′ biotinylated 8-oxoG oligonucleotide 37mer (8-OG) or control 37mer (CT) as in vitro substrates for RPS3, and subsequently asked whether p53 and or MDM2 could be captured in a complex with RPS3 bound to 8-OG. Briefly, anti-biotin antibody conjugated agarose beads were incubated with CT or 8-OG 37mers, then purified and subsequently used as substrates for purified RPS3 and/or p53 and/or MDM2 proteins. After 30 min incubation at room temperature, DNA-protein complexes bound to beads were collected by centrifugation, resuspended in loading buffer and subjected to SDS-PAGE and probed by western blotting. As expected, we found RPS3 bound to 8-OG, but not to CT 37mer (Fig. 5A). When p53 was tested, binding to 8-OG or CT 37mers was not detected (Fig. 5C). Similar results were found for MDM2 and its lack of binding to 8-OG or CT 37mers (Fig. 5B). Conversely, p53 (Fig. 5A), MDM2 (Fig. 5B), or both proteins together (Fig. 5C) were identified in the pull-down experiments with an 8-OG 37mer, but only when RPS3 was present and bound to 8-OG.
Fig. 5.
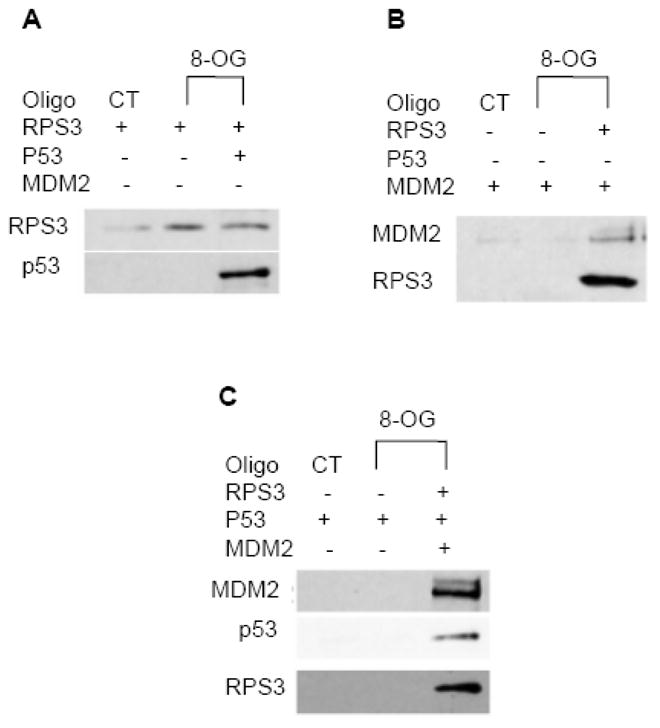
RPS3/p53 and RPS3/MDM2 interactions can localize at 8-oxodG sites as determined by in vitro 8-oxodG duplex 37mer pull-down assay. (A) 5′-biotinylated control (CT) or 8-oxodG (8-OG) 37mer oligonucleotides were hybridized by incubating with the complementary oligonucleotide in annealing buffer for 10 min at 75 °C. Equal amounts of duplexes were then immobilized onto anti-biotin antibody labeled agarose beads and then incubated with purified RPS3 and p53 proteins as shown. After washing the beads, DNA-protein complexes were eluted into Laemmli sample buffer and resolved on SDSPAGE followed by electrotransfer on to nitrocellulose membranes which were subsequently blotted with RPS3 and p53 antibodies. (B) Reactions containing CT or 8-OG duplex oligonucleotides and purified RPS3 and MDM2 proteins were processed as described in (A) and analyzed for interactions by immunoblotting the membranes with RPS3 and MDM2 antibodies. (C) DNA oligonucleotides pull-down assays were carried out by mixing CT or 8-OG duplex oligonucleotides with RPS3, MDM2 and p53 proteins as shown, and interactions were analyzed by immunoblotting the membrane with RPS3, MDM2 and p53 antibodies.
3.5. RPS3 protects p53 from MDM2-mediated ubiquitination in vitro
Given that RPS3 interacts with both p53 and MDM2, we next explored how RPS3 might affect these proteins in an in vitro assay that measures the ubiquitination of p53 by MDM2. As expected, purified MDM2 was able to ubiquitinate purified p53 as reflected by the numerous slower migrating bands shown by SDS-PAGE and western analysis (Fig. 6, lane 4). In reactions containing increasing amounts of purified RPS3, the ubiquitination of p53 by MDM2 significantly decreased. The levels of p53-ubiquitination in the presence of increasing amounts of RPS3 eventually produced a result similar to that seen for reactions containing the ubiquitination ligase defective mutant C464S of MDM2 (Fig. 6, lane 3).
Fig. 6.

Effect of RPS3 on in vitro ubiquitination of p53 by MDM2. For the in vitro ubiquitination assay, 500 ng of purified p53 (8 pmol) was mixed with 2 μg of recombinant MDM2 (wild type, 20 pmol) or MDM2 C464S (E3 ubiquitin ligase defective) proteins and added to the ubiquitination reaction containing E1 (4.5 μM), E2 (130 μM) and 1 mg of ubiquitin. The reactions were then incubated with increasing concentrations (2.5, 25, and 250 ng; 5 pmol) of RPS3. p53 ubiquitination was examined by resolving the reactions on SDS-PAGE, transferring on to PVDF membrane and immunoblotting with p53 DO-1 antibody. Light and dark (above and below) exposures of the western blot show p53 laddering, representing ubiquitination.
3.6. RPS3 knockdown results in decreased p53 and MDM2 levels
Since the presence of RPS3 appeared in vitro to protect p53 from MDM2 ubiquitination, we next determined the effect of RNAi-mediated knockdown of RPS3 on the protein levels of both MDM2 and p53. We used a siRNA that was previously reported by us to decrease the total amount of RPS3 protein by about 50% [14]. This level of knockdown has been shown to have no adverse affects on protein synthesis that might contribute to ribosomal instability. Furthermore, RNAi knockdown appeared to only affect cytosolic, ribosome-free RPS3 levels, whereas ribosomal-bound RPS3 was not affected [14].
Cells were transiently transfected with a siRNA construct specific for RPS3, or with a scrambled, non-specific siRNA control construct (NS), and exposed to oxidative stress for various periods of time for up to 24 h. Protein extracts were separated by SDS-PAGE and analyzed by western analysis. In control cells (NS), p53 levels increased roughly 60% when cells were exposed to oxidative stress (Fig. 7A). On the other hand, in cells transiently transfected with RPS3 siRNA, there was roughly 65% as much p53 that did not substantially change in the first 12 h after exposure to oxidative stress. The levels of MDM2 in control (NS) cells clearly changed in response to oxidative stress, in which there was a substantial 80% decrease within the first 6 h of exposure to hydrogen peroxide, and then an incremental increase over the next 18 h for cells transiently transfected with a scrambled siRNA (Fig. 7B). The changes in MDM2 levels in cells transiently transfected with RPS3 siRNA also showed a temporary 80% drop in MDM2 levels that incrementally increased after 24 h exposure to oxidative stress. However, western analysis revealed that the faster migrating band identified by our antibody cocktail to MDM2 was much fainter in cells expressing siRNA to knockdown RPS3 (Fig. 7B). Taken together, our results show decreases in both p53 and MDM2 levels (oxidative-stress dependant) in cells expressing siRNA for the knockdown of RPS3.
Fig. 7.

Effect of RPS3 knockdown on cellular MDM2 and p53 levels in HEK 293 Cells. (A) Cells were transfected with non-silencing iRNA (NS) or RPS3 specific iRNA (RPS3) for 24 h and exposed to 0.125 mM H2O2 (>80% survival) for various periods of time, as shown. Whole cell lysates were prepared and 20 μg aliquot of protein extract from each sample was immobilized by SDS-PAGE. After electro-transfer, cellular p53 levels were detected by immunoblot analysis utilizing the mouse monoclonal anti-p53 antibody. The same blot was stripped and re-probed with anti-GAPDH antibody to normalize for the protein loading. After densitometry, integrated density value (IDV) for each protein band was determined and normalized levels of p53 were calculated by dividing the IDV of a protein band by the IDV of the GAPDH within the same sample. (B) Normalized cellular MDM2 levels in the same lysates as in (A) were determined by immunoblotting with an anti-MDM2 antibody and densitometry analysis as described in (A).
4. Discussion
Even though the existence of the p53/MDM2 regulatory loop has been known for some time, the system most likely contains as yet identified components as our studies suggest. The unexpected finding that RPS3 was trapped using a biotinylated peptide of MDM2 as bait led us to examine whether RPS3 also interacted with p53, which in fact was observed using several different in vitro and in vivo assays.
RPS3 was originally identified as a component of the small ribosomal subunit where it is involved in protein synthesis [20,21]. We subsequently found that RPS3 is involved in an oxidative-stress signaling pathway that involves its translocation to the nucleus and co-localization with foci of 8-oxodG [12]. RPS3’s ability to also enhance the catalytic activity of BER proteins [22] pointed to the multifunctional roles RPS3 participates in. More recently, that view was underscored by the surprising discovery that RPS3 is a subunit of nuclear NF-κB complexes [14]. The translocation of RPS3 to the nucleus has been shown to occur by CD3/CD28 (TCR) and TNFα stimulation [14], as well as ERK1/2 mediated phosphorylation in response to oxidative stress [12].
Our findings show that RPS3 interacts with both p53 and MDM2. Furthermore, the interaction depends upon the KH domain possessed by RPS3. This domain was originally defined as an important ingredient for binding to single-stranded RNA and DNA [23]. As an example, hnRNP K has been found to be associated with RNA in RNP particles [24], as well DNA recognition [25,26]. Notably, hnRNP K has also been found to be an important co-activator of p53 in response to DNA damage [27]. More recently, the KH domain of RPS3 was found to be essential for the interaction between it and p65 in NF-κB complexes [14]. Thus, KH domains offer an important ingredient in DNA damage response and transcriptional activities.
Using the highly sensitive FRET assay for demonstrating protein:protein interactions at single-cell resolution, we observed a robust interaction between RPS3 and p53 that rose over two-fold when cells were exposed to oxidative stress. Similar observations were made using the equally sensitive in situ proximity ligation assay. In regard to the interaction between RPS3 and MDM2, it is clear using FRET that these proteins interact, but whether the association is enhanced upon exposure to oxidative stress is not as clear as that observed for the interaction between RPS3 and p53. The FRET results appear to suggest a moderate increase in the interaction between RPS3 and MDM2 under oxidative stress conditions, whereas the in situ proximity ligation assay clearly shows a robust increase in the intensity of interaction between RPS3 and MDM2 when cells were exposed to oxidative stress.
Our data for experiments performed in the presence of Nutlin-3 imply that the interaction mechanism between RPS3 and p53 is different than that between RPS3 and MDM2. For example, the presence of Nutlin-3 reduced over two-fold the nuclear interaction between p53 and RPS3 under oxidative-stress conditions. On the other hand, Nutlin-3 had little if any affect on the interaction between RPS3 and MDM2, regardless of oxidative conditions or subcellular location. These results therefore imply that the optimal nuclear interaction for RPS3 with p53 is likely more efficient in the added presence of MDM2.
Our in vitro and in vivo data suggest that at least one biological endpoint of the interaction of RPS3 with p53 and MDM2 is to protect p53 from MDM2-mediated ubiquitination. This was suggested in cells expressing a siRNA duplex to knockdown RPS3 that resulted in a 30% to 40% decrease for p53 and the loss of one of the domains possessed by MDM2. It is known that ribosomal instability can give rise to the binding of ribosomal proteins L5, L11, and L23 to MDM2 [28–30]. A question therefore arises as to whether the conditions chosen for the experiments presented in Fig. 7 was due to ribosomal stress brought on by the iRNA-generated partial decrease of endogenous RPS3 protein. This seems extremely unlikely based upon our previous results [14] using the same iRNA construct as was used here, in which we found using several methods that the partial knockdown (40–60%) of RPS3 had no effect on protein synthesis.
The results from our DNA pull-down assay offers the intriguing possibility that the presence of 8-oxodG in damaged DNA could act as staging position for the interaction of RPS3 with p53 and MDM2. What remains to be determined is how the interactions at 8-oxodG between RPS3, MDM2, and p53 are coordinated and controlled. Clearly, post-translational modifications of RPS3 could dictate its involvement in its interaction with both p53 and MDM2. It is known that p53 is post-translationally modified at roughly 30 different sites by phosphorylation, acetylation, methylation, O-glycosylation, neddylation, sumoylation and ubiquitination [31]. Similar post-translatioanal modifications for RPS3 have been identified, including ERK1/2 phosphorylation [12], Hrr25 phosphorylation [21], and neddylation [32]. We have also found that RPS3 is subject to ubiquitination and AKT phosphorylation (unpublished observations). Clearly, any or all of the above modifications could play an important role in the in vivo interactions established between RPS3, p53, and MDM2. What remains to be seen is how the interaction between RPS3, p53 or MDM2 affects genomic stability, especially at sites of 8-oxodG
Acknowledgments
We wish to thank Dr. Stu Linn for the suggestion that the moderation of DSB repair in human cells could involve the action of RPS3. We are grateful to Fengyi Wan and Michael Lenardo for providing us with the CFP and YPP vectors and GST-RPS3-ΔKH used in this study. This work utilized the facilities of the Cell Biology and Bioimaging Core that are supported in part by COBRE (NIH P20-RR021945) and CNRU (NIH 1P30-DK072476) center grants from the National Institutes of Health. This research was supported by a PBRC Pilot and Feasibility Project Award (S. Y.) and a National Institutes of Health Grant CA 109798 (W.A.D.).
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coutts AS, Adams CJ, La NB. Thangue p53 ubiquitination by Mdm2: A never ending tail? DNA Repair (Amst) 2009;8:483–490. doi: 10.1016/j.dnarep.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 3.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 4.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 5.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 6.Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006;13:927–934. doi: 10.1038/sj.cdd.4401912. [DOI] [PubMed] [Google Scholar]
- 7.Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–1303. doi: 10.1038/sj.onc.1205181. [DOI] [PubMed] [Google Scholar]
- 8.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. Embo J. 2004;23:1547–1556. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schon O, Friedler A, Bycroft M, Freund SM, Fersht AR. Molecular mechanism of the interaction between MDM2 and p53. J Mol Biol. 2002;323:491–501. doi: 10.1016/s0022-2836(02)00852-5. [DOI] [PubMed] [Google Scholar]
- 11.Mayo LD, Turchi JJ, Berberich SJ. Mdm-2 phosphorylation by DNA-dependent protein kinase prevents interaction with p53. Cancer Res. 1997;57:5013–5016. [PubMed] [Google Scholar]
- 12.Yadavilli S, Hegde V, Deutsch WA. Translocation of human ribosomal protein S3 to sites of DNA damage is dependant on ERK-mediated phosphorylation following genotoxic stress. DNA Repair (Amst) 2007;6:1453–1462. doi: 10.1016/j.dnarep.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 14.Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L, Hegde V, Lam LT, Staudt LM, Levens D, Deutsch WA, Lenardo MJ. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Centonze VE, Firulli BA, Firulli AB. Fluorescence Resonance Energy Transfer (FRET) as a method to calculate the dimerization strength of basic Helix-Loop-Helix (bHLH) proteins. Biol Proced Online. 2004;6:78–82. doi: 10.1251/bpo75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hegde V, Kelley MR, Xu Y, Mian IS, Deutsch WA. Conversion of the bifunctional 8-oxoguanine/beta-delta apurinic/apyrimidinic DNA repair activities of Drosophila ribosomal protein S3 into the human S3 monofunctional beta-elimination catalyst through a single amino acid change. J Biol Chem. 2001;276:27591–27596. doi: 10.1074/jbc.M101213200. [DOI] [PubMed] [Google Scholar]
- 17.Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell. 2006;23:251–263. doi: 10.1016/j.molcel.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 18.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 19.Hegde V, Wang M, Deutsch WA. Characterization of human ribosomal protein S3 binding to 7,8-dihydro-8-oxoguanine and abasic sites by surface plasmon resonance. DNA Repair (Amst) 2004;3:121–126. doi: 10.1016/j.dnarep.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Bommer UA, Lutsch G, Stahl J, Bielka H. Eukaryotic initiation factors eIF-2 and eIF-3: interactions, structure, and localization in ribosomal initiation complexes. Biochimie. 1991;73:1007–1019. doi: 10.1016/0300-9084(91)90142-n. [DOI] [PubMed] [Google Scholar]
- 21.Schafer T, Maco B, Petfalski E, Tollervey D, Bottcher B, Aebi U, Hurt E. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature. 2006;441:651–655. doi: 10.1038/nature04840. [DOI] [PubMed] [Google Scholar]
- 22.Hegde V, Wang M, Deutsch WA. Human ribosomal protein S3 interacts with DNA base excision repair proteins hAPE/Ref-1 and hOGG1. Biochemistry. 2004;43:14211–14217. doi: 10.1021/bi049234b. [DOI] [PubMed] [Google Scholar]
- 23.Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostareck-Lederer A, Ostareck DH. Control of mRNA translation and stability in haematopoietic cells: the function of hnRNPs K and E1/E2. Biol Cell. 2004;96:407–411. doi: 10.1016/j.biolcel.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Tomonaga T, Levens D. Heterogeneous nuclear ribonucleoprotein K is a DNA-binding transactivator. J Biol Chem. 1995;270:4875–4881. doi: 10.1074/jbc.270.9.4875. [DOI] [PubMed] [Google Scholar]
- 26.Tomonaga T, Levens D. Activating transcription from single stranded DNA. Proc Natl Acad Sci U S A. 1996;93:5830–5835. doi: 10.1073/pnas.93.12.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moumen A, Masterson P, O’Connor MJ, Jackson SP. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 28.Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004;24:7669–7680. doi: 10.1128/MCB.24.17.7669-7680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 30.Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol Cell Biol. 1994;14:7414–7420. doi: 10.1128/mcb.14.11.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kruse JP, Gu W. SnapShot: p53 posttranslational modifications. Cell. 2008;133:930–930. e931. doi: 10.1016/j.cell.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xirodimas DP, Sundqvist A, Nakamura A, Shen L, Botting C, Hay RT. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Rep. 2008;9:280–286. doi: 10.1038/embor.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]