

Abstract
Caloric restriction (CR) mitigates neurological damage arising from aging and a variety of other sources, including neuropathology in young adult mice that express single and double transgenic (tg) mutations associated with Alzheimer disease (AD). To evaluate the potential of CR to protect against relatively heavy AD-type pathology, middle-aged (13-14 month-old) mice that co-express two mutations related to familial AD, amyloid precursor protein (APP) and presenilin 1 (PS1), were fed balanced diets with 40% fewer calories than ad libitum-fed controls. Following 18 weeks of treatment, mice were killed and brains processed for quantification of total volume of amyloid-beta (Aβ) in the hippocampal formation and the overlying neocortex. Computerized stereology confirmed that CR reduced the total Aβ volume by about one-third compared to that in age-matched controls. Thus, CR appears to attenuate the accumulation of AD-type neuropathology in two cortical brain regions of middle-aged dtg APP/ PS1 mice. These findings support the view that CR could be a potentially effective, non-pharmacology strategy for reducing relatively heavy Aβ deposition in older adult dtg APP/ PS1 mice, and possibly afford similar protection against the onset and progression of AD in older adult humans.
Keywords: Caloric restriction, unbiased stereology, Stereologer, APP, PS1
Introduction
In the 1930s Mary Crowell and Clive McCay reported that rats submitted to calorie restriction (CR) live almost twice as long as non-restricted rats. Since that time, findings from a diverse range of species support the view that CR exerts beneficial effects on health and longevity [12, 15, 22, 31, 32]. In mammals, an inverse relationship exists between the degree of CR and morbidity associated with disease and early death [28, 30]. The mechanisms of these effects involve alterations in insulin production and sensitivity, body temperature, systolic blood pressure, serum triglycerides, glycation products, and oxidative damage to skeletal muscle [25, 28, 30, 31]. Using a variety of experimental models of aging and neurological disease, dietary factors appear to protect against deleterious changes in brain [3, 4, 10, 11, 14, 24-28, 40-49]. Rats treated with up to 40% CR exhibit neuroprotection against neuronal damage from kainic acid in a model of excitotoxicity [11]. In mice, 40% CR attenuates against production of protein carbonyls measured in homogenates of whole brain and different subregions, including cortex, striatum, midbrain and cerebellum, with significant increases in protein-bound sulfhydryls in whole brain, cortex, striatum, midbrain, cerebellum and hindbrain [10].
The diagnosis of Alzheimer's disease (AD), an age-related condition clinically characterized by progressive dementia with late middle-age onset, is confirmed by postmortem findings of widespread amyloid-beta (Aβ)-containing plaques and neurofibrillary tangles throughout cortical tissue [8, 34]. Elevated Aβ peptide levels precede the formation of neurofibrillary tangles in cortical brain regions, suggesting a primary role for Aβ in the neuropathology associated with AD [19]. Quantitative studies of structural changes in brains of AD cases and age-matched controls support the view that progressive amyloid deposition in cortical brain regions correlates with the severity of dementia [9, 19, 34, 36, 38].
About 10-15% of the approximately 5 million patients with AD in the U.S. possess mutations associated with an early-onset, familial form of the disease [13,17]. Transgenic expression of these human mutations led to the development of mouse models that express mutant Aβ peptides and form amyloid plaques in cortical brain areas that are histologically identical to those in AD [5-7, 20, 21, 33]. One line of double transgenic (dtg) mice that co-overexpresses two mutant AD-type proteins, amyloid precursor protein (APPA246E) and presenilin 1 (PS1ΔE9), is associated with unusually early, abundant cortical deposition of mutant Aβ-containing amyloid plaques in cortical tissue. By 5 months of age, these dtg APP/ PS1 mice demonstrate accelerated increases in Aβ1-42 production and formation of AD-type plaques compared to that in single transgenic APP mice [5, 6]. At 6 mos of age, mice in this line of dtg APP/ PS1 mice show no observable impairments in cognitive function compared to non-tg littermate controls [45]. By middle-age (13-18 mos), the accumulation of Aβ (amyloid load) in hippocampus significantly correlates with the degree of neuron loss in two populations of neurons (CA1 and LC) involved with normal cognitive function [1, 29, 38]. As these mice age to 18 mos, the accumulation of amyloid in cortical tissue correlates with episodic memory impairments [45]. Thus, studies in this line of dtg APP/ PS1 mice support the view that amyloid accumulation in cortical brain regions plays a critical role in the development of AD-type neuropathology and memory impairment.
Previous studies in which CR is started in relatively young mice (< 7 mos of age) with tg APP or dtg APP/ PS1 mutations, i.e., mice with relatively light amyloid loads, report that CR blocks the formation of mutant Aβ-containing amyloid plaques [40, 49]. To date, however, no studies have assessed the effects of CR to protect older dtg APP/ PS1 mice with amyloid accumulation analogous to that in middle-aged humans who develop AD. To address this question, we started middle-aged (13-14 mos-old) dtg APP/ PS1 mice on 18 weeks of treatment with 40% CR or ad libitum (AL) feeding, and then used computer-assisted stereology to assess amyloid load in hippocampus and neocortex.
Materials and methods
Mice for this study were male dtg APPswe/ PS1ΔE9 with a mixed strain background, primarily C57BL/6 and C3He/J, developed at The Johns Hopkins School of Medicine [5, 6] by David R. Borchelt and Michael K. Lee. Mice were reared, isotyped, and maintained at the Gerontology Research Center (GRC) of the National Institute on Aging (NIH) in Baltimore, MD. After weaning, mice were raised in standard plastic cages (four per cage) with wood chips for bedding, filtered water from an automated system, and under controlled environmental conditions of 22±1°C, 70±10% humidity and a 12h light/dark cycle. All procedures involving mice were approved by the Animal Care and Use Committee at the NIA, and carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Dtg APP/ PS1 mice were entered into this study at age 13-14 mos and maintained on a standard laboratory diet (NIH-07). Calorie intake was determined through measurement of food consumption by age-matched dtg APP/ PS1 mice fed AL once per week, with CR introduced at 10% increments per week. After an average of 4 weeks, mice reached a diet of 40% CR relative to AL-fed controls, which was maintained for the following 14 weeks prior to sacrifice. Body weight data during the period of CR are shown in Figure 1.
Figure 1.

Mean (SEM) body weight on male C57BL/6J mice begun on calorie restriction (CR) at 13-14 mo of age. Body weight was significantly reduced in CR mice beginning 4 weeks after initiation of 40% CR phased in with 10% restriction each week (two-tailed t-test comparisons at each week, ps <0.05). Mean (SEM) body weight on male C57BL/6J mice following CR starting at 13-14 mo of age.
After 18 weeks of treatment, a total of 6-8 mice per group were euthanized with isoflurane anesthesia (IsoFlo, Abbott Laboratories, North Chicago, IL) and transcardially perfused with 0.1M phosphate buffered saline (PBS) (pH 7.4) followed by 4% paraformaldehyde in 0.1M PBS. Brains were removed and fixed overnight in buffered fixative, transferred to a 30% sucrose/PBS solution until sinking, frozen in cooled isopentane, and then stored in cryoprotectant at -80°C until sectioning. Frozen sections were cut through the entire forebrain in the coronal plane at a microtome setting of 50 μm.
Stereology
A systematic-random set of sections was sampled from random right or left hemispheres through the two reference spaces for this study: (1) hippocampal formation (HF), which consists of Ammon's horn and the dentate gyrus; and (2) the neocortex (NTX) overlying the hippocampal formation. Every 6th section was sampled with a random start in the first interval of six sections, a total of 8-10 sections per reference space, and sections were stained by free-floating histochemistry for Congo red (Sigma, St Louis MO). Stained sections were mounted on subbed slides, dried for a minimum of 12 hr, and then processed through a dehydrating ethanol series (70%, 95%, 100% 95%, 70% for 2 min each), followed by a 1-min dH20 rinse. Sections were counterstained for 2 min with cresyl violet (Sigma, St. Louis, MO), and color was adjusted with 5% acetic acid followed by a final rinse in dH20. Sections were rehydrated in a reverse ethanol series, rinsed twice in Xylene for 10 min each, and then coverslipped with Cytoseal 60 (Richard-Allan Scientific, Kalamazoo MI). After all tissue processing, the average section thickness in the z-axis was 18 μm (+/- 0.2 μm).
An operator blind to treatment quantified total volume of HF and NTX using the Cavalieri-point counting method; and total volume of Aβ stained by Congo red (amyloid load), as previously reported using a computerized stereology system [Stereologer, Stereology Resource Center, Chester, MD; for review, see 35, 37]. Briefly, on each sampled section, the HF and NTX were outlined at low power (5×), and a uniform array of points (+) with known area per point [area per point = 500 um2] placed at systematic-random locations across each reference space. A trained operator selected points that intersected Aβ deposits (ΣPamyloid) and tissue sampled (ΣPsampled) at high magnification (100× oil, numerical aperture 1.4; Figure 2; area per point = 50 um2). Sampling was continued to a mean coefficient of error per group of 0.10 or less (CE < 0.10).
Figure 2.
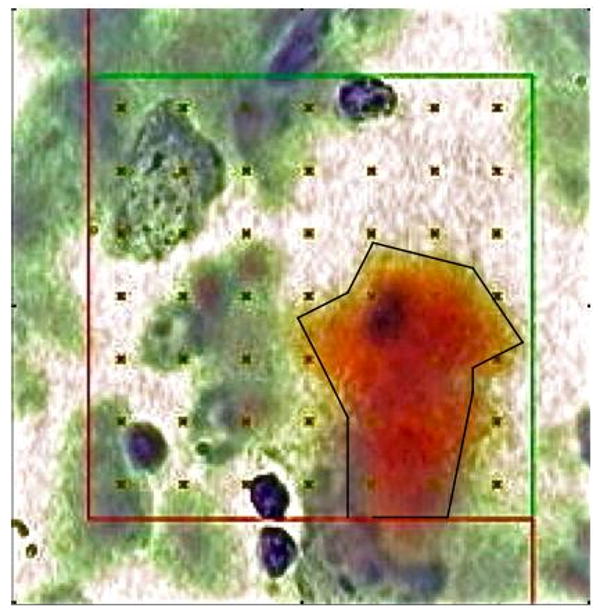
Congo red stained amyloid deposit with overlaid point-grid (area per point = 50 um2) for quantification of amyloid load (100× oil, n.a. 1.4).
The mean total volume of beta-amyloid (Aβ) deposits, Total VAβ, was quantified based on the Area Fraction estimated by point counting [18, 35, 37]. The Area Fraction, i.e., the ratio of amyloid area to sampled tissue area, was quantified from intersections between a randomly placed point-grid and Congo red-stained Aβ deposits through each reference space, as indicated by Figure 2.
| (1) |
where ΣPAβ = the sum of points intersecting Aβ; ΣPsampled= sum of points intersecting the sampled tissue; and a(p) = area per point = distance between points in x direction • distance between points in y direction on the point-grid. According to the Delesse principle, area fraction on random sections through tissue is equivalent to the volume fraction, i.e., AAβ/ Asampled = VAβ/ Vsampled. Finally, the Total VBrDU was calculated as the product of the volume fraction and the total volume of the reference space (Vref)
| (2) |
with Vref, determined by the Cavalieri-point counting method (area per point = 500 um2). Since both Vref and the denominator of VAβ /Vsampled undergo equivalent shrinkage from tissue processing, this shrinkage cancels in their product (Equation 2 above), leading to an unbiased estimate of Total VAβ (18, 35, 37).
Group means were statistically analyzed for protective effects of CR using ANOVA with statistical significance at p < 0.05.
Results
Body weights for dtg APP/ PS1 mice treated with CR mice shown showed expected reductions (Fig. 1) beginning at 4 weeks on diet. This reduction was maintained at about 30-35% lower than controls until time of sacrifice. Since some mice died during the experiment (2 CR, 1 AL), statistical analysis was carried out as individual t-tests for each week. At 18 months of age, Congo red-stained Aβ-containing plaques were distributed throughout the HF and NTX in all dtg APP/ PS1 mice. Amyloid deposits analyzed by point counting (Fig. 2) confirmed reductions of 33% in NTX and 32% in HF for the CR group compared to the same brain regions in AL-fed mice (Table 1).
Table 1.
Mean (SEM) for Aβ load (mm3) in hippocampal formation (HF) and neocortex (NTX) in dtg APPSWE/PS1ΔE9 mice. Treatment with 40% CR led to amyloid load reductions in NTX (p<0.05) and HF (p<0.057) compared to AL diet; % decrease = [(AL-CR)/AL].
| Groups | HF | NTX |
|---|---|---|
| AL | 1.032 (0.10) | 0.1325 (0.01) |
| CR | 0.6998 (0.17) | 0.0892 (0.02) |
| % decrease | 0.32 | 0.33 |
Discussion
Since AD patients are thought to accumulate heavy Aβ deposition by middle-age, the present study assessed whether CR could blunt the accumulation of AD-type neuropathology in the brains of older dtg APP/ PS1 mice. Using computerized stereology we find that 14 weeks of 40% CR initiated at age 13-14 months reduced amyloid load by about one-third in NTX (p < 0.05) and HF (p < 0.057) regions compared to AL-fed controls.
The primary significance of the present study is that 40% CR attenuates the accumulation of Aβ load in middle-aged dtg APP/ PS1 mice. After 5 mos of age, this line of dtg APP/ PS1 mice deposit heavier amounts of Aβ peptides, especially the more fibrillogenic Aβ1-42 peptides, leading to heavier deposits of amyloid in cortical tissue at earlier ages than mice expressing other dtg APP/ PS1 or single APP mutations [5, 6, 45]. By 18 mos of age the heavy amyloid accumulation is associated with cognitive impairments [45] together with substantial losses of neurons in brain regions associated with cognitive function, including the locus coeruleus [29, 39], the ventral tegmental area [29], and CA regions of the hippocampus [1]. Treatment with CR also blocks amyloid plaque formation in other mouse models with relatively less amyloid deposition compared to that in the present study. For instance, Patel et al. [40] reported that two durations of 40% CR in tg APPswe (CR for 6 weeks) and APPswe/ PS1M146L (CR for 14 weeks) decreased amyloid by 40% and 55%, respectively, compared to AL-fed controls. However, these mice were sacrificed at only 5 mos of age, i.e., when Aβ accumulation is minor compared to that in the present study [5, 6, 45]. In a second example of CR-mediated protection in younger mice, Wang et al. [49] treated 3-month old female Tg2576 mice for 9 mos with 30% CR, quantified amyloid load based on thioflavin-S immunofluoresence, and reported reductions of 75% and 85%, in hippocampus and neocortex, respectively, compared to that in AL-fed controls.
The important result in the present study is that CR reduces amyloid accumulation in middle-aged dtg APP/ PS1 mice. The promise of this finding is that similar intervention could become part of a diet-based regimen to protect against the onset and progression of AD in middle-aged humans. According to the amyloid cascade hypothesis, the primary pathogenesis in AD arises from the formation and deposition of Aβ in the forebrain [19]. Future studies are needed to better understand how reduced consumption of calories might delay the onset and/or reduce the accumulation of the amyloid pathology in the brains of aged dtg APP/ PS1 mice, and possibly block or delay the onset of AD. Hyperlipidemia, hypercholesterolemia, and obesity are all associated with increased accumulation of amyloid in AD and mouse models that form AD-type amyloid plaques. Epidemiological studies of obesity, a known risk factor for AD [3, 13], support the view that dietary factors influence the severity of dementia [3, 26, 42]. Excessive consumption of calories, particularly fat, opposes healthy brain aging though the precise mechanisms remain to be elucidated. Evidence from experimental studies in rabbits [43] and single tg mice [43, 46] suggest that increased deposition of AD-type Aβ occurs in association with cholesterol-enriched diets. Another potential mechanism for the observed effects of CR is anti-oxidative effects on memory-associated neuronal signal-regulated kinase that alter in sphingomyelin-specific phospholipase C activity [24]. Thus, CR may mitigate the formation of amyloid plaques through pathways involved in cholesterol synthesis.
As a potentially effective, non-pharmacological intervention, CR has several known benefits to a wide variety of other metabolic processes, raising the possibility that CR could supplement the therapeutic regimen in older adults to reduce or block the onset of AD. Like environmental enrichment in older dtg APP/ PS1 mice [2], CR appears to slow the progression of AD-type pathology, possibly by protection against oxidative damage, enhancement of neurotrophin expression and neurogenesis in adult mice [27], amelioration of the decline in neurogenesis through enhanced survival of new cells [3] and, either alone or in combination with exercise, a reduction in the age-related decline in neurogenesis [12]. Future studies will focus on discerning which combination of these mechanisms allows CR to blunt the formation of amyloid plaques in older dtg APP/ PS1 mice.
Acknowledgments
This research was supported in part by the Intramural Research Program (NIA/NIH) and extramural grants from the U.S. Public Health Service (NIH grant MH076541).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Edossa A, Kalifa S, Polston EK, Mouton PR, Manaye KF. AD-type degeneration of pyramidal cells in hippocampus of dtg APP/ PS1 mice. Soc Neurosci Abstr. 2008;638 [Google Scholar]
- 2.Arendash GW, Garcia MF, Costa DA, Cracchiolo JR, Wefes IM, Potter H. Environmental enrichment improves cognition in aged Alzheimer's transgenic mice despite stable B-amyloid deposition. Neuroreport. 2004;15:1751–1754. doi: 10.1097/01.wnr.0000137183.68847.4e. [DOI] [PubMed] [Google Scholar]
- 3.Barberger-Gateau P, Letenneur L, Larrieu S, Dartigues JF. Dietary fat and dementia: Data from PAQUID. J Neurol Sci. 2005;229:372–372. [Google Scholar]
- 4.Bondolfi L, Ermini F, Long JM. Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57bl/6 mice. Neurobiol Aging. 2004;25:333–340. doi: 10.1016/S0197-4580(03)00083-6. [DOI] [PubMed] [Google Scholar]
- 5.Borchelt DR, Thinakaran G, Eckman CB, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate A beta 1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 6.Borchelt DR, Ratovitski T, van Lare J, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 7.Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol. 2001;158:1173–1177. doi: 10.1016/s0002-9440(10)64064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa PT, Williams TF, Somerfield M. Recognition and initial assessment of Alzheimer's disease and related dementias. Rockville, MD: US Dept of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; AHCPR publication; 1996. pp. 97–702. [Google Scholar]
- 9.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 10.Dubey, Forster MJ, Lal H, Sohal RS. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch Biochem Biophys. 1996;333:189–197. doi: 10.1006/abbi.1996.0380. [DOI] [PubMed] [Google Scholar]
- 11.Duffy KB, Spangler EL, Devan BD, et al. A blueberry-enriched diet provides cellular protection against oxidative stress and reduces a kainate-induced learning impairment in rats. Neurobiol Aging. 2008;11:1680–9. doi: 10.1016/j.neurobiolaging.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Eadie BD, Redila VA, Christie BR. Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J Comp Neurol. 2005;23:39–47. doi: 10.1002/cne.20493. [DOI] [PubMed] [Google Scholar]
- 13.Evans DA. Estimated prevalence of Alzheimer's disease in the United States. Milbank Q. 1990;68:267–289. [PubMed] [Google Scholar]
- 14.Gillette-Guyonnet S, Vellas B. Caloric restriction and brain function. Curr Opin Clin Nutrit Metab Care. 2008;11:686–692. doi: 10.1097/MCO.0b013e328313968f. [DOI] [PubMed] [Google Scholar]
- 15.Goodrick CL, Ingram DK, Reynolds MA, Freeman JR, Cider NL. Effects of intermittent feeding upon growth and life span in rats. Gerontology. 1982;28:233–241. doi: 10.1159/000212538. [DOI] [PubMed] [Google Scholar]
- 16.Gordon MN, Holcomb LA, Jantzen P, DiCarlo G, Wilcock D, Connor K, Melachrino J, O'Callaghan J, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic mPS1+mAPP mouse. Exp Neurol. 2002;172:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- 17.Grant WB. Year 2000 prevalence of Alzheimer disease in the United States. Arch Neurol. 2004;61:802–3. doi: 10.1001/archneur.61.5.802-b. [DOI] [PubMed] [Google Scholar]
- 18.Gundersen HJ, Korbo L, Marcussen N, et al. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS. 1988;96:379–94. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- 19.Hardy JA, Higgins GA. Alzheimer's disease – the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 20.Holcomb LA, Gordon MN, McGowan E, et al. Accelerated Alzheimer phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin I transgenes. Nature Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 21.Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 22.Ingram DK, Anson RM, de Cabo R, et al. Development of calorie restriction mimetics as a prolongevity strategy. Ann NY Acad Sci. 2004;1019:412–423. doi: 10.1196/annals.1297.074. [DOI] [PubMed] [Google Scholar]
- 23.Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human B-amyloid (1-42) protects Alzheimer's transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130:667–684. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 24.Joseph J, Denisova N, Arendash GW, Gordon M, Diamond D, Shukitt-Hale B, Morgan D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- 25.Kruman I, Mouton PR, Emokpae R, Cutler RG, Mattson MP. Folate deficiency inhibits proliferation of adult hippocampal progenitors. Neuroreport. 2005;16:1055–9. doi: 10.1097/00001756-200507130-00005. [DOI] [PubMed] [Google Scholar]
- 26.Lane MA, Mattison JA, Roth GS, Brant LJ, Ingram DK. Effects of long-term diet restriction on aging and longevity in primates remain uncertain. J Gerontol A Biol Sci Med Sci. 2004;59:405–7. doi: 10.1093/gerona/59.5.b405. [DOI] [PubMed] [Google Scholar]
- 27.Lee J, Seroogy KB, Mattson MP. Dietary restriction enhances neurotrophin expression and neurogenesis in the hippocampus of adult mice. J Neuochem. 2002;80:539–547. doi: 10.1046/j.0022-3042.2001.00747.x. [DOI] [PubMed] [Google Scholar]
- 28.Levenson CW, Rich NJ. Eat less, live longer? New insights into the role of caloric restriction in the brain. Nutr Rev. 2007;65:412–415. doi: 10.1111/j.1753-4887.2007.tb00319.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Yoo MJ, Savonenko A, et al. Amyloid Pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattson MP, Wan RQ. Beneficial effects of intermittent fasting and caloric restriction on the cardiovascular and cerebrovascular systems. J Nutr Biochem. 2005;16:129–137. doi: 10.1016/j.jnutbio.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Masoro EJ. Caloric restriction and aging: an update. Exp Gerontol. 2000;35:299–305. doi: 10.1016/s0531-5565(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 32.McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of lifespan and upon ultimate body size. J Nutr. 1935;10:63–79. [PubMed] [Google Scholar]
- 33.McGowan E, Sanders S, Iwatsubo T, et al. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin-1 transgenes. Neurobiol Dis. 1999;6:231–244. doi: 10.1006/nbdi.1999.0243. [DOI] [PubMed] [Google Scholar]
- 34.Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer's disease. A primer for practicing pathologists. Arch Pathol Lab Med. 1993;117:132–44. [PubMed] [Google Scholar]
- 35.Mouton PR. Principles and Practices of Unbiased Stereology: An Introduction For Biomedical Scientists. The Johns Hopkins University Press; Baltimore: 2002. [Google Scholar]
- 36.Mouton PR, Martin LJ, Calhoun ME, Dal Forno G, Price DL. Cognitive decline strongly correlates with cortical atrophy in Alzheimer's dementia. Neurobiol Aging. 1998;19:371–7. doi: 10.1016/s0197-4580(98)00080-3. [DOI] [PubMed] [Google Scholar]
- 37.Mouton PR, Durgavich J, Ingram DK. Automatic estimation of size parameters using Verified Computerized Stereoanalysis. Image Anal Stereol. 2005;24:1–9. [Google Scholar]
- 38.Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid B-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 39.O'Neil JN, Mouton PR, Tizabi Y, Ottinger MA, Lei DL, Ingram DK, Manaye KF. Catecholaminergic neuron number in locus coeruleus of aged female dtg APP/ PS1 Mice. J Chem Neuroanat. 2007;34:102–7. doi: 10.1016/j.jchemneu.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, Finch CE. Caloric restriction attenuates Aβ-deposition in Alzheimer transgenic models. Neurobiology of Aging. 2005;26:995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Petot GJ, Friedland ROP. Lipids, diet, and Alzheimer disease: an extended summary. J Neurol Sci. 2004;226:31–33. doi: 10.1016/j.jns.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 42.Razay G, Vreugdenhil A. Obesity in middle age and future risk of dementia: midlife obesity increases risk of future dementia. 2005;33:455. doi: 10.1136/bmj.331.7514.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Aβ Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;4:321–31. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 44.Roth GS, Mattison JA, Ottinger MA, Chachich ME, Lane MA, Ingram DK. Aging in Rhesus monkeys: relevance to human health interventions. Science. 2004;305:1423–1426. doi: 10.1126/science.1102541. [DOI] [PubMed] [Google Scholar]
- 45.Savonenko A, Xu GM, Melnikova T, Morton JL, et al. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer's disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis. 2005;18:602–17. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 46.Shie FS, Jin LW, Cook DG, Leverenz JB, LeBoeuf RC. Diet-induced hypercholesterolemia enhances brain A beta accumulation in transgenic mice. Neuroreport. 2002;13:455–9. doi: 10.1097/00001756-200203250-00019. [DOI] [PubMed] [Google Scholar]
- 47.Solfrizzi V, Colacicco AM, D'Introno A, Capurso C, Torres F, Rizzo C, Capurso A, Panza F. Dietary intake of unsaturated fatty acids and age-related cognitive decline: A 8.5-year follow-up of the Italian Longitudinal Study on Aging. Neurobiol Aging. 2006;27:1694–704. doi: 10.1016/j.neurobiolaging.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 48.Sparks DL, Kuo YM, Roher A, Martin T, Lukas RJ. Alterations of Alzheimer's disease in the cholesterol-fed rabbit, including vascular inflammation: Preliminary observations. Ann N Y Acad Sci. 2000;903:335–44. doi: 10.1111/j.1749-6632.2000.tb06384.x. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Ho L, Qin W, et al. Caloric restriction attenuates β-amyloid neuropathology in a mouse model of Alzheimer's disease. FASEB. 2005;19:U483–U500. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]