

Abstract
Bile acids (BAs) have a long established role in fat digestion in the intestine by acting as tensioactives, due to their amphipathic characteristics. BAs are reabsorbed very efficiently by the intestinal epithelium and recycled back to the liver via transport mechanisms that have been largely elucidated. The transport and synthesis of BAs are tightly regulated in part by specific plasma membrane receptors and nuclear receptors. In addition to their primary effect, BAs have been claimed to play a role in gastrointestinal cancer, intestinal inflammation and intestinal ionic transport. BAs are not equivalent in any of these biological activities, and structural requirements have been generally identified. In particular, some BAs may be useful for cancer chemoprevention and perhaps in inflammatory bowel disease, although further research is necessary in this field. This review covers the most recent developments in these aspects of BA intestinal biology.
Keywords: Colitis, Bile acid absorption, Colon cancer
INTRODUCTION
Separate chapters in this series of minireviews are devoted to cover various aspects of bile acids (BAs) chemistry, physiology and pathophysiology, including the hepatic synthesis and handling of BAs and their implications in health and disease. Here we will deal with the normal and pathological roles of BAs in one of the traditionally known natural sites of action, i.e. the intestine. It is well known that BAs are secreted into the duodenal lumen after meals in order to act as tensioactives and facilitate fat digestion. This is possible because of the amphipathic characteristics of BAs, which are molecules with a highly hydrophobic core and a number of hydroxyl groups attached. Because these groups may extend to one of the two sides of the basically planar structure formed by the hydrocarbon rings of the BA molecule, its polarity is maximal when all hydroxyl groups are set out in the same side. Thus cholic acid (CA), deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA), all of which have alpha hydroxyl groups, are more efficient tensioactives than ursodeoxycholic acid (UDCA), which has hydroxyls with both alpha and beta conformation. In order to act efficiently as tensioactives BAs must work in coordination with other amphipathic compounds, namely phospholipids, which are another essential component of bile.
Once their physiological function is accomplished, most BA molecules are efficiently reabsorbed in the distal part of the small intestine and reach the liver via portal blood, where they are avidly taken up by hepatocytes. Thus BAs are not wasted but recycled with an almost perfect yield (approximately 95%). The molecular details of this enterohepatic cycle have been elucidated (Figure 1). The type and amount of BAs thus reabsorbed, as well as of those that pass into the colon, are not constant but subject to variation as a result of diet, transit time, drugs, disease, etc. This in turn has an impact on the effects of BAs because they are not equivalent in terms of bioactivity. Certain BAs have been involved in colon cancer, but also in other conditions such as intestinal inflammation, diarrhea, etc.
Figure 1.
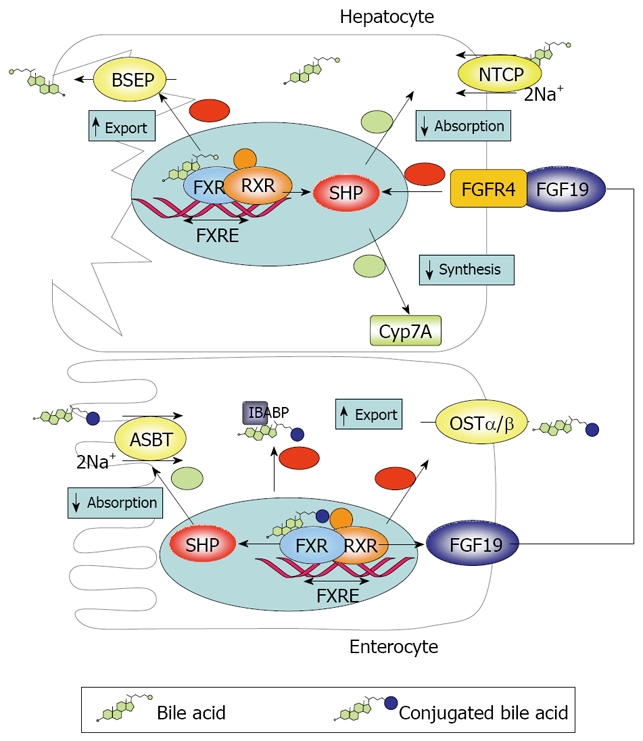
Diagram showing the mechanisms of BA transport and regulation at the intestinal and hepatic level.
The dynamics of BAs transport and metabolism is tightly regulated. This is necessary because intracellular accumulation of BAs, as happens in cholestasis, may result in cytotoxicity. Because BAs are synthesized in the liver, an important part of the control of their homeostasis takes place in hepatocytes, the details of these processes are covered in other chapters of this series. However, in part of the regulatory system the intestine plays important direct (transport/metabolism) and indirect (endocrine response of intestinal epithelium to BAs) roles.
BA IN THE INTESTINE
As mentioned above, the liver synthesizes BAs at the expense of cholesterol and also retrieves reabsorbed BAs from the blood. From hepatocytes they are secreted against steep concentration gradients into bile, together with cholesterol and phospholipids. Thus, between meals, most of the pool of BAs resides in the gallbladder ready to be used at short notice. The mechanisms whereby hepatocytes take up BAs from the bloodstream and synthetize and secrete them into bile have been reviewed in other chapters of this series. When food is ingested (more precisely if the meal is rich in fat) the gallbladder contracts in response to cholecystokinin. It has been proposed that intraduodenal BAs exert a negative feedback control on postprandial cholecystokinin release and the resulting gallbladder contraction. Thus the acute (but not chronic) intraduodenal bile salt depletion with cholestyramine affects gallbladder and also antroduodenal motility, possibly by enhanced motilin release[1].
The mix of BAs contained in bile represents a balance of primary and secondary compounds. Primary BAs are those synthesized as such by the liver, and comprise predominantly CA and CDCA. These are secreted to bile mainly conjugated with glycine and taurine, thus having enhanced water solubility. Secondary BAs are derived from primary BAs by modifications carried out by intestinal bacteria. The main modifications are deconjugation, oxidation of hydroxyl groups in 3, 7 and 12 positions, and 7-dehydroxylation[2]. The main secondary BAs are lithocholic acid (LCA) and DCA. The overall result is an increase in the hydrophobicity of BA pool. The transformation of BAs by bacterial enzymes has several important consequences. First, it favors passive absorption in the colon of those BAs escaping the active uptake that takes place in the ileum. If both mechanisms operate normally, only 1%-3% of the amount of BA that is secreted by the liver is ultimately excreted in faeces (deconjugated and otherwise transformed). Second, it increases the potentiality of BAs to cause noxious effect, like carcinogenicity and cholesterol gallstone disease[3]. Third, the composition of the BA pool will vary when the conditions of biotransformation are altered, for instance by changes in transit time or alterations in the microbiota brought about by drugs, diet, etc.[3,4]. For instance, a high fat diet and a long (slowed) transit time favor DCA generation from CA and absorption, which in turn is associated with higher risk of cholesterol gallstones and cancer (see below). In addition, diets rich in fat and poor in fiber can increase more than 10-fold the amount of taurine conjugated BAs reaching the colon, due to higher conjugation and production (higher conjugation reduces ileal absorption)[5,6]. Taurine dietary intake (meat, seafood) also contributes to this result. Overweight and accelerated intestinal transit reduce BA absorption and may cause idiopathic BA malabsorption[7]. Drugs such as cholestiramine bind BAs and reduce absorption.
Owing to the fact that 7-dehydroxylation cannot be reversed by the host enzymatic machinery, LCA and DCA tend to accumulate in the BA pool. However, LCA is 3-sulfated and conjugated at C-24 by the liver, resulting in a derivative that is poorly absorbed from the colonic mucosa, and consequently LCA is not present in significant amounts in the bile[8]. Thus major BAs in human bile are CA, CDCA and DCA, which are accompanied by minor amounts of UDCA, LCA and other BAs, whereas faeces contain mainly DCA, LCA, minor amounts of CDCA, CA and UDCA and a variety of bacteria transformed derivatives[2]. Concentrations of BAs in the intestinal lumen are variable but usually high, estimated in the medium millimolar range. This is consistent with their critical micellar concentration (e.g. 6-10 mmol/L for TCA), i.e. the concentration corresponding to spontaneous formation of micelles[9].
BA TRANSPORT BY EPITHELIAL CELLS
As mentioned above, BAs are efficiently taken up from the lumen of the ileal segment, leaving only approximately 5% (or approximately 0.5 g/d) in the lumen[10]. This fraction is in part passively absorbed in the colon, a process facilitated by bacterial deconjugation, and in part transformed and extruded with faeces. In contrast, ileal uptake is predominantly an active process carried out by the apical sodium-dependent BA transporter (ASBT, gene symbol SLC10A2), which imports BAs coupled to Na+ absorption (1:2 stoichiometry)[11]. ASBT is highly homologous to the hepatocyte Na+/taurocholate cotransporting polypeptide transporter (NTCP, gene symbol SLC10A1), which plays a pivotal role in BA uptake by the liver from the portal bloodstream. BAs species are not equally transported by ASBT. Thus, conjugated (more hydrophilic) BAs are transported more efficiently than unconjugated forms[12,13]. This is physiologically consistent with the fact that deconjugation normally takes place in the colon. The affinity of ASBT is also higher for dihydroxy BAs (CDCA and DCA) than for trihydroxy BAs (CA, taurocholic acid-TCA-, glycocholic acid-GCA-)[12].
BAs are believed to be transferred directly from ASBT to an intracellular 14 kDa protein called ileal BA binding protein (IBABP/FABP6) through the formation of a 2:1 stoichiometric complex[14]. IBABP is supposed to facilitate transport of BAs within the cell to the basolateral membrane. This is suggested by data showing coordinated expression of both ASBT and IBABP in the postnatal development in the intestine and in cholangyocytes, as well as by the fact that IBABP and ASBT form complexes with a defined stoichiometry[15]. It should be noted however that ASBT is expressed in the kidney without being accompanied by IBABP[16], and also that FXR knockout mice, which do not express IBABP, show enhanced rather than inhibited intestinal BA absorption, suggesting that IBABP may function as a negative regulator of intestinal BA reabsorption, at least in the mouse[17]. Interestingly, low IBABP expression has been linked to the risk of necrotizing enterocolitis in an animal model, suggesting that inefficient transfer of BA to the basolateral membrane may ultimately result in epithelial damage and inflammation[18].
Finally, BAs exit the enterocyte via the recently characterized OSTα/β transporter[19], an obligate heterodimer which functions in a Na+-independent manner and also transports prostaglandin E2, estrone-3-sulfate, dehydroepiandrosterone sulfate[13].
Developmentally ileal transport has been described to be preceded by active colonic absorption in rabbits[20]. This differs markedly from adult animals, which show net colonic secretion. The expression of IBABP and ABST is known to be subjected to distinct changes early in life, which are dependent on the species and protein considered. Thus, in rats and mice, ASBT is highly expressed in the ileum of the fetus before birth but is downregulated or entirely absent in the newborn and later upregulated again. IBABP shows a similar ontogenic profile in mice, while in rats it first appears postnatally[20]. The induction of ABST after birth is stimulated by thyroxine in rats[21].
It is interesting to examine the effects of interference with normal transporter function in the intestine. Genetic disruption of ASBT activity or pharmacological inhibition with SC-435 results in BA malabsorption and diarrhea[22-24]. Conversely, Ostα -/- mice show reduced intestinal capacity to take up BAs but unaffected fecal BA output, which is secondary to a marked shrinkage of the BA pool[19]. On the other hand, there are significant numbers of patients with idiopathic ileal BA malabsoprtion who suffer of unexplained chronic diarrhea[25].
The exact role of other transporters is controversial. They may play a minor role in BA handling by the intestine. These include MRP3 and an alternatively spliced form of ASBT, t-ASBT[13].
REGULATION OF INTESTINAL BA TRANSPORT
The intestinal (ileal) absorption of BAs is tightly regulated to meet physiological demands. In addition, intestinal BA uptake has direct and indirect impact on hepatic BA homeostasis. The main factor involved in both functions is the farnesoid X receptor (FXR/NR1H4), which was originally identified as an orphan nuclear receptor that was activated by farnesol, an intermediate in the mevalonate biosynthetic pathway[26]. FXR is expressed in ileal enterocytes and also in the liver, as well as in other tissues, such as the adrenal gland and the kidney[27]. Interestingly, the intestine seems to have the most intense FXR expression in the body[28]. Agonists of the FXR include BAs, particularly CDCA, followed by DCA, LCA and many other BAs with minor efficacy (conjugation does not affect binding)[29]. The α-position of OH groups in BA molecule is very important for interaction with FXR[30]. Upon activation, FXR modulates gene transcription acting in concert with another nuclear receptor, the retinoid X receptor alpha (RXRα), by recognizing a specific promoter sequence called the FXR responsive element. FXR is pivotal in the BA regulation both in the liver and in the intestine. In hepatocytes FXR increases expression of the bile salt export pump (BSEP) and downregulates the expression of NTCP and CYP7A. Since NTCP and BSEP mediate BA uptake from blood and export to bile in hepatocytes and CYP7A catalyzes the limiting-rate step in the classical BA biosynthetic pathway, this leads to reduced BA uptake, decreased synthesis and enhanced export to bile. Thus BA accumulation in hepatocytes tends to be self-limiting. In enterocytes, FXR is coupled to reduced ASBT and increased IBABP and OSTα/β expression, resulting in inhibition of intestinal absorption of BAs and prevention of intracellular BA accumulation.
The repressing effects of FXR are mediated by the transcription factor SHP (Small Heterodimer Partner), which is induced by FXR but lacks a DNA binding domain[31,32]. Instead, SHP binds to other nuclear receptors such as RXR/RAR (retinoic acid receptor), LRH1 (liver receptor homolog 1) and LXR (liver X receptor), inhibiting their transcriptional effects[11,33]. IBABP seems to interact with ASBT and FXR to promote FXR transcription[34].
An important feature of FXR role in regulation of BA homeostasis is that it is not limited to local effects. Rather, sensing of the enterocyte BA pool by FXR affects the liver by way of the endocrine factor FGF19 (Fgf15 in mice)[32,33]. FGF19 is released to the portal circulation and activates fibroblast growth factor receptor 4 (FGFR4) in hepatocytes, which results in downregulation of CYP7A1 and therefore inhibition of the classical BA synthetic pathway, both by SHP induction and possibly other pathways[33]. Thus BAs modulate their own synthesis both by local hepatic and remote intestinal negative feedback. Tissue specific FXR gene knockdown experiments suggest that both pathways are similarly important[27]. The importance of the latter pathway is exemplified by the fact that administration of TCA downregulates CYP7A1 in the liver only when administered intraduodenally, but not after intravenous or portal instillation[35]. However, alternative pathways for feedback control have been proposed[33].
Physiologically, lack of BA uptake by ASBT inhibition leads to increased fecal BA (and diarrhea) and reduced FXR stimulation, lower FGF19 synthesis, and consequently enhanced BA synthesis, expanding the BA pool and lowering plasma cholesterol[24]. In contrast, Ostα -/- mice do not exhibit increased fecal BA output and have downregulated Cyp7a1 expression and a reduced BA pool[19]. This is due to increased Fgf15, secondary to FXR activation by “trapped” BAs.
In addition, FXR activation has been claimed to participate in the regulation of bacterial growth within the intestine. This hypothesis is supported by the findings that cholestasis results in bacterial overgrowth in the small intestine and increased translocation, which are counteracted in experimental models by oral BAs[36,37]. The FXR mediated induction by BAs of antibacterial genes such as angiogenin, carbonic anhydrase 12 and inducible nitric oxide synthase may account for this effect[38].
Other nuclear factors are regulated by BAs, including the pregnane X receptor (PXR/NR1I2), the Vitamin D receptor (VDR/NR1I1) and the androstane constitutive receptor (CAR)[33]. Thus LCA binds and activates intestinal and systemic VDR[39]. It has been proposed that the effects brought about by LCA are essentially local, directed to induce CYP3A genes and to aid in detoxification[40], but at any rate it can substitute for Vitamin D systemically. PXR and CAR activation leads to the upregulation of secondary BA transporters such as MRP2, MRP3 and MDR1. For instance, CA induces MRP2 and MRP3 in the intestine[41]. In addition, BAs have been reported to activate a G-protein associate receptor, named after the fact G protein-coupled BA receptor 1 (also known as TGR5), which is expressed in many tissues including the gastrointestinal tract[42].
IBABP is also regulated by PPARα/β in humans but not mice[43], and can also be indirectly up-regulated by cholesterol through the activation of sterol-responsive element-binding protein 1c (SREBP1c) by LXR[44]. ASBT is also regulated by PPARα[45]. These changes are expected to increase BA uptake and possibly reduce cholesterol absorption, a putative mechanism of action of the hypolipemic drugs fibrates[43]. Corticoids are also known to upregulate ASBT expression[46]. Because BA ileal uptake is inhibited in intestinal inflammation and probably contributes to diarrhea, corticoid treatment may be specifically useful in this setting.
Among the pathological conditions affecting BA homeostasis, cholestasis downregulates ASBT expression[47,48]. The mechanism is unclear, but it may be related to PPARα inhibition by BAs (possibly because of high blood levels), given that PPARα has been reported to transactivate ASBT transcription, as mentioned above[45,49]. Intestinal MRP2 but not MRP3 is decreased by cholestasis in rats[50] and in humans[48,51], although the significance of these findings is uncertain. In contrast, increased absorption has been reported in primary biliary cirrhosis, thus contributing to cholestasis in this condition[52]. Hypertriglicerydemia also reduces ASBT expression and inhibits BA absorption[53], an effect which in turn might exacerbate hypertriglyceridemia[54]. Interestingly, gallstones have been associated with lower intestinal ASBT and IBABP expression in normal weight but not overweight women[55]. These changes are accounted for by lower hepatic FXR and thereby increased BA synthesis.
BA AND COLORECTAL CANCER
There is wide epidemiological evidence linking BA exposure (for instance due to high fat diet) and gastrointestinal (specially colorectal) cancer[56-58]. Patients with colorectal adenomas and carcinomas exhibit high blood and fecal levels of secondary BAs[59,60]. Diets rich in fat are powerful stimulants of BA secretion, as mentioned above. Thus many investigators have studied the effects of BAs, particularly secondary BAs (DCA and LCA) on intestinal epithelial cell proliferation, apoptosis and mutagenesis in vitro, as well as on cancer promotion in vivo. Paradoxically DCA, but not CA or UDCA, exhibit proapoptotic effects on cell lines, which appear to depend on a variety of mechanisms[61,62]. The ability of BAs to induce apoptosis has been linked to their hydrophobicity, so that unconjugated DCA and CDCA are the most powerful inducers[63]. This makes sense, since only hydrophobic BAs can gain access to colonic cells via passive diffusion.
Different mechanisms have been involved in the proapoptotic effect of BAs. Direct increase in mitochondrial membrane permeability has been suggested, leading to mitochondrial swelling, release of cytochrome c and apoptosis[64]. Alterations in plasma membrane composition with subsequent up-regulation of caveolin-1 may underlie also the activation of protein kinase C by BAs[65]. DCA and UDCA have opposing effects on PKC translocation, affecting a number of isoenzymes including PKC alpha, epsilon and beta1[66,67]. DCA also activates NF-κB and AP-1 in colonic epithelial cells, downstream of PKC stimulation[68]. As expected, UDCA has the opposite effect. ERK activation has been involved in DCA proapoptotic effects, inasmuch as genetic or pharmacological inhibition blocks them[69]. In addition, DCA (and CDCA) induce c-Fos and COX2 in intestinal epithelial cells[70].
Despite these observations, it is important to note that epithelial cells may develop resistance to DCA induced apoptosis, which is achieved partly via the NO pathway[71] and is correlated with shifted expression of multiple proteins, as assessed by proteomic analysis[72]. Alternatively, additional factors may protect against DCA induced apoptosis, such as glutathione-S-transferase P1-1[73]. The expected result would be the selection of transformed cells, favoring the formation of adenomas and predisposing to subsequent development of cancer.
BAs also exert direct actions that can lead to tumorigenesis. Thus, DCA has genotoxic effects, which are believed to be secondary to induction of oxidative stress in the cell[74], and suppresses the p53 response to DNA damage, an action that is at least partly dependent on ERK signaling[75]. Moreover, inhibition of BRCA-1 by relatively high DCA concentrations contributes to defective DNA repair[76]. Recently DCA and LCA (in conjugated form) were shown to elicit transactivation of the epidermal growth factor receptor via interaction with muscarinic receptors and phosphorylation of ERK[77]. Another gene target of DCA via ERK is the tumor marker EphA2 receptor protein tyrosine kinase[78]. In general, these actions are not shared by UDCA and may be opposed by it[61,79,80]. Taken together, these data indicate that DCA may behave as a co-carcinogenic and/or cancer promoter agent, which may potentiate the activity of any primary carcinogen or cancer initiator. In addition, DCA may increase tumor invasiveness by activation of beta-catenin signaling[81]. An interesting observation is that FXR expression is diminished in colon cancer[82]. Under these circumstances, FXR-mediated mechanisms involved in the prevention of BA accumulation in these cells could be expected to be completely or partly inactive, thus exacerbating BA-induced effects.
We also count on substantial in vivo evidence about the effects of BAs on colorectal cancer. Thus colonic grafts from mice with an APC gene mutation do not develop adenomas if they are removed from the fecal stream[83]. In the standard Min mouse model UDCA produces a dose dependent decrease in the number of intestinal tumors, showing synergism with the cycloxygenase 2 inhibitor sulindac[84]. In the azoxymethane model of cancer associated to chronic colitis, UDCA lowered the multiplicity of colonic adenocarcinoma, while sulfasalazine had no significant effect[85]. Similar results were obtained in the regular (without colitis) azoxymethane model[86]. The chemopreventive effect of UDCA is associated with decreased Ras activation and COX2 expression[87].
This type of observations can be extended to human disease. Thus, a study carried out in patients with primary biliary cirrhosis undergoing surveillance by colonoscopy revealed a non-significant reduction in the prevalence of colorectal adenomas and, more importantly, a lower probability of recurrence (7% vs 28% at 3 years, P = 0.04)[88]. UDCA lowers cancer mortality (but not incidence) in ulcerative colitis patients with sclerosing cholangitis[89]. In a clinical trial on the secondary prevention of colorectal cancer, UDCA caused a non-significant 12% decrease in recurrence rate but a significant reduction (39%) in the subgroup with high-grade dysplasia[90]. In clinical trials of cancer associated to inflammatory bowel disease, a condition which increases the risk of developing cancer, UDCA has been shown to be beneficial, ranging from a mild chemoprotective effect[91] to a clear decrease in the relative risk of developing colorectal dysplasia or cancer[92]. Mechanistically, UDCA has been reported to reduce mucosal proliferation in cancer naive patients[88] but to have no effect in adenoma patients[93]. From a pharmacokinetic point of view, the main effect of UDCA administration in humans is an increase of luminal (fecal) UDCA/DCA ratio, although DCA absolute levels remain unaltered[94].
An intriguing possibility is that taurine, which is bound to a substantial fraction of the BA pool, contributes to cancer risk. Taurine, which can be released in the intestinal lumen from conjugated BAs due to the metabolic activity of several bacterial strains, is metabolized by the intestinal flora yielding hydrogen sulfite, which increases colonocyte turnover and inhibits butyrate metabolism. Although these cells oxidize efficiently this compound to thiosulfate, taurine derived hydrogen sulfite may be involved in carcinogenesis. In fact, defects in the hydrogen sulfite detoxification pathway may increase the risk of UC, a significant risk factor for colon cancer[95,96]. It is interesting to note that taurine conjugation and sulfite production are increased in meat consumers, thus providing another link to colon cancer[2]. In addition, sulfite promotes DCA generation by bacteria through stimulation of 7α-dehydroxylation.
EFFECT OF BA ON ION TRANSPORT
It is well known that BAs elicit fluid secretion and also increased permeability in the gastrointestinal tract[97-101]. BAs also affect intestinal motility, although this field of study has received relatively little attention[1,102]. The effect of BAs on permeability is primarily due to their detergent action on tight junctions, which is reversible. However, at high BA concentrations epithelial lesions may occur. This effect is to a large extent indirect, induced by intramural reflexes containing nicotinic receptors, but probably it does not involve histamine or nitric oxide pathways[97,103,104]. However, this question is controversial, because in some cases histamine has been suggested to be involved[98].
The secretory effect of BAs has been studied in the small and large intestine, and in both cases the mechanism of secretion appears to be largely indirect[99]. In the small intestine BAs elicit serotonin release by enterochromaffin cells in the mucosa by a Ca2+-dependent mechanism, initiating a neural reflex that stimulates ion secretion, as well as inhibited absorption[105]. In ileal perfusion experiments permeability and fluid transport were studied in parallel, finding that the effect is dependent on nicotinic receptors in both cases[97,103]. On the other hand, in the colon BA-induced secretion has been claimed to be prostaglandin dependent[106]. More specifically, prostaglandins may account for the early response to BA stimulation observed in vitro[98]. Mast cells have also been involved in this process[98]. Indeed, BAs have been shown to induce prostaglandin[107] and histamine secretion[108] in vitro. Some of these actions may be secondary to direct mucosal injury[100,109]. Considering the variety of mediators proposed to participate in these events, it is not surprising that multiple intracellular signaling pathways have also been involved.
As with other biological activities of BAs, not all molecular species are equivalent. Dihydroxy BAs, and in particular DCA and CDCA, exhibit prosecretory/antiabsorptive and mucosal damaging effects in the colon, particularly in their unconjugated forms[100,101]. Conversely, other important BAs such as CA and UDCA are generally considered to have no significant bioactivity at this regard. In the small intestine, these differential species-dependent effects are not so well characterized, but they certainly differ from those in the colon. For instance, CDCA is a relatively poor secretagogue in the ileum[110-113]. It should be noted that ileal BA absorption is electrogenic, even in the absence of chloride/bicarbonate secretion, due to the 2:1 stoichiometry of ASBT-mediated Na+:BA symport[111,114,115].
However, BAs also exert direct actions on intestinal epithelial cells. Using the prototypic T84 cell line TDCA was shown to elicit chloride secretion by a Ca2+ dependent mechanism[116]. This pertains to the actions of BAs in the large intestine, since T84 cells have a colonic epithelium phenotype. More recently, BA were described to induce ion secretion via transactivation of the cystic fibrosis transmembrane conductance regulator (CFTR)[113]. CFTR is the main chloride/bicarbonate channel in intestinal epithelial cells and is pivotal to ion secretion in the gastrointestinal tract, among other tissues. Transactivation requires apical colocalization of both CFTR and ASBT, which does occur in the distal ileum but also in cholangiocytes. Moreover, CFTR also plays a role in bile flow, as suggested the presence of plugging and dilatation of bile ducts in cystic fibrosis. Interestingly, BA ileal uptake is compromised in this condition, leading to BA waste and diarrhea[113,114,117], suggesting that CFTR has a reciprocal influence on ASBT. Moreover, this provides an additional meaningful link to inhibited BA absorption in intestinal inflammation, where CFTR has been shown to be downregulated, as is ASBT itself[118,119]. Although the relationship between the expression of CFTR and that of ASBT has been previously demonstrated also for other transporters[120], the underlying mechanism has not been elucidated yet.
One interesting question arises as to what is the physiological role of BA-induced ileal and colonic secretion. The simplest explanation is that ileal secretion, which is evoked in normal conditions, may be useful to aid in intestinal propulsion or to prevent the formation of micelles and the consequent epithelial damage during the absorptive process[113]. In contrast, colonic secretion occurs only in pathologic conditions and may be part of a nonspecific mechanism aimed to eliminate invading microorganisms. In addition, BA ileal absorption is compromised in conditions such as irritable bowel syndrome, Crohn’s disease, cystic fibrosis and surgical resection (short bowel syndrome)[117], producing diarrhea because of the presence of high (millimolar) concentrations in the colonic lumen. BAs (CDCA, UDCA) can also induce diarrhea in their own right when given to gallstone patients[121,122].
BA AND INTESTINAL INFLAMMATION
Certain BAs have been shown to exert intestinal antiinflammatory actions in vivo. Thus UDCA reduces intestinal permeability and oxidative stress in the indomethacin model of ileitis in the rat[123]. Similarly, UDCA counteracts ibuprofen intestinal ulceration in rats[124]. These effects may be related to the actions of BAs on intestinal epithelial cells. Thus DCA induces IL-8 and activates NF-κB in HT29 cells, actions that are opposed by taurine-conjugated UDCA[125,126]. The mechanism for IL-8 induction is probably via the classical NF-κB pathway for DCA and via RelA phosphorylation in the case of TDCA[126]. The stimulatory effect of DCA is reproduced in other[127,128] but not all cell lines[129]. Thus UDCA might exert antiinflammatory actions in the intestine by inhibiting epithelial stimulation. Conversely, DCA is predicted to aggravate inflammation, but this has not been tested. On the other hand, BAs have also been described to enhance epithelial wound healing, an action dependent on NF-κB activation and the release of transforming growth factor β[130]. This effect is shared by TDCA, DCA and TCA. Unfortunately UDCA was not investigated[130]. It cannot be ruled out that this effect may also form part of the mechanism of action of this beneficial compound.
INTESTINAL APPLICATIONS OF BA
The main clinical application of BAs is in the management of gallstones and cholestasis (primary biliary cirrhosis, cystic fibrosis liver disease, drug induced cholestasis)[88,131,132]. UDCA and to a lesser extent CDCA have been used. Cholylsarcosine is an artificial derivative that has been proposed as an alternative. However, BAs have no current application for intestinal conditions. UDCA has been studied in the clinical setting for cancer chemoprevention as discussed above. Specific artificial BA derivatives have also been studied for chemopreventive application[133]. Interestingly, it may be feasible to increase UDCA intestinal exposure by the use of a specific type of probiotic (living bacteria) that epimerizes CDCA to UDCA within the intestinal lumen[134]. BAs have been used in some cases as a replacement to reduce steatorrhea due to short bowel syndrome or secondary to metabolic genetic diseases, frequently at the price of enhanced diarrhea[135,136]. Conversely, diarrhea without steatorrhea benefits from treatment with ion exchange resins such as cholestiramine that bind and act as sequestrant of BAs during their intestinal transit[137].
Moreover, BAs are being considered as galenic agents to improve intestinal absorption of drugs compounds such as nucleotides, heparin or insulin[138-140].
CONCLUSION
Owing to their tensioactive properties BAs play an important role in the intestine, facilitating fat digestion and the absorption of lipids and liposoluble vitamins. Efficient intestinal uptake, mainly at the ileum, permits to recover most of the secreted BA molecules, which are sent back to the liver with the portal blood. The existence of the enterohepatic circulation maintains appropriate levels of BAs ready to be used after meals and prevents exposure of other tissues to high levels of these dangerous detergents. In this respect, due to the potential toxicity of BAs and, at the same time, their biological relevance the homeostasis of BAs is tightly regulated, in part by specific plasma membrane receptors and nuclear receptors. The function, transport and regulatory mechanisms regarding BAs and the intestine have been elucidated in great detail, although some questions remain unanswered, such as the exact physiological role of IBABP. Some BA species have peculiar biological or pharmacological effects, which have been characterized to a great extent. Nevertheless, their role in colon cancer and intestinal inflammation requires further study, which is especially interesting considering the potential therapeutical applications.
Footnotes
Supported by Grants from the Instituto de Salud Carlos III (PI051625 and PI051651). The group is member of the Network for Cooperative Research on Membrane Transport Proteins (REIT), co-funded by the Ministerio de Educación y Ciencia, Spain and the European Regional Development Fund (Grant BFU2005-24983-E/BFI). The CIBER-EHD is funded by the Instituto de Salud Carlos III
Peer reviewer: Serhan Karvar, MD, Assistant Professor of Medicine, University of Southern California, Keck School of Medicine, Division of Gastrointestinal & Liver Diseases, 2011 Zonal Avenue, HMR 101, Los Angeles, CA 90089, United States
S- Editor Li DL E- Editor Ma WH
References
- 1.Portincasa P, Peeters TL, van Berge-Henegouwen GP, van Solinge WW, Palasciano G, van Erpecum KJ. Acute intraduodenal bile salt depletion leads to strong gallbladder contraction, altered antroduodenal motility and high plasma motilin levels in humans. Neurogastroenterol Motil. 2000;12:421–430. doi: 10.1046/j.1365-2982.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 2.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotrans-formations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Thomas LA, Veysey MJ, Bathgate T, King A, French G, Smeeton NC, Murphy GM, Dowling RH. Mechanism for the transit-induced increase in colonic deoxycholic acid formation in cholesterol cholelithiasis. Gastroenterology. 2000;119:806–815. doi: 10.1053/gast.2000.16495. [DOI] [PubMed] [Google Scholar]
- 4.Thomas LA, Veysey MJ, Murphy GM, Russell-Jones D, French GL, Wass JA, Dowling RH. Octreotide induced prolongation of colonic transit increases faecal anaerobic bacteria, bile acid metabolising enzymes, and serum deoxycholic acid in patients with acromegaly. Gut. 2005;54:630–635. doi: 10.1136/gut.2003.028431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadler J, Stern HS, Yeung KS, McGuire V, Furrer R, Marcon N, Bruce WR. Effect of high fat consumption on cell proliferation activity of colorectal mucosa and on soluble faecal bile acids. Gut. 1988;29:1326–1331. doi: 10.1136/gut.29.10.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardison WG. Hepatic taurine concentration and dietary taurine as regulators of bile acid conjugation with taurine. Gastroenterology. 1978;75:71–75. [PubMed] [Google Scholar]
- 7.Sadik R, Abrahamsson H, Ung KA, Stotzer PO. Accelerated regional bowel transit and overweight shown in idiopathic bile acid malabsorption. Am J Gastroenterol. 2004;99:711–718. doi: 10.1111/j.1572-0241.2004.04139.x. [DOI] [PubMed] [Google Scholar]
- 8.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703–722. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 9.Roda A, Hofmann AF, Mysels KJ. The influence of bile salt structure on self-association in aqueous solutions. J Biol Chem. 1983;258:6362–6370. [PubMed] [Google Scholar]
- 10.Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem. 2002;277:3801–3804. doi: 10.1074/jbc.R100057200. [DOI] [PubMed] [Google Scholar]
- 11.Pellicoro A, Faber KN. Review article: The function and regulation of proteins involved in bile salt biosynthesis and transport. Aliment Pharmacol Ther. 2007;26 Suppl 2:149–160. doi: 10.1111/j.1365-2036.2007.03522.x. [DOI] [PubMed] [Google Scholar]
- 12.Craddock AL, Love MW, Daniel RW, Kirby LC, Walters HC, Wong MH, Dawson PA. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am J Physiol. 1998;274:G157–G169. doi: 10.1152/ajpgi.1998.274.1.G157. [DOI] [PubMed] [Google Scholar]
- 13.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24:1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 14.Tochtrop GP, Bruns JL, Tang C, Covey DF, Cistola DP. Steroid ring hydroxylation patterns govern cooperativity in human bile acid binding protein. Biochemistry. 2003;42:11561–11567. doi: 10.1021/bi0346502. [DOI] [PubMed] [Google Scholar]
- 15.Kramer W, Wess G, Bewersdorf U, Corsiero D, Girbig F, Weyland C, Stengelin S, Enhsen A, Bock K, Kleine H, et al. Topological photoaffinity labeling of the rabbit ileal Na+/bile-salt-cotransport system. Eur J Biochem. 1997;249:456–464. doi: 10.1111/j.1432-1033.1997.00456.x. [DOI] [PubMed] [Google Scholar]
- 16.Gong YZ, Everett ET, Schwartz DA, Norris JS, Wilson FA. Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol. Proc Natl Acad Sci USA. 1994;91:4741–4745. doi: 10.1073/pnas.91.11.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kok T, Hulzebos CV, Wolters H, Havinga R, Agellon LB, Stellaard F, Shan B, Schwarz M, Kuipers F. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J Biol Chem. 2003;278:41930–41937. doi: 10.1074/jbc.M306309200. [DOI] [PubMed] [Google Scholar]
- 18.Halpern MD, Dvorak B. Does abnormal bile acid metabolism contribute to NEC? Semin Perinatol. 2008;32:114–121. doi: 10.1053/j.semperi.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao A, Haywood J, Craddock AL, Belinsky MG, Kruh GD, Dawson PA. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci USA. 2008;105:3891–3896. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weihrauch D, Kanchanapoo J, Ao M, Prasad R, Piyachaturawat P, Rao MC. Weanling, but not adult, rabbit colon absorbs bile acids: flux is linked to expression of putative bile acid transporters. Am J Physiol Gastrointest Liver Physiol. 2006;290:G439–G450. doi: 10.1152/ajpgi.00163.2005. [DOI] [PubMed] [Google Scholar]
- 21.Monteiro I, David ES, Ferraris RP. Ontogenetic development of rat intestinal bile acid transport requires thyroxine but not corticosterone. Pediatr Res. 2004;55:611–621. doi: 10.1203/01.PDR.0000112126.07230.9A. [DOI] [PubMed] [Google Scholar]
- 22.Wong MH, Oelkers P, Dawson PA. Identification of a mutation in the ileal sodium-dependent bile acid transporter gene that abolishes transport activity. J Biol Chem. 1995;270:27228–27234. doi: 10.1074/jbc.270.45.27228. [DOI] [PubMed] [Google Scholar]
- 23.Dawson PA, Haywood J, Craddock AL, Wilson M, Tietjen M, Kluckman K, Maeda N, Parks JS. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278:33920–33927. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Xu G, Shang Q, Pan L, Shefer S, Batta AK, Bollineni J, Tint GS, Keller BT, Salen G. Inhibition of ileal bile acid transport lowers plasma cholesterol levels by inactivating hepatic farnesoid X receptor and stimulating cholesterol 7 alpha-hydroxylase. Metabolism. 2004;53:927–932. doi: 10.1016/j.metabol.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Balesaria S, Pell RJ, Abbott LJ, Tasleem A, Chavele KM, Barley NF, Khair U, Simon A, Moriarty KJ, Brydon WG, et al. Exploring possible mechanisms for primary bile acid malabsorption: evidence for different regulation of ileal bile acid transporter transcripts in chronic diarrhoea. Eur J Gastroenterol Hepatol. 2008;20:413–422. doi: 10.1097/MEG.0b013e3282f41b82. [DOI] [PubMed] [Google Scholar]
- 26.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 27.Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–2672. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Houten SM, Volle DH, Cummins CL, Mangelsdorf DJ, Auwerx J. In vivo imaging of farnesoid X receptor activity reveals the ileum as the primary bile acid signaling tissue. Mol Endocrinol. 2007;21:1312–1323. doi: 10.1210/me.2007-0113. [DOI] [PubMed] [Google Scholar]
- 29.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 30.Shea HC, Head DD, Setchell KD, Russell DW. Analysis of HSD3B7 knockout mice reveals that a 3alpha-hydroxyl stereochemistry is required for bile acid function. Proc Natl Acad Sci USA. 2007;104:11526–11533. doi: 10.1073/pnas.0705089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neimark E, Chen F, Li X, Shneider BL. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology. 2004;40:149–156. doi: 10.1002/hep.20295. [DOI] [PubMed] [Google Scholar]
- 32.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Lee FY, Lee H, Hubbert ML, Edwards PA, Zhang Y. FXR, a multipurpose nuclear receptor. Trends Biochem Sci. 2006;31:572–580. doi: 10.1016/j.tibs.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Nakahara M, Furuya N, Takagaki K, Sugaya T, Hirota K, Fukamizu A, Kanda T, Fujii H, Sato R. Ileal bile acid-binding protein, functionally associated with the farnesoid X receptor or the ileal bile acid transporter, regulates bile acid activity in the small intestine. J Biol Chem. 2005;280:42283–42289. doi: 10.1074/jbc.M507454200. [DOI] [PubMed] [Google Scholar]
- 35.Nagano M, Kuroki S, Mizuta A, Furukawa M, Noshiro M, Chijiiwa K, Tanaka M. Regulation of bile acid synthesis under reconstructed enterohepatic circulation in rats. Steroids. 2004;69:701–709. doi: 10.1016/j.steroids.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Ding JW, Andersson R, Soltesz V, Willen R, Bengmark S. The role of bile and bile acids in bacterial translocation in obstructive jaundice in rats. Eur Surg Res. 1993;25:11–19. doi: 10.1159/000129252. [DOI] [PubMed] [Google Scholar]
- 37.Lorenzo-Zuniga V, Bartoli R, Planas R, Hofmann AF, Vinado B, Hagey LR, Hernandez JM, Mane J, Alvarez MA, Ausina V, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37:551–557. doi: 10.1053/jhep.2003.50116. [DOI] [PubMed] [Google Scholar]
- 38.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nehring JA, Zierold C, DeLuca HF. Lithocholic acid can carry out in vivo functions of vitamin D. Proc Natl Acad Sci USA. 2007;104:10006–10009. doi: 10.1073/pnas.0703512104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jurutka PW, Thompson PD, Whitfield GK, Eichhorst KR, Hall N, Dominguez CE, Hsieh JC, Haussler CA, Haussler MR. Molecular and functional comparison of 1,25-dihydroxyvitamin D(3) and the novel vitamin D receptor ligand, lithocholic acid, in activating transcription of cytochrome P450 3A4. J Cell Biochem. 2005;94:917–943. doi: 10.1002/jcb.20359. [DOI] [PubMed] [Google Scholar]
- 41.Kamisako T, Ogawa H, Yamamoto K. Effect of cholesterol, cholic acid and cholestyramine administration on the intestinal mRNA expressions related to cholesterol and bile acid metabolism in the rat. J Gastroenterol Hepatol. 2007;22:1832–1837. doi: 10.1111/j.1440-1746.2007.04910.x. [DOI] [PubMed] [Google Scholar]
- 42.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 43.Landrier JF, Thomas C, Grober J, Zaghini I, Petit V, Poirier H, Niot I, Besnard P. The gene encoding the human ileal bile acid-binding protein (I-BABP) is regulated by peroxisome proliferator-activated receptors. Biochim Biophys Acta. 2005;1735:41–49. doi: 10.1016/j.bbalip.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 44.Landrier JF, Grober J, Demydchuk J, Besnard P. FXRE can function as an LXRE in the promoter of human ileal bile acid-binding protein (I-BABP) gene. FEBS Lett. 2003;553:299–303. doi: 10.1016/s0014-5793(03)01033-0. [DOI] [PubMed] [Google Scholar]
- 45.Jung D, Fried M, Kullak-Ublick GA. Human apical sodium-dependent bile salt transporter gene (SLC10A2) is regulated by the peroxisome proliferator-activated receptor alpha. J Biol Chem. 2002;277:30559–30566. doi: 10.1074/jbc.M203511200. [DOI] [PubMed] [Google Scholar]
- 46.Jung D, Fantin AC, Scheurer U, Fried M, Kullak-Ublick GA. Human ileal bile acid transporter gene ASBT (SLC10A2) is transactivated by the glucocorticoid receptor. Gut. 2004;53:78–84. doi: 10.1136/gut.53.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sauer P, Stiehl A, Fitscher BA, Riedel HD, Benz C, Kloters-Plachky P, Stengelin S, Stremmel W, Kramer W. Downregulation of ileal bile acid absorption in bile-duct-ligated rats. J Hepatol. 2000;33:2–8. doi: 10.1016/s0168-8278(00)80152-x. [DOI] [PubMed] [Google Scholar]
- 48.Hruz P, Zimmermann C, Gutmann H, Degen L, Beuers U, Terracciano L, Drewe J, Beglinger C. Adaptive regulation of the ileal apical sodium dependent bile acid transporter (ASBT) in patients with obstructive cholestasis. Gut. 2006;55:395–402. doi: 10.1136/gut.2005.067389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinal CJ, Yoon M, Gonzalez FJ. Antagonism of the actions of peroxisome proliferator-activated receptor-alpha by bile acids. J Biol Chem. 2001;276:47154–47162. doi: 10.1074/jbc.M107000200. [DOI] [PubMed] [Google Scholar]
- 50.Kamisako T, Ogawa H. Alteration of the expression of adenosine triphosphate-binding cassette transporters associated with bile acid and cholesterol transport in the rat liver and intestine during cholestasis. J Gastroenterol Hepatol. 2005;20:1429–1434. doi: 10.1111/j.1440-1746.2005.03950.x. [DOI] [PubMed] [Google Scholar]
- 51.Dietrich CG, Geier A, Salein N, Lammert F, Roeb E, Oude Elferink RP, Matern S, Gartung C. Consequences of bile duct obstruction on intestinal expression and function of multidrug resistance-associated protein 2. Gastroenterology. 2004;126:1044–1053. doi: 10.1053/j.gastro.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 52.Lanzini A, De Tavonatti MG, Panarotto B, Scalia S, Mora A, Benini F, Baisini O, Lanzarotto F. Intestinal absorption of the bile acid analogue 75Se-homocholic acid-taurine is increased in primary biliary cirrhosis, and reverts to normal during ursodeoxycholic acid administration. Gut. 2003;52:1371–1375. doi: 10.1136/gut.52.9.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duane WC, Hartich LA, Bartman AE, Ho SB. Diminished gene expression of ileal apical sodium bile acid transporter explains impaired absorption of bile acid in patients with hypertriglyceridemia. J Lipid Res. 2000;41:1384–1389. [PubMed] [Google Scholar]
- 54.Duane WC. Abnormal bile acid absorption in familial hypertriglyceridemia. J Lipid Res. 1995;36:96–107. [PubMed] [Google Scholar]
- 55.Bergheim I, Harsch S, Mueller O, Schimmel S, Fritz P, Stange EF. Apical sodium bile acid transporter and ileal lipid binding protein in gallstone carriers. J Lipid Res. 2006;47:42–50. doi: 10.1194/jlr.M500215-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Armstrong B, Doll R. Environmental factors and cancer incidence and mortality in different countries, with special reference to dietary practices. Int J Cancer. 1975;15:617–631. doi: 10.1002/ijc.2910150411. [DOI] [PubMed] [Google Scholar]
- 57.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Chen YQ, Edwards IJ, Kridel SJ, Thornburg T, Berquin IM. Dietary fat-gene interactions in cancer. Cancer Metastasis Rev. 2007;26:535–551. doi: 10.1007/s10555-007-9075-x. [DOI] [PubMed] [Google Scholar]
- 59.Wynder EL, Reddy BS, McCoy GD, Weisburger JH, Williams GM. Diet and cancer of the gastrointestinal tract. Adv Intern Med. 1977;22:397–419. [PubMed] [Google Scholar]
- 60.Bayerdorffer E, Mannes GA, Richter WO, Ochsenkuhn T, Wiebecke B, Kopcke W, Paumgartner G. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology. 1993;104:145–151. doi: 10.1016/0016-5085(93)90846-5. [DOI] [PubMed] [Google Scholar]
- 61.Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, Payne CM, Powell MB, Gerner EW, Earnest DL. Different bile acids exhibit distinct biological effects: the tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer. 1998;31:111–118. doi: 10.1080/01635589809514689. [DOI] [PubMed] [Google Scholar]
- 62.Yui S, Saeki T, Kanamoto R, Iwami K. Characteristics of apoptosis in HCT116 colon cancer cells induced by deoxycholic acid. J Biochem. 2005;138:151–157. doi: 10.1093/jb/mvi106. [DOI] [PubMed] [Google Scholar]
- 63.Powell AA, LaRue JM, Batta AK, Martinez JD. Bile acid hydrophobicity is correlated with induction of apoptosis and/or growth arrest in HCT116 cells. Biochem J. 2001;356:481–486. doi: 10.1042/0264-6021:3560481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wachs FP, Krieg RC, Rodrigues CM, Messmann H, Kullmann F, Knuchel-Clarke R, Scholmerich J, Rogler G, Schlottmann K. Bile salt-induced apoptosis in human colon cancer cell lines involves the mitochondrial transmembrane potential but not the CD95 (Fas/Apo-1) receptor. Int J Colorectal Dis. 2005;20:103–113. doi: 10.1007/s00384-004-0616-2. [DOI] [PubMed] [Google Scholar]
- 65.Akare S, Martinez JD. Bile acid induces hydrophobicity-dependent membrane alterations. Biochim Biophys Acta. 2005;1735:59–67. doi: 10.1016/j.bbalip.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 66.Shah SA, Looby E, Volkov Y, Long A, Kelleher D. Ursodeoxycholic acid inhibits translocation of protein kinase C in human colonic cancer cell lines. Eur J Cancer. 2005;41:2160–2169. doi: 10.1016/j.ejca.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 67.Looby E, Long A, Kelleher D, Volkov Y. Bile acid deoxycholate induces differential subcellular localisation of the PKC isoenzymes beta 1, epsilon and delta in colonic epithelial cells in a sodium butyrate insensitive manner. Int J Cancer. 2005;114:887–895. doi: 10.1002/ijc.20803. [DOI] [PubMed] [Google Scholar]
- 68.Shah SA, Volkov Y, Arfin Q, Abdel-Latif MM, Kelleher D. Ursodeoxycholic acid inhibits interleukin 1 beta [corrected] and deoxycholic acid-induced activation of NF-kappaB and AP-1 in human colon cancer cells. Int J Cancer. 2006;118:532–539. doi: 10.1002/ijc.21365. [DOI] [PubMed] [Google Scholar]
- 69.Qiao D, Stratagouleas ED, Martinez JD. Activation and role of mitogen-activated protein kinases in deoxycholic acid-induced apoptosis. Carcinogenesis. 2001;22:35–41. doi: 10.1093/carcin/22.1.35. [DOI] [PubMed] [Google Scholar]
- 70.Jurek D, Fleckl E, Marian B. Bile acid induced gene expression in LT97 colonic adenoma cells. Food Chem Toxicol. 2005;43:87–93. doi: 10.1016/j.fct.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 71.Payne CM, Waltmire CN, Crowley C, Crowley-Weber CL, Dvorakova K, Bernstein H, Bernstein C, Holubec H, Garewal H. Caspase-6 mediated cleavage of guanylate cyclase alpha 1 during deoxycholate-induced apoptosis: protective role of the nitric oxide signaling module. Cell Biol Toxicol. 2003;19:373–392. doi: 10.1023/b:cbto.0000013331.70391.0e. [DOI] [PubMed] [Google Scholar]
- 72.Bernstein H, Payne CM, Kunke K, Crowley-Weber CL, Waltmire CN, Dvorakova K, Holubec H, Bernstein C, Vaillancourt RR, Raynes DA, et al. A proteomic study of resistance to deoxycholate-induced apoptosis. Carcinogenesis. 2004;25:681–692. doi: 10.1093/carcin/bgh072. [DOI] [PubMed] [Google Scholar]
- 73.Nobuoka A, Takayama T, Miyanishi K, Sato T, Takanashi K, Hayashi T, Kukitsu T, Sato Y, Takahashi M, Okamoto T, et al. Glutathione-S-transferase P1-1 protects aberrant crypt foci from apoptosis induced by deoxycholic acid. Gastroenterology. 2004;127:428–443. doi: 10.1053/j.gastro.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 74.Powolny A, Xu J, Loo G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int J Biochem Cell Biol. 2001;33:193–203. doi: 10.1016/s1357-2725(00)00080-7. [DOI] [PubMed] [Google Scholar]
- 75.Qiao D, Gaitonde SV, Qi W, Martinez JD. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis. 2001;22:957–964. doi: 10.1093/carcin/22.6.957. [DOI] [PubMed] [Google Scholar]
- 76.Romagnolo DF, Chirnomas RB, Ku J, Jeffy BD, Payne CM, Holubec H, Ramsey L, Bernstein H, Bernstein C, Kunke K, et al. Deoxycholate, an endogenous tumor promoter and DNA damaging agent, modulates BRCA-1 expression in apoptosis-sensitive epithelial cells: loss of BRCA-1 expression in colonic adenocarcinomas. Nutr Cancer. 2003;46:82–92. doi: 10.1207/S15327914NC4601_11. [DOI] [PubMed] [Google Scholar]
- 77.Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharmacol. 2005;70:1035–1047. doi: 10.1016/j.bcp.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 78.Li Z, Tanaka M, Kataoka H, Nakamura R, Sanjar R, Shinmura K, Sugimura H. EphA2 up-regulation induced by deoxycholic acid in human colon carcinoma cells, an involvement of extracellular signal-regulated kinase and p53-independence. J Cancer Res Clin Oncol. 2003;129:703–708. doi: 10.1007/s00432-003-0493-z. [DOI] [PubMed] [Google Scholar]
- 79.Im E, Akare S, Powell A, Martinez JD. Ursodeoxycholic acid can suppress deoxycholic acid-induced apoptosis by stimulating Akt/PKB-dependent survival signaling. Nutr Cancer. 2005;51:110–116. doi: 10.1207/s15327914nc5101_15. [DOI] [PubMed] [Google Scholar]
- 80.Im E, Martinez JD. Ursodeoxycholic acid (UDCA) can inhibit deoxycholic acid (DCA)-induced apoptosis via modulation of EGFR/Raf-1/ERK signaling in human colon cancer cells. J Nutr. 2004;134:483–486. doi: 10.1093/jn/134.2.483. [DOI] [PubMed] [Google Scholar]
- 81.Pai R, Tarnawski AS, Tran T. Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell. 2004;15:2156–2163. doi: 10.1091/mbc.E03-12-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Gottardi A, Touri F, Maurer CA, Perez A, Maurhofer O, Ventre G, Bentzen CL, Niesor EJ, Dufour JF. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig Dis Sci. 2004;49:982–989. doi: 10.1023/b:ddas.0000034558.78747.98. [DOI] [PubMed] [Google Scholar]
- 83.Gould KA, Dove WF. Action of Min and Mom1 on neoplasia in ectopic intestinal grafts. Cell Growth Differ. 1996;7:1361–1368. [PubMed] [Google Scholar]
- 84.Jacoby RF, Cole CE, Hawk ET, Lubet RA. Ursodeoxycholate/Sulindac combination treatment effectively prevents intestinal adenomas in a mouse model of polyposis. Gastroenterology. 2004;127:838–844. doi: 10.1053/j.gastro.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 85.Kohno H, Suzuki R, Yasui Y, Miyamoto S, Wakabayashi K, Tanaka T. Ursodeoxycholic acid versus sulfasalazine in colitis-related colon carcinogenesis in mice. Clin Cancer Res. 2007;13:2519–2525. doi: 10.1158/1078-0432.CCR-06-2727. [DOI] [PubMed] [Google Scholar]
- 86.Narahara H, Tatsuta M, Iishi H, Baba M, Uedo N, Sakai N, Yano H, Ishiguro S. K-ras point mutation is associated with enhancement by deoxycholic acid of colon carcinogenesis induced by azoxymethane, but not with its attenuation by all-trans-retinoic acid. Int J Cancer. 2000;88:157–161. doi: 10.1002/1097-0215(20001015)88:2<157::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 87.Khare S, Cerda S, Wali RK, von Lintig FC, Tretiakova M, Joseph L, Stoiber D, Cohen G, Nimmagadda K, Hart J, et al. Ursodeoxycholic acid inhibits Ras mutations, wild-type Ras activation, and cyclooxygenase-2 expression in colon cancer. Cancer Res. 2003;63:3517–3523. [PubMed] [Google Scholar]
- 88.Serfaty L, De Leusse A, Rosmorduc O, Desaint B, Flejou JF, Chazouilleres O, Poupon RE, Poupon R. Ursodeoxycholic acid therapy and the risk of colorectal adenoma in patients with primary biliary cirrhosis: an observational study. Hepatology. 2003;38:203–209. doi: 10.1053/jhep.2003.50311. [DOI] [PubMed] [Google Scholar]
- 89.Wolf JM, Rybicki LA, Lashner BA. The impact of ursodeoxycholic acid on cancer, dysplasia and mortality in ulcerative colitis patients with primary sclerosing cholangitis. Aliment Pharmacol Ther. 2005;22:783–788. doi: 10.1111/j.1365-2036.2005.02650.x. [DOI] [PubMed] [Google Scholar]
- 90.Alberts DS, Martinez ME, Hess LM, Einspahr JG, Green SB, Bhattacharyya AK, Guillen J, Krutzsch M, Batta AK, Salen G, et al. Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence. J Natl Cancer Inst. 2005;97:846–853. doi: 10.1093/jnci/dji144. [DOI] [PubMed] [Google Scholar]
- 91.Sjoqvist U, Tribukait B, Ost A, Einarsson C, Oxelmark L, Lofberg R. Ursodeoxycholic acid treatment in IBD-patients with colorectal dysplasia and/or DNA-aneuploidy: a prospective, double-blind, randomized controlled pilot study. Anticancer Res. 2004;24:3121–3127. [PubMed] [Google Scholar]
- 92.Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889–893. doi: 10.1053/gast.2003.50156. [DOI] [PubMed] [Google Scholar]
- 93.Ochsenkuhn T, Marsteller I, Hay U, Diebold J, Paumgartner G, Goke B, Sackmann M. Does ursodeoxycholic acid change the proliferation of the colorectal mucosa?. A randomized, placebo-controlled study. Digestion. 2003;68:209–216. doi: 10.1159/000075927. [DOI] [PubMed] [Google Scholar]
- 94.Hess LM, Krutzsch MF, Guillen J, Chow HH, Einspahr J, Batta AK, Salen G, Reid ME, Earnest DL, Alberts DS. Results of a phase I multiple-dose clinical study of ursodeoxycholic Acid. Cancer Epidemiol Biomarkers Prev. 2004;13:861–867. [PubMed] [Google Scholar]
- 95.Levine J, Ellis CJ, Furne JK, Springfield J, Levitt MD. Fecal hydrogen sulfide production in ulcerative colitis. Am J Gastroenterol. 1998;93:83–87. doi: 10.1111/j.1572-0241.1998.083_c.x. [DOI] [PubMed] [Google Scholar]
- 96.Levitt MD, Furne J, Springfield J, Suarez F, DeMaster E. Detoxification of hydrogen sulfide and methanethiol in the cecal mucosa. J Clin Invest. 1999;104:1107–1114. doi: 10.1172/JCI7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fihn BM, Sjoqvist A, Jodal M. Involvement of enteric nerves in permeability changes due to deoxycholic acid in rat jejunum in vivo. Acta Physiol Scand. 2003;178:241–250. doi: 10.1046/j.1365-201X.2003.01144.x. [DOI] [PubMed] [Google Scholar]
- 98.Gelbmann CM, Schteingart CD, Thompson SM, Hofmann AF, Barrett KE. Mast cells and histamine contribute to bile acid-stimulated secretion in the mouse colon. J Clin Invest. 1995;95:2831–2839. doi: 10.1172/JCI117988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karlstrom L. Evidence of involvement of the enteric nervous system in the effects of sodium deoxycholate on small-intestinal transepithelial fluid transport and motility. Scand J Gastroenterol. 1986;21:321–330. doi: 10.3109/00365528609003082. [DOI] [PubMed] [Google Scholar]
- 100.Chadwick VS, Gaginella TS, Carlson GL, Debongnie JC, Phillips SF, Hofmann AF. Effect of molecular structure on bile acid-induced alterations in absorptive function, permeability, and morphology in the perfused rabbit colon. J Lab Clin Med. 1979;94:661–674. [PubMed] [Google Scholar]
- 101.Mekjian HS, Phillips SF, Hofmann AF. Colonic secretion of water and electrolytes induced by bile acids: perfusion studies in man. J Clin Invest. 1971;50:1569–1577. doi: 10.1172/JCI106644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fang P, Dong L, Zhang WJ, Luo JY. Relationship between entero-hepatic bile acid circulation and interdigestive migrating myoelectrical activity in rats. World J Gastroenterol. 2005;11:5377–5380. doi: 10.3748/wjg.v11.i34.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karlstrom L, Cassuto J, Jodal M, Lundgren O. The importance of the enteric nervous system for the bile-salt-induced secretion in the small intestine of the rat. Scand J Gastroenterol. 1983;18:117–123. doi: 10.3109/00365528309181570. [DOI] [PubMed] [Google Scholar]
- 104.Karlstrom L, Jodal M, Lundgren O. Blood flow distribution, lymph flow, villus tissue osmolality and fluid and electrolyte transport after exposing the cat small intestine to sodium deoxycholate. Acta Physiol Scand. 1986;128:83–96. doi: 10.1111/j.1748-1716.1986.tb07953.x. [DOI] [PubMed] [Google Scholar]
- 105.Bossmann B, Haschen RJ. Release of enzymes from rat jejunal mucosa by bile salts. J Clin Chem Clin Biochem. 1983;21:1–9. doi: 10.1515/cclm.1983.21.1.1. [DOI] [PubMed] [Google Scholar]
- 106.Binder HJ, Filburn C, Volpe BT. Bile salt alteration of colonic electrolyte transport: role of cyclic adenosine monophosphate. Gastroenterology. 1975;68:503–508. [PubMed] [Google Scholar]
- 107.Zhu Y, Hua P, Rafiq S, Waffner EJ, Duffey ME, Lance P. Ca2+- and PKC-dependent stimulation of PGE2 synthesis by deoxycholic acid in human colonic fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;283:G503–G510. doi: 10.1152/ajpgi.00525.2001. [DOI] [PubMed] [Google Scholar]
- 108.Quist RG, Ton-Nu HT, Lillienau J, Hofmann AF, Barrett KE. Activation of mast cells by bile acids. Gastroenterology. 1991;101:446–456. doi: 10.1016/0016-5085(91)90024-f. [DOI] [PubMed] [Google Scholar]
- 109.Camilleri M, Murphy R, Chadwick VS. Dose-related effects of chenodeoxycholic acid in the rabbit colon. Dig Dis Sci. 1980;25:433–438. doi: 10.1007/BF01395507. [DOI] [PubMed] [Google Scholar]
- 110.Hardcastle J, Hardcastle PT, Chapman J, Taylor CJ. Taurocholic acid-induced secretion in normal and cystic fibrosis mouse ileum. J Pharm Pharmacol. 2001;53:711–719. doi: 10.1211/0022357011775839. [DOI] [PubMed] [Google Scholar]
- 111.Hardcastle J, Hardcastle PT, Chapman J, Taylor CJ. Ursodeoxycholic acid action on the transport function of the small intestine in normal and cystic fibrosis mice. J Pharm Pharmacol. 2001;53:1457–1467. doi: 10.1211/0022357011777990. [DOI] [PubMed] [Google Scholar]
- 112.Volpe BT, Binder HJ. Bile salt alteration of ion transport across jejunal mucosa. Biochim Biophys Acta. 1975;394:597–604. doi: 10.1016/0005-2736(75)90145-5. [DOI] [PubMed] [Google Scholar]
- 113.Bijvelds MJ, Jorna H, Verkade HJ, Bot AG, Hofmann F, Agellon LB, Sinaasappel M, de Jonge HR. Activation of CFTR by ASBT-mediated bile salt absorption. Am J Physiol Gastrointest Liver Physiol. 2005;289:G870–G879. doi: 10.1152/ajpgi.00226.2005. [DOI] [PubMed] [Google Scholar]
- 114.Hardcastle J, Harwood MD, Taylor CJ. Absorption of taurocholic acid by the ileum of normal and transgenic DeltaF508 cystic fibrosis mice. J Pharm Pharmacol. 2004;56:445–452. doi: 10.1211/0022357022881. [DOI] [PubMed] [Google Scholar]
- 115.Wall MJ, Baker RD. Effect of taurocholate on electrical potential difference across rat small intestine. Life Sci I. 1972;11:375–386. doi: 10.1016/0024-3205(72)90047-1. [DOI] [PubMed] [Google Scholar]
- 116.Devor DC, Sekar MC, Frizzell RA, Duffey ME. Taurodeoxycholate activates potassium and chloride conductances via an IP3-mediated release of calcium from intracellular stores in a colonic cell line (T84) J Clin Invest. 1993;92:2173–2181. doi: 10.1172/JCI116819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Balistreri WF, Heubi JE, Suchy FJ. Bile acid metabolism: relationship of bile acid malabsorption and diarrhea. J Pediatr Gastroenterol Nutr. 1983;2:105–121. [PubMed] [Google Scholar]
- 118.Sanchez de Medina F, Perez R, Martinez-Augustin O, Gonzalez R, Lorente MD, Galvez J, Zarzuelo A. Disturbances of colonic ion secretion in inflammation: role of the enteric nervous system and cAMP. Pflugers Arch. 2002;444:378–388. doi: 10.1007/s00424-002-0807-z. [DOI] [PubMed] [Google Scholar]
- 119.Chen F, Ma L, Sartor RB, Li F, Xiong H, Sun AQ, Shneider B. Inflammatory-mediated repression of the rat ileal sodium-dependent bile acid transporter by c-fos nuclear translocation. Gastroenterology. 2002;123:2005–2016. doi: 10.1053/gast.2002.37055. [DOI] [PubMed] [Google Scholar]
- 120.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 121.Cotting J, Lentze MJ, Reichen J. Effects of ursodeoxycholic acid treatment on nutrition and liver function in patients with cystic fibrosis and longstanding cholestasis. Gut. 1990;31:918–921. doi: 10.1136/gut.31.8.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Talamini MA, Gadacz TR. Gallstone dissolution. Surg Clin North Am. 1990;70:1217–1230. doi: 10.1016/s0039-6109(16)45280-1. [DOI] [PubMed] [Google Scholar]
- 123.Bernardes-Silva CF, Damiao AO, Sipahi AM, Laurindo FR, Iriya K, Lopasso FP, Buchpiguel CA, Lordello ML, Agostinho CL, Laudanna AA. Ursodeoxycholic acid ameliorates experimental ileitis counteracting intestinal barrier dysfunction and oxidative stress. Dig Dis Sci. 2004;49:1569–1574. doi: 10.1023/b:ddas.0000043365.39251.6e. [DOI] [PubMed] [Google Scholar]
- 124.Lloyd-Still JD, Beno DW, Uhing MR, Jiyamapa-Serna VA, Kimura RE. Ursodeoxycholic acid ameliorates ibuprofen-induced enteropathy in the rat. J Pediatr Gastroenterol Nutr. 2001;32:270–273. doi: 10.1097/00005176-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 125.Lee DK, Park SY, Baik SK, Kwon SO, Chung JM, Oh ES, Kim HS. [Deoxycholic acid-induced signal transduction in HT-29 cells: role of NF-kappa B and interleukin-8] Korean J Gastroenterol. 2004;43:176–185. [PubMed] [Google Scholar]
- 126.Muhlbauer M, Allard B, Bosserhoff AK, Kiessling S, Herfarth H, Rogler G, Scholmerich J, Jobin C, Hellerbrand C. Differential effects of deoxycholic acid and taurodeoxycholic acid on NF-kappa B signal transduction and IL-8 gene expression in colonic epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;286:G1000–G1008. doi: 10.1152/ajpgi.00338.2003. [DOI] [PubMed] [Google Scholar]
- 127.Payne CM, Crowley C, Washo-Stultz D, Briehl M, Bernstein H, Bernstein C, Beard S, Holubec H, Warneke J. The stress-response proteins poly(ADP-ribose) polymerase and NF-kappaB protect against bile salt-induced apoptosis. Cell Death Differ. 1998;5:623–636. doi: 10.1038/sj.cdd.4400395. [DOI] [PubMed] [Google Scholar]
- 128.Strauch ED, Bass BL, Rao JN, Vann JA, Wang JY. NF-kappaB regulates intestinal epithelial cell and bile salt-induced migration after injury. Ann Surg. 2003;237:494–501. doi: 10.1097/01.SLA.0000060459.03270.E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hirano F, Tanada H, Makino Y, Okamoto K, Hiramoto M, Handa H, Makino I. Induction of the transcription factor AP-1 in cultured human colon adenocarcinoma cells following exposure to bile acids. Carcinogenesis. 1996;17:427–433. doi: 10.1093/carcin/17.3.427. [DOI] [PubMed] [Google Scholar]
- 130.Strauch ED, Yamaguchi J, Bass BL, Wang JY. Bile salts regulate intestinal epithelial cell migration by nuclear factor-kappa B-induced expression of transforming growth factor-beta. J Am Coll Surg. 2003;197:974–984. doi: 10.1016/S1072-7515(03)00720-8. [DOI] [PubMed] [Google Scholar]
- 131.Lazaridis KN, Gores GJ, Lindor KD. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J Hepatol. 2001;35:134–146. doi: 10.1016/s0168-8278(01)00092-7. [DOI] [PubMed] [Google Scholar]
- 132.Paumgartner G. Medical treatment of cholestatic liver diseases: From pathobiology to pharmacological targets. World J Gastroenterol. 2006;12:4445–4451. doi: 10.3748/wjg.v12.i28.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Park SE, Choi HJ, Yee SB, Chung HY, Suh H, Choi YH, Yoo YH, Kim ND. Synthetic bile acid derivatives inhibit cell proliferation and induce apoptosis in HT-29 human colon cancer cells. Int J Oncol. 2004;25:231–236. [PubMed] [Google Scholar]
- 134.Lepercq P, Hermier D, David O, Michelin R, Gibard C, Beguet F, Relano P, Cayuela C, Juste C. Increasing ursodeoxycholic acid in the enterohepatic circulation of pigs through the administration of living bacteria. Br J Nutr. 2005;93:457–469. doi: 10.1079/bjn20041386. [DOI] [PubMed] [Google Scholar]
- 135.Kapral C, Wewalka F, Praxmarer V, Lenz K, Hofmann AF. Conjugated bile acid replacement therapy in short bowel syndrome patients with a residual colon. Z Gastroenterol. 2004;42:583–589. doi: 10.1055/s-2004-813059. [DOI] [PubMed] [Google Scholar]
- 136.Akobeng AK, Clayton PT, Miller V, Super M, Thomas AG. An inborn error of bile acid synthesis (3beta-hydroxy-delta5-C27-steroid dehydrogenase deficiency) presenting as malabsorption leading to rickets. Arch Dis Child. 1999;80:463–465. doi: 10.1136/adc.80.5.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Eusufzai S. Bile acid malabsorption: mechanisms and treatment. Dig Dis. 1995;13:312–321. doi: 10.1159/000171511. [DOI] [PubMed] [Google Scholar]
- 138.Tsutsumi K, Li SK, Hymas RV, Teng CL, Tillman LG, Hardee GE, Higuchi WI, Ho NF. Systematic studies on the paracellular permeation of model permeants and oligonucleotides in the rat small intestine with chenodeoxycholate as enhancer. J Pharm Sci. 2008;97:350–367. doi: 10.1002/jps.21093. [DOI] [PubMed] [Google Scholar]
- 139.Lane ME, O’driscoll CM, Corrigan OI. Quantitative estimation of the effects of bile salt surfactant systems on insulin stability and permeability in the rat intestine using a mass balance model. J Pharm Pharmacol. 2005;57:169–175. doi: 10.1211/0022357055434. [DOI] [PubMed] [Google Scholar]
- 140.Lee DY, Lee J, Lee S, Kim SK, Byun Y. Liphophilic complexation of heparin based on bile acid for oral delivery. J Control Release. 2007;123:39–45. doi: 10.1016/j.jconrel.2007.07.013. [DOI] [PubMed] [Google Scholar]