

Abstract
AIM: To determine whether HBV with the same characteristics causes dissimilar mutations in different hosts.
METHODS: Full-length HBV genome was amplified and linked with pMD T18 vector. Positive clones were selected by double-restriction endonuclease digestion (EcoRI and HindIII) and PCR. Twenty seven clones were randomly selected from an asymptomatic mother [at two time points: 602 (1 d) and 6022 (6 mo)] and her son [602 (S)], and the phylogenetic and mutational analysis was performed using BioEditor, Clustal X and MEGA software. Potential immune epitopes were determined by the Stabilized Matrix Method (SMM), SMM-Align Method and Emini Surface Accessibility Prediction.
RESULTS: All of the 27 sequences were genotype C, the divergence between the mother and son was 0%-0.8%. Compared with another 50 complete sequences of genotype C, the mother and her son each had 13 specific nucleotides that differed from the other genotype C isolates. AA 1-11 deletion in preS1 was the dominant mutation in the mother (14/18). The 1762T/1764A double mutation existed in all clones of the mother, 3 of them were also coupled with G1896A mutation, but none were found in the son. 17 bp deletion starting at nucleotide 2330 was the major mutation (5/9) in the son, which caused seven potential HLA class I epitopes and one B cell epitope deletion, and produced a presumptive new start codon, downstream from the original one of the P gene.
CONCLUSION: The HBV strain in the son came from his mother, and discrepant mutation occurred in the mother and her son during infection.
Keywords: Hepatic B virus, Vertical transmission, Full genome, Mutation, Phylogenetic, Deletion
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a significant global public health problem. Approximately 3-4 billion people worldwide have been infected with HBV, and 350-400 million persons have chronic HBV infection. Vertical transmission, especially mother-to-infant transmission is the primary pathway of hepatitis B virus infection, in approximately 90% of the infants born to HBeAg positive HBsAg carrier mothers, if no immunoprophylaxis is given. HBV with the same characteristics in the mother and child interacts with the immune system and may result in different mutations to escape immune pressure.
In the present study, we isolated the full genomic sequence of HBV in an asymptomatic mother and her son by direct sequencing. Phylogenetic and mutational analysis were performed to obtain evidence of mother to infant HBV transmission, and to elucidate distinctive HBV mutations in the different hosts.
MATERIALS AND METHODS
Patient
A 31-year-old female who was first seen at the First People’s Hospital of Yunnan Province, Kunming, Yunnan, China in 2005 because of allergic purpura was reevaluated. She was HBsAg and HBeAg positive since at least 1999. Her sera were obtained from two time points: 602 (1 d) and 6022 (6 mo later). When seen again, 6 mo after her initial visit, her 7-year old son’s serum [602 (S)] was collected. The alanine aminotransferase (ALT) level and abdominal ultrasound examination of the mother and son were normal. There was no history of alcohol abuse, parenteral drug use and hepatotoxin exposure. Neither the mother nor her son had received immunoprophylaxis or immunotherapy.
Serological markers and HBV DNA
HBV serological markers were evaluated using commercially available enzyme-linked immunosorbent assay (ELISA) kit (Kehua Bio-Engineering Co. LTD, Shanghai, China). HBV DNA in the sera was determined using a commercially available real-time fluorescence quantitative PCR (FQ-PCR) kit (Da An Gene Diagnostic Center, Shenzheng, China). Both subjects were also tested for hepatitis C virus (HCV), hepatitis A virus (HAV) and hepatitis E virus (HEV) infection.
Amplification of full-length HBV genome
HBV DNA was extracted from the sera using proteinase K digestion followed by phenol/chloroform extraction. The complete HBV genome was amplified by polymerase chain reaction (PCR) as described previously[1]. Amplification was performed in a 96-well cycler (Bio-RAD, USA), and 25 μL PCR mixture containing 2.5 mmol MgCl2, 200 μmol/L dNTP, 400 nmol/L of each primer, and 2 U of Ex Taq polymerase (Takara BioTech). The PCR reaction was performed using the following cycles: 94°C pre-denature for 5 min, 30 cycles of 94°C for 1 min, 56°C for 1 min, 72°C for 2 min; and 72°C for 10 min as a final extension step. The full-length amplicon were purified using a gel extraction kit (HuaShun Bio-Engineering).
Cloning and sequencing of the full-length HBV genome
Purified full-length HBV-DNA was directly linked with pMD18-T vector (TaKaRa Bio-Tech), using the standard cloning techniques. White colonies were picked and the genomic length of insertions was confirmed with PCR and double-restriction endonuclease digestion with EcoRI and HindIII. DNA sequencing analysis of the correct recombinants was performed with BigDye Terminator v3.1 and 3130 Genetic Analyzer (Applied Biosystems). For each patient and time point, 9 recombinants were randomly selected and sequenced for the full-length genome of HBV.
Sequence analysis
Thirty three HBV complete genome sequences (genotypes A to H) were used in as reference[2]. Additionally, 40 complete HBV genome sequences of genotype C from GenBank were used for phylogenetic analysis. The complete genome sequences were assembled from the sequencing data, using the BioEdit sequence alignment editor software, version 7.0.5.2[3]. Alignment was performed using Clustal X software, version 1.83[4]. Phylogenetic and molecular evolutionary analysis and nucleotide differences between the isolate sequences were carried out by the MEGA program, version 3.1[5]. The genetic distance was estimated using the Kimura two-parameter algorithm[6]. The phylogenetic trees were constructed using the neighbor-joining method[7]. Bootstrap re-sampling and reconstruction was carried out 1000 times to confirm the reliability of the phylogenetic trees[8]. The HBV genotype was assigned according to the classification system reported previously.
Potential immune epitopes prediction
For a new deletion fragment (17 bp deletion in Core gene region from nt 2330), Stabilized Matrix Method (SMM) and SMM-Align method were used to predict the potential major histocompatibility classes I/II[9,10] (Predicted IC50 < 200), and the Emini Surface Accessibility Prediction was used to predict the potential B cell epitope[11].
RESULTS
Serological markers and HBV DNA quantification
Both the mother and son were positive for HBsAg, HBeAg and anti-HBc. They were seronegative for HAV, HCV and HEV. The HBV viral load was 2.4 × 108 copy/mL in the mother (6022) and 4.2 × 108 copy/mL in the son [602(S)]. The 602 sera were not quantified due to insufficient material.
Genotypic and serotypic relatedness
The 27 sequenced clones (Gene Bank accession No: DQ377159-377165, EU306713- EU306729, EU439005- 439007) ranged in length from 3036bp to 3254 bp. The divergence of the complete genome of 18 clones for the mother was 0%-0.8%, while the divergence of the 9 clones for the son was 0.1%-0.6 %, and the divergence of the 27 clones for the mother and child was 0%-0.8%. For the 27 clones, the homogenous of S gene was 98.5%-100%. The 27 clones were 51H, 54E, 62A, 73G, 125T and 227S in the large S gene, 136A, 142K, 233R, 236S, 248P, 252R, 304H, and 354H in the P gene, and 122K,160R and 159A in the small S gene. Compared with the 33 global complete genome sequences of HBV from GenBank, phylogenetic analysis showed that with bootstrap values of 100%, the clones were classified into genotype C and C2 subgroup (Figure 1)[12]. The serotype was classified as adrq+[13].
Figure 1.

Phylogenetic tree constructed on the complete genome (A) and S gene (B) [Mother: 602 (1 d), 6022 (6 mo); Son: 602 (S)]. Bootstrap values are shown at the beginning of each main node. The length of the horizontal bar indicates the number of nucleotide substitutions per site. The origin of each strain has also been shown.
Characterization of the nucleotides and the deduced amino acid sequence
Compared with the other 50 complete sequences of genotype C, there were 13 specific point mutations in the 27 clones, the nucleotide positions were at 105T, 346C, 855C, 861T, 930G, 951T, 1110T, 1341C, 1972C, 2215C, 2246C, 2480T and 3102A. Except for C105T and G3102A which caused mutation of amino acid A158V in the large S gene and D266N in the P gene, respectively, the other substitutions resulted in the same sense mutation. Within the pre-S1 region, 14 clones of the mother had 18bp deletion from the start codon, which resulted in deletion at codons 1-11. The C-terminal truncated S protein was found in both the mother and son, which caused random point mutation in or near the hydrophilic region of S gene (nt 200, nt361, nt455, and nt565, respectively). Within the X region, 1762T/1764A double mutation was observed in all clones in the mother, but none was found in the son. The hypervariable region (HVR) sequence at positions 1751 to 1755, and the AT-rich region sequence at 1789 to 1795 in the X-ORF was conserved in all of the clones. In the precore and core regions, 3 clones of the mother had G1896A mutation and one clone had 19bp deletion, resulting in a novel stop codon of the C gene. In the P region, the YMDD motif was conserved in all the clones, 5 clones of the son had a 17 bp deletion from nt2330, and produced a 43 aa residue in the N-terminal of polymerase (Figure 2). Other random mutations occurred throughout the sequenced genomes, like a “G” insertion at nt 761 [602 (S)-39] was observed in one clone of the son and a “TT” deletion at nt2012 in one clone of the mother.
Figure 2.
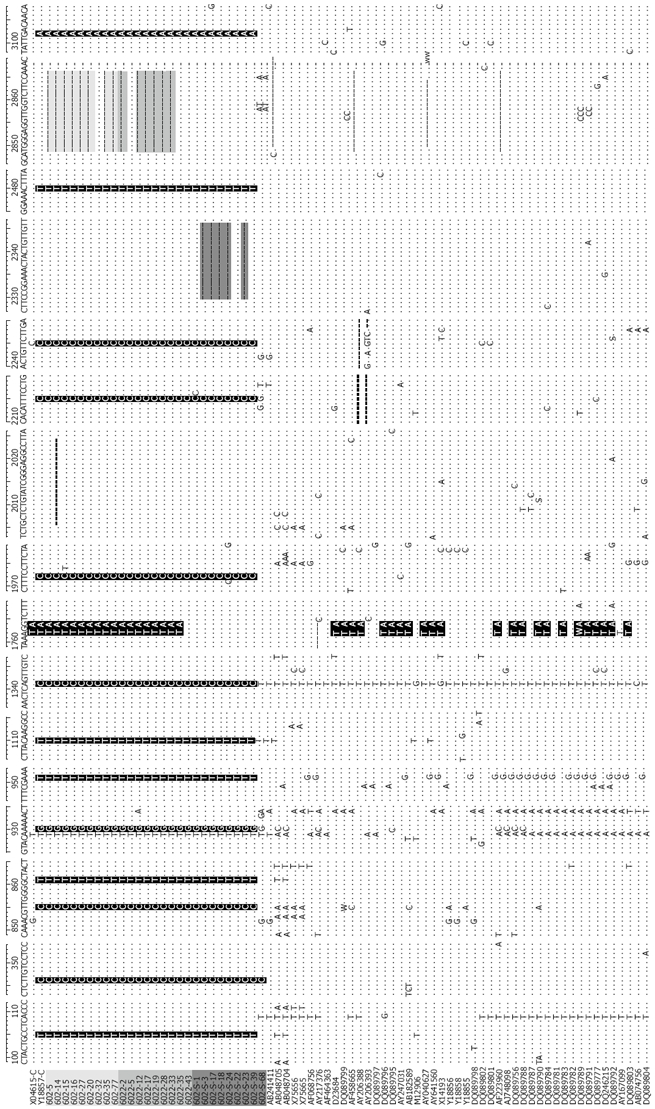
Nucleotide sequences of the 27 HBV/C isolates and 50 other complete genome sequences of genotype C [light gray, Mother: 602 (1 d); Gray, 6022 (6 mo); Dark gray, Son: 602-S; Sequences: Gray (deletion), black (substitution of nucleotides)]. Compared with the 50 complete genome sequences of genotype C, nucleotide positions at 105T, 346C, 855C,861T, 930G, 951T, 1110T, 1341C, 1972C, 2215C, 2246C,2480T and 3102A were specific for the 27 samples. 14 clones of the mother had an 18bp deletion from start codon; 5 clones of the son had a 17nt deletion from nt2330; 1762T/1764A double mutations were found in all clones of the mother, but none in the sequenced clones of the son. Nucleotides A1762A and G1764A double mutation comprised 54.5% of the total genotype C (42/77).
Potential immune epitopes caused by 17 bp deletion from nt2330
The 17bp deletion mutant from nt2330 in 5 clones of the son led to the deletion of aa144-183 of the HBcAg, as reported in this study after performing Blastn. By the immune epitope prediction method, potential clustal immune epitopes were located in the deletion region of Core gene, including 7 HLA class I epitopes and one B cell epitope (PRRRRSQ; Table 1, Figures 3 and 4).
Table 1.
MHC-I binding predictions of 144-183 residue of HbcAg, processed by the SMM method
| Allele | Position (aa) | Pep length | Sequence | IC50 (nm)1 |
| HLA A*3101 | 148-157 | 10 | VVRRRGRSPR | 30.9 |
| HLA A*3101 | 150-159 | 10 | RRRGRSPRRR | 13.7 |
| HLA A*3101 | 157-166 | 10 | RRRTPSPRRR | 19.2 |
| HLA A*3101 | 159-167 | 9 | RTPSPRRRR | 38.4 |
| HLA A*3101 | 165-174 | 10 | RRRSQSPRRR | 19.2 |
| HLA A*3101 | 166-175 | 10 | RRSQSPRRRR | 35.4 |
| HLA A*3101 | 167-175 | 9 | RSQSPRRRR | 13.7 |
| HLA A*3201 | 159-167 | 9 | RTPSPRRRR | 48.6 |
| HLA A*6801 | 145-154 | 9 | ETTVVRRRGR | 23 |
| HLA A*6801 | 146-154 | 9 | TTVVRRRGR | 22.3 |
| HLA B*0702 | 162-170 | 9 | SPRRRRSQS | 21.3 |
| HLA B*0702 | 170-178 | 9 | SPRRRRSQS | 21.3 |
| HLA B*0702 | 155-163 | 9 | SPRRRTPSP | 40.4 |
| HLA B*0801 | 162-170 | 9 | SPRRRRSQS | 39.5 |
| HLA B*0801 | 170-178 | 9 | SPRRRRSQS | 39.5 |
| HLA B*2705 | 150-159 | 10 | RRRGRSPRRR | 49.5 |
| HLA B*2705 | 164-173 | 10 | RRRRSQSPRR | 14.7 |
| HLA B*2705 | 166-175 | 10 | RRSQSPRRRR | 33.9 |
IC50 (nM) < 50 was considered as high affinity. Only IC50 (nM) has been shown in the Table.
Figure 3.

Potential HLA class I epitopes of the deletion fragment in HBcAg (aa residue144-183), determined by the SMM method [only IC50 (nM) < 50]. Blue line: HLA A*3101 (aa 148-175); Black line: HLA A*3201(aa 159-167); Orange line: HLA A*6801 (aa 145-154); Pink line: HLA B*0702 (aa 155-178); Brown line: HLA B*0801 (aa 162-178); Green line: HLA B*2705 (aa 150-159, 164-175).
Figure 4.
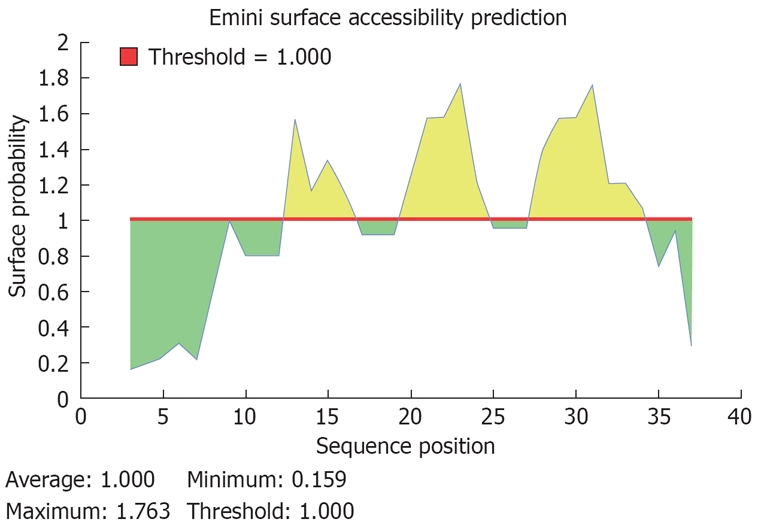
Predicted peptides of the potential B cell epitope of the deleted fragment in HBcAg (aa residue 144-183). A potential sequence of B cell epotide in aa residue 144-183 of HBcAg was PRRRRSQ, start position at aa 171, end position at aa 177.
DISCUSSION
Using phylogenetic and mutational analysis, clones of the mother obtained at two time points were conserved. The dominant mutants in the mother obtained at two time points were the same (18 bp deletion in preS1 region and BCP double mutation), except for the scattering mutation. Although, the son was infected from the mother, the divergence of the mother and the son was 0%-0.8%, and the dominant mutants in the son were different from the mother.
Universal infant immunization has reduced HBV infection rate in highly endemic countries with a relatively homogenous population. However, HBV transmission continues to occur via vertical (mother-to-child) and horizontal (sexual, parenteral and household) routes. In Asia, where genotypes B and C predominate, vertical transmission is common. In the present study, we isolated HBV strains from an asymptomatic mother and her son and analyzed the sequences phylogenetically. All the 27 clones were of the C2 subtype and adrq+ serotype. Compared with 50 other complete sequences of genotype C, there were 13 specific nucleotides belonging to the mother and her son. It should be noted that there was no history of alcohol abuse, parenteral drug use or hepatotoxin exposure, and no immunoprophylaxis or immunotherapy was given to the mother and her son. The son’s father was seronegative for HBV markers. Based on these findings combined with the data of divergence in the complete viral genome and homogeneity in the S gene, it can be concluded that the son became infected via a mother-to-infant transmission.
The pre-S1 domain is the essential binding site for hepatocyte receptors, and mutations at this region may directly influence HBV infection, and progression of liver disease. In the present study, 14 clones of the mother had aa 1-11 deletion in preS1 caused by a 18bp deletion from the original start codon of preS1.This deletion mutant has previously been reported in different clinical conditions, especially hepatocellular carcinoma (HCC) and chronic hepatitis B (CHB) infection with genotypes C and D, and fulminant hepatitis B (FHB) with genotype A in Africa. It has also been observed in isolates obtained from nonhuman primates (Table 2)[14-32]. Interestingly, there are no reports of this deletion mutation in genotypes B, E, F and G. In a heart transplant recipient who died from fulminate hepatitis B transmitted by the donor, the 18bp deletion was detected in the recipient, but not in the donor[20]. In our case, the 18bp deletion mutants in preS1 were present in 14 clones of the mother (78%), but in none of the sequenced clones in the son (0%). This phenomenon may imply that host immune pressure was the primary cause of aa 1-11 deletion in preS1. In a subsequent study (2 years later), we observed that the 18bp deletion in preS1 had a tendency for substitution by large fragment deletions in preS1, and laboratory tests in the mother showed abnormal values (ALT > 2 times of reference values) (detailed discussion reported in a separate publication). However, whether the deletion of aa 1-11 mutation in preS1 was a precursor of large fragment deletion mutation or was an isolated event under immune pressure remains to be determined. To our knowledge, no previous study has reported such deletion mutants. This deletion, which disrupts the preS1 start codon, may play an important role in enhancing the progression of chronic liver disease.
Table 2.
Deletion of aa 1-11 in preS1 in the different HBV genotypes
| Genotype | GenBank No. | Clinic status | Countries/district | Ref. |
| Human | ||||
| Genotype C | AF223959 | CHB | Vietnam | Erik et al, 2001 |
| AY217372 | CHB | China | Luo et al, 2004 | |
| AB195945 | CHB | Japan | Horiike et al, 2007 | |
| EF533714 | HCC | China | 1Gao et al, 2007 | |
| AY206389 | HCC | China | 1Lin et al, 2002 | |
| AB014395 | HCC | Japan | Takahashik et al, 1998 | |
| AY641563 | HCC | South Korea | Song et al, 2005 | |
| D1666 | AHB | Japan | Uchida et al, 1995 | |
| X98076 | FHB | Switzerland | Pult et al, 1997 | |
| Genotype D | AB033559 | CHB | Papua New Guinea | Okamoto et al, 1988 |
| AB119256 | CHB | Japan | Michitaka et al, 2006 | |
| M32138 | Anti-HBe | France | Tong et al, 1990 | |
| AY902777 | FHB | Montana | Garfein et al, 2004 | |
| Genotype A | AF297621 | FHB | Africa | Owiredu et al, 2001 |
| U87743 | AHB | South Africa | Bowyer et al, 1997 | |
| X69458 | AHB | Zimbabwe | Chirara et al, 1994 | |
| Ape | ||||
| Gibbon | AB037928 | East Asia | Aiba et al, 2003 | |
| Chimpanzee | D00220 | HBsAg (+) | London | Vaudin et al, 1988 |
| Gorila | AJ131567 | HBsAg (+) | Japan | Grethe et al, 2000 |
| Orangutan | AF193864 | CHB | Bornean | Verschoor et al, 2001 |
| Recombination | ||||
| Human-Gibbon | AB048705 | HBsAg (+) | Australian Aborigines | Sugauchi et al, 2001 |
CHB: Chronic hepatitis B; AHB: Acute hepatitis B; FHB: Fulminant hepatitis B.
Sequences were submitted directly to the GenBank.
The 18 bp deletion mutants in preS1, located in the overlapping region of S/P gene region, caused six amino acid deletions, from 183 to 187 of the HBV polymerase. In the present study, all of these deletion mutants coexisted with the wild-type full-length genome. Transfection experiments in human hepatocells (HuH-7) demonstrated that the polymerase gene function was not affected by the large pre-S1 deletions, and mutant viral genomes were capable of replication[33]. This finding may explain the above observation, although deletion mutation of pre S1 was the major mutation in the mother, and the viral load in the serum remained high.
Cloning and sequence analysis showed that 3 clones of the mother and one clone of the son had premature S protein, caused by random points mutation. These mutations could affect not only the spatial structure, but also the immune reactivity. Some reports have found that C-terminal truncated S proteins exist in patients with chronic hepatitis B infection and hepatocellular carcinoma[34]. The shorted S proteins can stimulate gene expression from the endogenous HBV enhancer I and can also deregulate the expression of oncogenes such as c-myc[35], which may help clarify the tumor development mechanism of HBV. Whether the truncated S proteins transactivate the oncogenes and/or worsen the clinical status of the subjects remains to be established.
The double mutation of nucleotide A1762T/G1764A in basal core promoter (BCP), and G1986A in the precore region are frequently observed in HBV sequences isolated from patients with chronic HBV infection, fulminant hepatitis, HCC, and in reactivation of HBV with a fulminant course[36-38], which results in mutations at two codons in the carboxyl functional region of X protein (K130M and V131I), and a stop codon at aa 28 of HBeAg, respectively. At present, there are conflicting opinions regarding 1762T/1764A hotspot mutations. Some studies suggest that these mutations decrease HBeAg expression and slightly increase viral DNA replication, and are mostly found in patients who seroconvert to anti-HBe[39,40]. By contrast, other studies indicate that this mutation is not associated with HBeAg/anti-HBe status or HBV DNA[41,42]. The lack of influence of BCP mutation on HBeAg expression was mainly indicated by its higher prevalence in genotype C than genotype B patients. In the present study, all clones of the mother from two time points had these double mutations (Figure 2); genotype C has a tendency for higher prevalence of 1762T/1764A double mutation (42 vs 35, 54.5%). Based on our data from two time points, we did not observe any HBeAg/anti-HBeAg seroconversion, and we did not detect the impact of BCP mutation on HBV DNA load, due to insufficient quantity of 602 sera. Besides, the 1762T/1764A double mutation was present in all clones of the mother, 3 of whom were also coupled with G1896A. The G1896A mutation is supposed to increase the stability of the stem-loop structure. Recent reports suggest that HBV subgenotypes Ba, C1, and C2 have an intermediate frequency of 1896A mutation[43]. Interestingly, although the son was infected via the mother, none of the BCP and 1896A mutants were found in the son (0/9). This finding is consistent with previous studies in which mutants with 1896A were seldom transmitted to the infant via the mother[44]. Whether the BCP and precore mutations in the mother were related to infection with genotype C, or whether she was seroconverting to anti-HBeAg, or may develop severe disease exacerbations remains to be determined.
A heterogeneous population of core antigen internal deletions (CID) has been found to be highly prevalent in chronic HBV carriers[45,46], HCC patients[47] and immune- suppressed patients[48]. Normally, CID coincides with a potent T/B-cell epitope, and is almost always found in the presence of HBV with an apparent full-length core gene[49-51]. HBcAg has been shown to be a major target of T-cell immunity[52]. However, deletion type HBc did not show any antibody response[53].
In the present study, the wild type precore/core protein and deletion type precore/core protein co-existed in the sera of both the mother and son. Out of the 27 clones, one clone in the mother had 19nt deletion at position nt2006, and 5 clones in the son had 17bp deletion at position nt2330, which caused in-frame shift and resulted in a C-terminal truncated preCore/Core protein. To the best of our knowledge, this is the first report on 17bp deletion mutant, resulting in immune epitope deletion based on the immune epitope prediction tools. Interestingly, none of these mutants were found in the mother. Therefore, we concluded that under host immune pressure, due to the loss of HLA class and B cell I epitopes, this deletion variant may evade the immune system and became dominant in the host.
The 17 bp deletion mutation occurred in the overlapping region of P/C gene, and produced 43 aa peptides of HBV polymerase, possibly resulting in a new start codon, downstream of the original one. P-ORF encodes the terminal protein (TP), polymerase (pol) and RNase H. Previous studies have identified that in a protein-primed reaction, the minimal domain of TP ranged from amino acid 20 to 175[54]. In the present report, these 5 clones had incomplete TP, based on the minimal function domain of TP. These mutants have little or no activity. Interestingly, the viral load in the son was high (4.2 × 108 copy/mL), and no similar deletion mutants were found in his mother. Whether this phenomenon coexisted with the wild-type genome, or was the function domain of TP remains to be examined.
In addition, there were other point mutations and deletion/insertion mutations scattered in the complete genome, many of these were located in the T and B lymphocytes epitope, such as P46L, C69R and F73L in X gene[55], and P54T and M132T in C gene[56], while others resulted in truncated surface or preCore/Core proteins (T361A, TT deletion at nt2012, etc.). Because these mutants were dissimilar in different clones, they may have been caused by spontaneous mutation or immune pressure during evolution.
In summary, the HBV strain in the son was transmitted via maternal-fetal transmission, based on the presence of 13 specific and common nucleotides in both the mother and son. The dominant HBV mutants were different in the mother and son, because of different immune-environments. These mutants were capable of escaping immune-pressure and were present in HBV carriers. These newly discovered variants were located within or close to the functional domains of viral proteins. Whether they can exist “silently” in the hosts, or will cause liver disease remains to be determined.
COMMENTS
Background
Mother-to-infant transmission is the main pathway of hepatitis B virus (HBV) infection. Approximately 90% infants of HBeAg positive HBsAg carrier mothers become carriers, if no immunoprophylaxis is given. HBV with the same characteristics in the child and the mother interacts with the immune system, resulting in the development of different mutations designed to escape the immune pressure.
Research frontiers
Biosoftwares are powerful tools designed to perform virologic and phylogenetic analysis, and to predict immune epitopes. Combining this knowledge with clinical data on HBV-infected patients, may provide new information on the prevention and treatment of HBV infection.
Innovations and breakthroughs
The results of our study suggest that the preS1 deletion of HBV may be associated with disease progression. Using immune epitope prediction tools, the immune epitope deletion was found to occur in the C gene region.
Applications
Data obtained by virologic and phylogenetic analysis may provide clues to the prevention and treatment of chronic HBV infection.
Peer review
Virologic and phylogenetic analysis provides evidence of mother to infant transmission, and indicates that HBV with the same characteristics has dissimilar mutations in different hosts. Follow-up studies should greatly enhance the significance of the present report.
Footnotes
Supported by The Natural Science Foundation of Yunnan Province, No.200300172; Hospital Science Foundation of The First People’s Hospital of Yunnan Province (2004)
Peer reviewer: David R Gretch, MD, PhD, Viral Hepatitis Laboratory, Room 706, UW Research and Training Building, Harborview Medical Center, Box 359690, 325 Ninth Avenue, Seattle, Washington 98104, United States
S- Editor Zhong XY L- Editor Anand BS E- Editor Lin YP
References
- 1.Gunther S, Sommer G, Von Breunig F, Iwanska A, Kalinina T, Sterneck M, Will H. Amplification of full-length hepatitis B virus genomes from samples from patients with low levels of viremia: frequency and functional consequences of PCR-introduced mutations. J Clin Microbiol. 1998;36:531–538. doi: 10.1128/jcm.36.2.531-538.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol. 2007;13:14–21. doi: 10.3748/wjg.v13.i1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.BioEdit. Available from URL: http://www.mbio.ncsu.edu/BioEdit/BioEdit.html.
- 4.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar S, Tamura K, Jakobsen IB, Nei M. MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001;17:1244–1245. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- 6.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 7.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 8.Felsenstein J. Estimating effective population size from samples of sequences: a bootstrap Monte Carlo integration method. Genet Res. 1992;60:209–220. doi: 10.1017/s0016672300030962. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.IEDB Analysis Resource. Available from URL: http://tools.immuneepitope.org/main/jsp/menu.jsp.
- 11.Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol. 1985;55:836–839. doi: 10.1128/jvi.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huy TT, Ushijima H, Quang VX, Win KM, Luengrojanakul P, Kikuchi K, Sata T, Abe K. Genotype C of hepatitis B virus can be classified into at least two subgroups. J Gen Virol. 2004;85:283–292. doi: 10.1099/vir.0.19633-0. [DOI] [PubMed] [Google Scholar]
- 13.Margeridon S, Lachaux A, Trepo C, Zoulim F, Kay A. A quasi-monoclonal anti-HBs response can lead to immune escape of 'wild-type' hepatitis B virus. J Gen Virol. 2005;86:1687–1693. doi: 10.1099/vir.0.80810-0. [DOI] [PubMed] [Google Scholar]
- 14.Alestig E, Hannoun C, Horal P, Lindh M. Phylogenetic origin of hepatitis B virus strains with precore C-1858 variant. J Clin Microbiol. 2001;39:3200–3203. doi: 10.1128/JCM.39.9.3200-3203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo K, Liu Z, He H, Peng J, Liang W, Dai W, Hou J. The putative recombination of hepatitis B virus genotype B with pre-C/C region of genotype C. Virus Genes. 2004;29:31–41. doi: 10.1023/B:VIRU.0000032787.77837.09. [DOI] [PubMed] [Google Scholar]
- 16.Horiike N, Duong TN, Michitaka K, Joko K, Hiasa Y, Konishi I, Yano M, Onji M. Characteristics of lamivudine-resistant hepatitis B virus (HBV) strains with and without breakthrough hepatitis in patients with chronic hepatitis B evaluated by serial HBV full-genome sequences. J Med Virol. 2007;79:911–918. doi: 10.1002/jmv.20915. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi K, Akahane Y, Hino K, Ohta Y, Mishiro S. Hepatitis B virus genomic sequence in the circulation of hepatocellular carcinoma patients: comparative analysis of 40 full-length isolates. Arch Virol. 1998;143:2313–2326. doi: 10.1007/s007050050463. [DOI] [PubMed] [Google Scholar]
- 18.Song BC, Kim H, Kim SH, Cha CY, Kook YH, Kim BJ. Comparison of full length sequences of hepatitis B virus isolates in hepatocellular carcinoma patients and asymptomatic carriers of Korea. J Med Virol. 2005;75:13–19. doi: 10.1002/jmv.20230. [DOI] [PubMed] [Google Scholar]
- 19.Uchida T, Gotoh K, Shikata T. Complete nucleotide sequences and the characteristics of two hepatitis B virus mutants causing serologically negative acute or chronic hepatitis B. J Med Virol. 1995;45:247–252. doi: 10.1002/jmv.1890450303. [DOI] [PubMed] [Google Scholar]
- 20.Pult I, Chouard T, Wieland S, Klemenz R, Yaniv M, Blum HE. A hepatitis B virus mutant with a new hepatocyte nuclear factor 1 binding site emerging in transplant-transmitted fulminant hepatitis B. Hepatology. 1997;25:1507–1515. doi: 10.1002/hep.510250633. [DOI] [PubMed] [Google Scholar]
- 21.Okamoto H, Tsuda F, Sakugawa H, Sastrosoewignjo RI, Imai M, Miyakawa Y, Mayumi M. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J Gen Virol. 1988;69(Pt 10):2575–2583. doi: 10.1099/0022-1317-69-10-2575. [DOI] [PubMed] [Google Scholar]
- 22.Michitaka K, Tanaka Y, Horiike N, Duong TN, Chen Y, Matsuura K, Hiasa Y, Mizokami M, Onji M. Tracing the history of hepatitis B virus genotype D in western Japan. J Med Virol. 2006;78:44–52. doi: 10.1002/jmv.20502. [DOI] [PubMed] [Google Scholar]
- 23.Tong SP, Li JS, Vitvitski L, Trepo C. Active hepatitis B virus replication in the presence of anti-HBe is associated with viral variants containing an inactive pre-C region. Virology. 1990;176:596–603. doi: 10.1016/0042-6822(90)90030-u. [DOI] [PubMed] [Google Scholar]
- 24.Garfein RS, Bower WA, Loney CM, Hutin YJ, Xia GL, Jawanda J, Groom AV, Nainan OV, Murphy JS, Bell BP. Factors associated with fulminant liver failure during an outbreak among injection drug users with acute hepatitis B. Hepatology. 2004;40:865–873. doi: 10.1002/hep.20383. [DOI] [PubMed] [Google Scholar]
- 25.Owiredu WK, Kramvis A, Kew MC. Molecular analysis of hepatitis B virus genomes isolated from black African patients with fulminant hepatitis B. J Med Virol. 2001;65:485–492. [PubMed] [Google Scholar]
- 26.Bowyer SM, van Staden L, Kew MC, Sim JG. A unique segment of the hepatitis B virus group A genotype identified in isolates from South Africa. J Gen Virol. 1997;78(Pt 7):1719–1729. doi: 10.1099/0022-1317-78-7-1719. [DOI] [PubMed] [Google Scholar]
- 27.Chirara MM, Chetsanga CJ. Variant of hepatitis B virus isolated in Zimbabwe. J Med Virol. 1994;42:73–78. doi: 10.1002/jmv.1890420114. [DOI] [PubMed] [Google Scholar]
- 28.Aiba N, Nishimura H, Arakawa Y, Abe K. Complete nucleotide sequence and phylogenetic analyses of hepatitis B virus isolated from two pileated gibbons. Virus Genes. 2003;27:219–226. doi: 10.1023/a:1026387614162. [DOI] [PubMed] [Google Scholar]
- 29.Vaudin M, Wolstenholme AJ, Tsiquaye KN, Zuckerman AJ, Harrison TJ. The complete nucleotide sequence of the genome of a hepatitis B virus isolated from a naturally infected chimpanzee. J Gen Virol. 1988;69(Pt 6):1383–1389. doi: 10.1099/0022-1317-69-6-1383. [DOI] [PubMed] [Google Scholar]
- 30.Grethe S, Heckel JO, Rietschel W, Hufert FT. Molecular epidemiology of hepatitis B virus variants in nonhuman primates. J Virol. 2000;74:5377–5381. doi: 10.1128/jvi.74.11.5377-5381.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verschoor EJ, Warren KS, Langenhuijzen S, Heriyanto , Swan RA, Heeney JL. Analysis of two genomic variants of orang-utan hepadnavirus and their relationship to other primate hepatitis B-like viruses. J Gen Virol. 2001;82:893–897. doi: 10.1099/0022-1317-82-4-893. [DOI] [PubMed] [Google Scholar]
- 32.Sugauchi F, Mizokami M, Orito E, Ohno T, Kato H, Suzuki S, Kimura Y, Ueda R, Butterworth LA, Cooksley WG. A novel variant genotype C of hepatitis B virus identified in isolates from Australian Aborigines: complete genome sequence and phylogenetic relatedness. J Gen Virol. 2001;82:883–892. doi: 10.1099/0022-1317-82-4-883. [DOI] [PubMed] [Google Scholar]
- 33.Melegari M, Bruno S, Wands JR. Properties of hepatitis B virus pre-S1 deletion mutants. Virology. 1994;199:292–300. doi: 10.1006/viro.1994.1127. [DOI] [PubMed] [Google Scholar]
- 34.Dong J, Cheng J, Wang QH, Wang G, Shi SS, Liu Y, Xia XB, Si CW. Cloning and sequence analysis of truncated S gene from circulation of patients with chronic hepatitis B virus infection. Zhonghua Ganzangbing Zazhi. 2001;9:163–165. [PubMed] [Google Scholar]
- 35.Banerjee A, Datta S, Chandra PK, Roychowdhury S, Panda CK, Chakravarty R. Distribution of hepatitis B virus genotypes: phylogenetic analysis and virological characteristics of genotype C circulating among HBV carriers in Kolkata, Eastern India. World J Gastroenterol. 2006;12:5964–5971. doi: 10.3748/wjg.v12.i37.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chauhan R, Kazim SN, Bhattacharjee J, Sakhuja P, Sarin SK. Basal core promoter, precore region mutations of HBV and their association with e antigen, genotype, and severity of liver disease in patients with chronic hepatitis B in India. J Med Virol. 2006;78:1047–1054. doi: 10.1002/jmv.20661. [DOI] [PubMed] [Google Scholar]
- 37.Dai MS, Lu JJ, Chen YC, Perng CL, Chao TY. Reactivation of precore mutant hepatitis B virus in chemotherapy-treated patients. Cancer. 2001;92:2927–2932. doi: 10.1002/1097-0142(20011201)92:11<2927::aid-cncr10109>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 38.Huang YH, Wu JC, Chang TT, Sheen IJ, Huo TI, Lee PC, Su CW, Lee SD. Association of core promoter/precore mutations and viral load in e antigen-negative chronic hepatitis B patients. J Viral Hepat. 2006;13:336–342. doi: 10.1111/j.1365-2893.2005.00688.x. [DOI] [PubMed] [Google Scholar]
- 39.Lin CL, Liao LY, Liu CJ, Chen PJ, Lai MY, Kao JH, Chen DS. Hepatitis B genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J Gastroenterol. 2002;37:283–287. doi: 10.1007/s005350200036. [DOI] [PubMed] [Google Scholar]
- 40.Preikschat P, Gunther S, Reinhold S, Will H, Budde K, Neumayer HH, Kruger DH, Meisel H. Complex HBV populations with mutations in core promoter, C gene, and pre-S region are associated with development of cirrhosis in long-term renal transplant recipients. Hepatology. 2002;35:466–477. doi: 10.1053/jhep.2002.30698. [DOI] [PubMed] [Google Scholar]
- 41.Yoo BC, Park JW, Kim HJ, Lee DH, Cha YJ, Park SM. Precore and core promoter mutations of hepatitis B virus and hepatitis B e antigen-negative chronic hepatitis B in Korea. J Hepatol. 2003;38:98–103. doi: 10.1016/s0168-8278(02)00349-5. [DOI] [PubMed] [Google Scholar]
- 42.Orito E, Mizokami M, Sakugawa H, Michitaka K, Ishikawa K, Ichida T, Okanoue T, Yotsuyanagi H, Iino S. A case-control study for clinical and molecular biological differences between hepatitis B viruses of genotypes B and C. Japan HBV Genotype Research Group. Hepatology. 2001;33:218–223. doi: 10.1053/jhep.2001.20532. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Tanaka Y, Huang Y, Kurbanov F, Chen J, Zeng G, Zhou B, Mizokami M, Hou J. Clinical and virological characteristics of hepatitis B virus subgenotypes Ba, C1, and C2 in China. J Clin Microbiol. 2007;45:1491–1496. doi: 10.1128/JCM.02157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Candotti D, Danso K, Allain JP. Maternofetal transmission of hepatitis B virus genotype E in Ghana, west Africa. J Gen Virol. 2007;88:2686–2695. doi: 10.1099/vir.0.83102-0. [DOI] [PubMed] [Google Scholar]
- 45.Okamoto H, Tsuda F, Mayumi M. Defective mutants of hepatitis B virus in the circulation of symptom-free carriers. Jpn J Exp Med. 1987;57:217–221. [PubMed] [Google Scholar]
- 46.Melegari M, Scaglioni PP, Wands JR. The small envelope protein is required for secretion of a naturally occurring hepatitis B virus mutant with pre-S1 deleted. J Virol. 1997;71:5449–5454. doi: 10.1128/jvi.71.7.5449-5454.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hosono S, Tai PC, Wang W, Ambrose M, Hwang DG, Yuan TT, Peng BH, Yang CS, Lee CS, Shih C. Core antigen mutations of human hepatitis B virus in hepatomas accumulate in MHC class II-restricted T cell epitopes. Virology. 1995;212:151–162. doi: 10.1006/viro.1995.1463. [DOI] [PubMed] [Google Scholar]
- 48.Gunther S, Li BC, Miska S, Kruger DH, Meisel H, Will H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J Virol. 1995;69:5437–5444. doi: 10.1128/jvi.69.9.5437-5444.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan TT, Lin MH, Chen DS, Shih C. A defective interference-like phenomenon of human hepatitis B virus in chronic carriers. J Virol. 1998;72:578–584. doi: 10.1128/jvi.72.1.578-584.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan TT, Lin MH, Qiu SM, Shih C. Functional characterization of naturally occurring variants of human hepatitis B virus containing the core internal deletion mutation. J Virol. 1998;72:2168–2176. doi: 10.1128/jvi.72.3.2168-2176.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milich DR, McLachlan A, Moriarty A, Thornton GB. Immune response to hepatitis B virus core antigen (HBcAg): localization of T cell recognition sites within HBcAg/HBeAg. J Immunol. 1987;139:1223–1231. [PubMed] [Google Scholar]
- 52.Lee YS, Yoon SJ, Kwon TK, Kim YH, Woo JH, Suh MH, Suh SI, Baek WK, Kim HJ, Ahn SY, et al. Immune response induced by immunization with Hepatitis B virus core DNA isolated from chronic active hepatitis patients. Immunol Lett. 2001;78:13–20. doi: 10.1016/s0165-2478(01)00230-9. [DOI] [PubMed] [Google Scholar]
- 53.Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lanford RE, Kim YH, Lee H, Notvall L, Beames B. Mapping of the hepatitis B virus reverse transcriptase TP and RT domains by transcomplementation for nucleotide priming and by protein-protein interaction. J Virol. 1999;73:1885–1893. doi: 10.1128/jvi.73.3.1885-1893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stemler M, Weimer T, Tu ZX, Wan DF, Levrero M, Jung C, Pape GR, Will H. Mapping of B-cell epitopes of the human hepatitis B virus X protein. J Virol. 1990;64:2802–2809. doi: 10.1128/jvi.64.6.2802-2809.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heathcote J, McHutchison J, Lee S, Tong M, Benner K, Minuk G, Wright T, Fikes J, Livingston B, Sette A, et al. A pilot study of the CY-1899 T-cell vaccine in subjects chronically infected with hepatitis B virus. The CY1899 T Cell Vaccine Study Group. Hepatology. 1999;30:531–536. doi: 10.1002/hep.510300208. [DOI] [PubMed] [Google Scholar]