

Synopsis
Staphylococcus aureus is the most abundant cause of bacterial infections in the United States. As such, the pathogen has devised means to circumvent destruction by the innate immune system. Neutrophils are a critical component of innate immunity and the primary cellular defense against S. aureus infections. Herein we review human neutrophil function in the context of S. aureus virulence mechanisms, and provide an overview of community-associated methicillin resistant S. aureus (CA-MRSA) pathogenicity.
Keywords: staphylococcus, neutrophil, innate immunity, virulence
Staphylococcus aureus has been a leading cause of human infections throughout history. During 1997–1999, S. aureus was reported as the most abundant cause of bloodstream, skin and soft tissue, and lower respiratory tract infections in the United States, Canada, Europe, Latin America, and the Western Pacific [1]. The pathogen is currently the leading cause of hospital-associated infections in the United States [2], and was associated with a remarkable economic burden of $14.5 billion in 2003 [3]. A high percentage of hospital infections are caused by MRSA [4]. Notably, there is a relatively high mortality rate (∼20%) associated with invasive MRSA infections, the majority of which are healthcare-associated [5]. This finding may be related in part to the prior health status of the patient, since these infections typically occur in individuals with predisposing risk factors, such as those who have had surgery, or in patients who are immunocompromised or have granulocyte defects.
By comparison, community-associated (CA) S. aureus cause infections in otherwise healthy individuals. Historically, community S. aureus infections were almost always caused by methicillin-susceptible S. aureus (MSSA) rather than MRSA [4], but this distribution has changed dramatically in the United States over the past 10 years [6]. Two reports in the late 1990s marked the beginning of a new era in MRSA epidemiology [7,8]. Isolates classified as pulsed-field gel electrophoresis type USA400 emerged as the prototype CA-MRSA genotype [9,10]. A complete genome sequence is available for MW2, a representative USA400 clinical isolate that caused fatal septicemia in 1998 [10]. Although USA400 remained a significant cause of CA-MRSA infections through 2005 [11,12], it has been replaced almost completely by a genotype known as USA300 [6,13], which is now epidemic in the United States. The current CA-MRSA epidemic is due to clonal emergence of USA300 isolates that have enhanced virulence or a “hypervirulence” phenotype [14,15]. Hypervirulence, defined here as the ability of CA-MRSA to cause widespread infections in otherwise healthy individuals, is likely related in part to the ability of USA300 and USA400 to circumvent killing by human polymorphonuclear leukocytes (PMNs) and cause rapid destruction of these host cells [16,17].
In general, the ability of bacteria to cause disease in humans is due to evasion of innate host defense, which includes resistance to antimicrobial peptides (AMPs) and killing by phagocytic leukocytes. Inasmuch as PMNs (also called neutrophils or granulocytes) constitute the greatest number of leukocytes in humans, they are the primary cellular defense against S. aureus infections. Here we review critical components of neutrophil function as they relate to S. aureus infection as well as staphylococcal virulence factors that contribute to immune evasion, including those produced by prominent CA-MRSA strains.
PMNs in the innate immune response
Neutrophil recruitment, chemotaxis, and priming
A first step in the eradication of invading microorganisms is active recruitment of PMNs to the site of infection by chemotaxis (reviewed by Cicchetti et al. [18]). This is a multistep process whereby neutrophils are mobilized from peripheral blood and/or bone marrow in response to host- and pathogen-derived chemotactic factors. Host molecules, such as interleukin-8 (IL-8, CXCL8), GROα (CXCL1), granulocyte chemotactic protein 2 (GCP2, CXCL6), and complement component C5a, recruit neutrophils to the site of infection.
S. aureus has been shown to elicit production of numerous chemotactic factors in vitro and in vivo. For example, S. aureus lipoteichoic acid (LTA) and capsular polysaccharide induce production of IL-8 by peripheral blood monocytes [19] and epithelial and endothelial cells [20], respectively. S. aureus-activated endothelial cells produce IL-8 that promotes transmigration of neutrophils [21]. Freely secreted virulence molecules of S. aureus, including toxic shock syndrome toxin-1 (TSST1), enterotoxin A (SEA), or enterotoxin B (SEB), also elicit production of IL-8 by human monocytic cells [22]. Recent studies have demonstrated that stimulation of CD4+ T-cells by S. aureus capsular polysaccharide leads to production of chemokines that recruit neutrophils to the site of infection [23,24]. Further, S. aureus cell surface components, primarily peptidoglycan (PGN), have long been known to elicit production of C5a [25], a potent chemotactic molecule for PMNs. S. aureus also produces molecules that directly recruit PMNs (e.g., phenol soluble modulin-like peptides, PSMs; see below) [26]. Although the pathogen elicits a robust proinflammatory response, it generates a number of molecules that block chemotaxis and these are discussed below (also see Table 1).
Table 1.
S. aureus molecules that contribute to immune evasion or alter host immune function
| Gene(s) | Protein or molecule | Function/effect on immune system |
|---|---|---|
| ahpC, ahpF | Alkyl hydroperoxide reductase subunits C and F, AhpC and AhpF | Promotes resistance to ROS |
| aur | Zinc metalloproteinase aureolysin, Aur | Degrades LL-37 |
| cap5 or cap8 genes | Capsular polysaccharide | Inhibits phagocytosis |
| katA | Catalase, KatA | Detoxifies hydrogen peroxide |
| chp | Chemotaxis inhibitory protein of S. aureus, CHIPS | Inhibits chemotaxis |
| clfA | Clumping factor A, ClfA | Inhibits phagocytosis, causes platelet activation |
| crtM, crtN | Carotenoid pigment, staphyloxanthin | Promotes resistance to ROS |
| dlt operon | Dlt operon, DltABCD | Promotes resistance to cationic AMPs and group IIA phospholipase A2 |
| eap | Extracellular adherence protein, Eap | Inhibits leukocyte adhesion |
| ecb | Extracellular complement-binding protein, Ecb | Inhibits C5a generation |
| efb | Extracellular fibrinogen-binding protein, Efb | Inhibits C5a generation |
| fnbA, fnbB | Fibronectin-binding proteins A and B, FnbA and FnbB | Cause platelet activation |
| hla, hly | Alpha-hemolysin (α-hemolysin), Hla | Causes host cell lysis |
| hld | Delta-hemolysin, Hld | Causes host cell lysis |
| hlgA, hlgB, hlgC | Gamma-hemolysin subunits A, B, and C; HlgA, HlgB, HlgC; two-component leukocidin | Causes leukocyte and erythrocyte lysis |
| icaA, icaD, icaB, icaC, icaR | Polysaccharide intercellular adhesin, PIA | Resistance to cationic AMPs |
| isdA, isdB | Iron-regulated surface determinants of S. aureus, IsdA and IsdB | Resistance to AMPs, skin fatty acids, and neutrophil ROS |
| lukS-PV, lukF-PV | Leukocidin S-PV and F-PV subunits; LukS/F-PV; PVL; two-component leukocidin | Causes phagocyte lysis |
| lukD, lukE | Leukocidin D and E; LukD and LukE; two-component leukocidin | Causes leukocyte lysis |
| mprF | Multiple peptide resistance factor, MprF | Promotes resistance to cationic AMPs |
| psm | Phenol-soluble modulin-like peptides, PSMs | Cause leukocyte lysis |
| sak | Staphylokinase | Inhibits host α-defensins |
| sbi | IgG-binding protein, Sbi | Sequesters host IgG |
| scn | Staphylococcal inhibitor of complement, SCIN | Inhibits complement |
| sea, seb, secn, sed, see, seg, seh, sei, sej, sek, sel, sep | Staphylococcal enterotoxins; SEA, SEB, SECn, SED, SEE, SEG, SEH, SEI, SEJ, SEK, SEL, and SEP | Activate T-cells |
| sodA, sodM | Superoxide dismutase, SodA, SodM | Promotes resistance to ROS |
| spa | Protein A | Sequesters host IgG, inhibits phagocytosis |
| ssl5 | Staphylococcal superantigen-like 5, SSL5 | Binds PSGL-1 and inhibits neutrophil rolling |
| ssl7 | Staphylococcal superantigen-like 7, SSL7 | Binds to C5a and IgA |
| tst | Toxic shock syndrome toxin-1, TSST1 | Activates T-cells |
Function of each molecule was determined based upon published studies (available on PubMed, http://www.ncbi.nlm.nih.gov/sites/entrez/). See also the review by T.J. Foster [57].
Many chemoattractants are priming agents (rather than activating agents) for neutrophils. Neutrophil “priming” was first described as the ability of a primary agonist, typically at sub-stimulatory concentration, to enhance superoxide production elicited by a second stimulus [27]. Neutrophils can be primed for enhanced adhesion, phagocytosis, production of reactive oxygen species (ROS), cytokine secretion, leukotriene synthesis, degranulation, and bactericidal activity (see ref. [28] for a comprehensive list of priming agents). Many neutrophil priming agents are host-derived molecules such as cytokines, chemokines, and growth factors [28]. Cell-cell contact and adhesion also prime PMNs for enhanced function. Some bacteria-derived priming agents, such as S. aureus LTA, are Toll-like receptor (TLR) agonists and TLRs are components important to the pathogen-mediated priming process (see below). There is a distinction between primed neutrophils and those that are fully activated. Priming includes mobilization of secretory vesicles (and thus up-regulation of specific cell surface receptors, e.g., CD11b/CD18) and secretion of cytokines, but fails to trigger release of azurophilic granules or elicit production of superoxide [29]. Importantly, chemotactic/priming agents ultimately promote efficient clearance of invading microorganisms.
Pathogen recognition and phagocytosis
Phagocytosis is a process whereby neutrophils bind and ingest invading microorganisms (Fig. 1). It is a critical step in the removal of bacteria during infection. Also, opsonophagocytosis is the means by which vaccines prevent bacterial disease. PMNs recognize many surface-bound or freely secreted molecules produced by bacteria, including PGN, lipoproteins, LTA, lipopolysaccharide (LPS), CpG-containing DNA, and flagellin. These conserved molecules, known as pathogen-associated molecular patterns (PAMPs), interact with pattern recognition receptors expressed on the neutrophil cell surface, including TLRs (reviewed in [30]). Neutrophil TLRs activate signal transduction pathways that prolong cell survival [31], promote and/or enhance adhesion, phagocytosis, release of cytokines, chemokines and ROS [31,32], and trigger granule exocytosis [33], thereby contributing to microbicidal activity. As an example of the importance of the TLRs in host defense, mice deficient in TLR2 are more susceptible to S. aureus infection compared with wild-type mice [34].
Fig. 1.
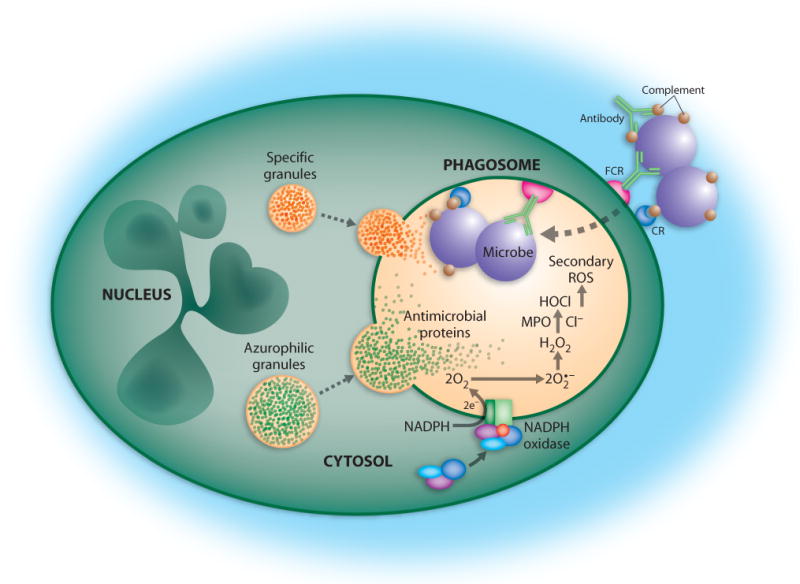
PMN phagocytosis and microbicidal activity. Bacteria are destroyed by NADPH oxidase-derived ROS and antimicrobial proteins released from granules after phagocytosis by neutrophils. FCR, Fc receptor; CR, complement receptor; MPO, myeloperoxidase. Reproduced with permission, from M.T. Quinn, M.C.B. Ammons and F.R. DeLeo, 2006, Clinical Science, 111, 1-20 (ref. [39]). © the Biochemical Society.
Peptidoglycan recognition protein (PGRP) is a secreted host protein that contributes to intracellular killing of Gram-positive bacteria by neutrophils [35]. There are four reported isoforms of PGRP in mammals, and neutrophils express PGRP-short (PGRP-S) [36]. In contrast to TLRs, which promote recognition of bacteria, PGRP-S contributes directly to bactericidal activity [35].
NOD-like receptors (NLRs) are cytoplasmic proteins that detect intracellular microbial components (reviewed by Nunez and colleagues [37]). NOD2 senses muramyl dipeptide derived from S. aureus PGN and ultimately promotes transcription of NF-κB target genes in the nucleus [37]. Phagocytosis is also facilitated by host pattern recognition molecules known as collectins, such as mannose-binding lectin, and these molecules are reviewed elsewhere [38].
Although pattern recognition receptors are important for detection of microbes by phagocytes, the efficiency of phagocytosis (i.e., uptake or ingestion) is enhanced if bacteria are opsonized with serum host proteins, such as complement and/or antibody. Complement-opsonized microbes are bound by complement surface receptors on PMNs, including ClqR, CD35 (CR1), CD11b/CD18 (CR3) and CD11c/CD18 (CR4). Antibody-coated microbes are recognized by PMN antibody-Fc receptors, namely CD16 (FcγRIIIb, IgG receptor), CD23 (FcεRI, IgE receptor), CD32 (FcγRIIa, IgG receptor), CD64 (FcγRI, IgG receptor), and CD89 (FcαR, IgA receptor). The concerted action of pattern recognition receptors/molecules and antibody and complement receptors promotes efficient phagocytosis of microbes (Fig. 1).
Neutrophil microbicidal activity
NADPH oxidase and the production of ROS
Human neutrophils employ oxygen-dependent and oxygen-independent mechanisms to kill ingested microorganisms. Phagocytosis of microorganisms activates a membrane-bound NADPH-dependent oxidase that generates high levels of superoxide, a process traditionally called “respiratory burst” (reviewed by Quinn et al. [39]). Superoxide anion is short-lived and dismutates rapidly to hydrogen peroxide and forms other secondary reactive products. These secondarily-derived products, including hypochlorous acid, hydroxyl radical, chloramines, and singlet oxygen, are effective microbicidal compounds [40-43]. The importance of NADPH oxidase and ROS for host defense is exemplified by a hereditary disorder known as chronic granulomatous disease (CGD) [44], in which there is a defect in NADPH oxidase. Individuals with CGD have recurrent bacterial and fungal infections, especially infections caused by S. aureus [44].
Degranulation and neutrophil antimicrobial peptides (AMPs) and proteins
Concomitant with the production of ROS, neutrophil cytoplasmic granules fuse with bacteria-containing phagosomes, a process called degranulation [45]. Fusion of azurophilic granules (also called primary granules) with phagosomes enriches the vacuole lumen with numerous antimicrobial peptides (AMPs) and antimicrobial proteins, including α-defensins, cathepsins, proteinase-3, elastase, and azurocidin [45,46] (Fig. 1). Neutrophil α-defensins comprise up to 50% of the protein in azurophilic granules and have potent antimicrobial activity [47]. Defensins are cationic polypeptides of 3-5 kDa that interact with negatively-charged molecules at the pathogen surface and permeabilize bacterial membranes. Degranulation also enriches phagosomes with components of the specific granules, such as lactoferrin, further augmenting antimicrobial potential [48].
Although killing of bacteria by neutrophils occurs primarily after phagocytosis, the process can be enhanced by extracellular molecules. For instance, group IIA phospholipase A2 (gIIA-PLA2), a small (∼14 kDa) cationic antimicrobial protein found in extracellular fluids, synergizes with the neutrophil NADPH oxidase to promote digestion of S. aureus phospholipids [49]. More recently, studies by Corbin et al. found that neutrophil calprotectin (S100A8/A9) inhibits S. aureus growth by sequestering nutrient Mn2+ and Zn2+ within abscesses [50].
Neutrophil extracellular traps (NETs)
Work by Zychlinsky and colleagues identified structures called neutrophil extracellular traps (NETs), composed of chromatin, histones, and azurophilic granule proteins, which have the capacity to kill bacteria, including S. aureus [51]. Whether these structures are produced by live cells or simply represent a byproduct of host cell lysis remains unclear. Uncontrolled release of granule proteins and other cytotoxic molecules into host tissues would likely be problematic for the resolution of the inflammatory response (see below and Fig. 2). It is possible that formation of NETs is a relatively infrequent process, thus limiting host tissue damage. In any case, NETs have significant microbicidal activity toward a number of organisms and further research is needed to better understand these interesting structures.
Fig. 2.
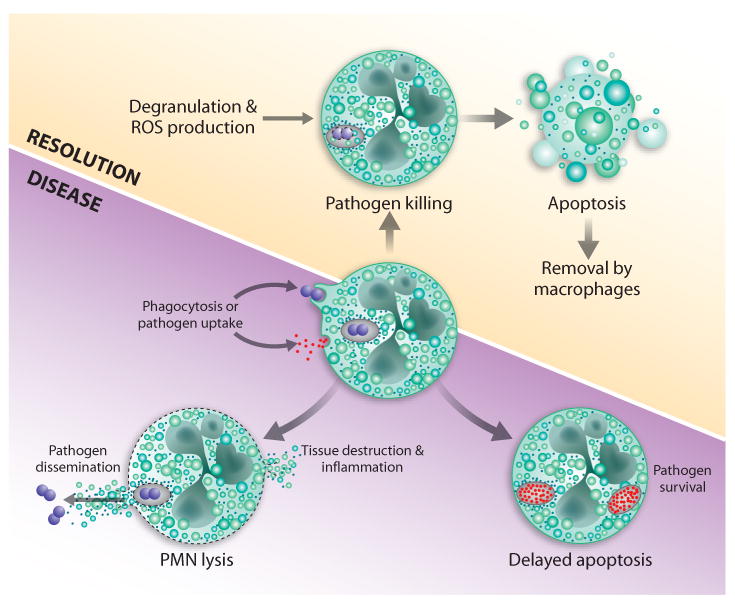
Two possible outcomes of bacteria-neutrophil interaction. Phagocytosis of bacteria triggers production of ROS and degranulation. These processes work collectively to kill ingested bacteria, after which neutrophils undergo apoptosis and are removed by macrophages. This process promotes healthy resolution of infection (top panel). Alternatively, bacterial pathogens cause neutrophil lysis or delay apoptosis, and thereby survive and cause disease (bottom panel).Reproduced with permission, from M.T. Quinn, M.C.B. Ammons and F.R. DeLeo, 2006, Clinical Science, 111, 1-20 (ref. [39]). © the Biochemical Society.
Neutrophil apoptosis and the resolution of inflammation
Bacterial infections are typically accompanied by tremendous influx of neutrophils to the affected tissues. Although neutrophils kill most ingested microorganisms efficiently, host tissues can be damaged by the inadvertent release of cytotoxic components from PMNs. Thus, neutrophil turnover must be highly regulated during infection. To that end, normal turnover of aging neutrophils occurs by spontaneous apoptosis and in the absence of an activating agent [52]. On the other hand, neutrophil apoptosis is accelerated significantly following phagocytosis [53,54] and this phenomenon appears critical to the resolution of the inflammatory response (Fig. 2). Therefore, one might predict that bacterial pathogens have evolved mechanisms to exploit normal neutrophil turnover and apoptosis.
Indeed, bacterial pathogens have devised mechanisms to alter apoptosis and promote pathogenesis [55,56]. For example, USA300 and USA400 cause rapid PMN lysis and/or accelerate bacteria-induced apoptosis to the point of secondary lysis [16,17]. Although these strains produce cytolytic leukotoxins known to cause destruction of human neutrophils, the contribution (if any) of these toxins to PMN lysis after phagocytosis remains to be determined [16]. A model schematic based upon much of the published data suggests that there are two possible outcomes for neutrophil-bacteria interactions (Fig. 2). On one hand, phagocytosis and killing of bacteria culminate with induction of neutrophil apoptosis (also called phagocytosis-induced cell death) and subsequent removal by macrophages, ultimately resulting in the resolution of infection. Alternatively, pathogens such as CA-MRSA alter the normal progression to apoptosis–in this case by causing PMN lysis–to survive, and thereby disseminate and cause disease.
Staphylococcus aureus immune evasion
Inhibition of phagocyte function
S. aureus has an astounding repertoire of immune evasion factors that frequently show functional redundancy in subverting the same host defense mechanism. The particularly crucial role of innate host defense in eliminating invading S. aureus is reflected by the abundance of mechanisms that the bacterium uses to evade killing by phagocytes [57] (Table 1).
Phagocyte function may be subverted at many different stages. S. aureus may simply hide from recognition by producing protective coats, such as capsular polysaccharide or biofilm. Further, they produce or secrete specific molecules to block phagocyte receptor function. After ingestion, the bacteria use mechanisms to decrease the efficiency of antimicrobial mechanisms, which likely account for noted post-phagocytosis survival [17,58]. Finally, they often produce toxins that lyse phagocytes, thus using the same kind of weapon that neutrophils use to kill bacteria.
S. aureus strains have the capacity to produce several exopolymers, which together make up the “camouflage coat” that protects from recognition by the immune system. Many strains are encapsulated by a polysaccharide capsule that has strain-specific chemical composition and protects from phagocytosis [59]. Polysaccharide intercellular adhesion (PIA) is a biofilm-related extracellular matrix substance [60], predominantly characterized in S. epidermidis but produced by most S. aureus strains, whose most important and unique feature is a positive net charge [61]. It has been shown to protect from neutrophil phagocytosis and AMPs [62].
As described above, receptors on the surface of neutrophils and other phagocytes play a key role in recognizing bacteria and secreted bacterial molecules, promoting phagocyte chemotaxis and activation. S. aureus produces a molecule called CHIPS (chemotaxis inhibitory protein of S. aureus) that blocks receptor-mediated recognition of formylated peptides [63], a PAMP secreted by bacteria and central for phagocyte detection of bacterial invaders. Other secreted S. aureus molecules block the complement system, thereby reducing phagocytosis after opsonization. The C3 convertase blocker SCIN (staphylococcal complement inhibitor) is but one of a series of S. aureus factors with the task of inhibiting complement function [64]. In fact, complement inhibition is an excellent example of the functional redundancy of S. aureus molecules interfering with the same immune defense mechanism [65] (Table 1).
After being ingested, S. aureus uses even more manifold weaponry of immune evasion molecules. Catalase and superoxide dismutase eliminate harmful ROS (Table 1). ROS also trigger a broad response of S. aureus to circumvent innate host defense mechanisms, which includes production of many toxins and mechanisms involved in the uptake of iron [66]. In addition, the characteristic yellow pigment of S. aureus, a carotenoid pigment called staphyloxanthin, has recently been shown to play an additional, crucial role in protecting from ROS [67]. Moreover, S. aureus secretes a series of proteins aimed at moderating the oxygen-independent killing mechanisms of the innate immune system, which include AMPs. First, relatively non-specific proteases are released to digest any protein-based antimicrobial effector [68]. In addition, S. aureus senses the presence of AMPs and reacts by an up-regulation of mechanisms to interfere specifically with the activity of cationic AMPs, using a dedicated 3-component AMP sensing system [69,70]. The regulated resistance mechanisms include D-alanylation of teichoic acids and incorporation of cationic phospholipid lysyl-phosphatidyl glycerol in the cytoplasmic membrane. These two mechanisms are aimed to reduce the negative net charge of the bacterial surface, thereby decreasing binding of cationic AMPs, and are encoded by the dlt operon and the mprF locus, respectively [71,72]. Furthermore, expression of the VraFG transporter is increased upon AMP exposure [70]. VraFG has a demonstrated role in AMP resistance [69] and the putative task of removing AMPs from the cell or cytoplasmic membrane.
The immune evasion mechanisms described so far are rather “passive”, enabling the bacteria to hide from recognition or blocking receptors or effectors involved in the elimination of the bacteria. However, S. aureus also produces toxins that directly attack human white and red blood cells. These toxins include the large family of leukocidins and α-toxin (also known as α-hemolysin), recently discovered phenol-soluble modulins (PSMs), and other hemolysins. The β-barrel structured leukocidins form pores almost exclusively in leukocytes, via a mechanism similar to that used by α-toxin [73]. Although the biochemical function of leukocidins–that is, lysis of leukocytes–has clearly been established, their role in S. aureus virulence and the biological reason for the presence of many similar toxins is not understood. This is also true for Panton-Valentine leukocidin (PVL), in which there has been recent renewed interest due to its epidemiological correlation with community-associated MRSA (see below). Finally, PSMs are short, α-helical and amphipathic peptides that have the capacity to lyse human neutrophils [26].
Moderating the acquired immune response
Protein A is probably the best known S. aureus protein due to its use in the laboratory for antibody purification, which is based on the interaction of protein A with the Fc part of IgG molecules [74]. During pathogenesis, this feature enables S. aureus to sequester non-specific antibodies on its surface, which protects efficiently from attacks by the innate and acquired immune systems [75]. Protein A also has a more specific role in the pathogenesis of airway infections by interacting with the tumor-necrosis-factor-alpha receptor on airway epithelia [76].
Potentiation of the immune response by superantigenic toxins
Potentiation or over-stimulation of the immune response represents a way of interfering with the human immune system clearly opposite to the mechanisms described so far, but equally as effective. S. aureus produces many superantigenic toxins, a class of secreted toxins that activate T-cells without the need for the presence of an antigen on an antigen-presenting cell [77] (Table 1). Activation of T-cells by superantigenic toxins is accomplished by cross-linking the T-cell receptor with the major histocompatibility complex (MHC) class II [77]. These toxins include the toxic shock syndrome toxin (TSST), exfoliative toxins involved in staphylococcal scalded skin syndrome, and the staphylococcal enterotoxins (SEs) [78]. Toxic shock syndrome (TSS) is a severe acute disease and may be of menstrual or non-menstrual origin. Menstrual TSS is caused by S. aureus colonization of tampons [79]. Non-menstrual TSS may also be caused by enterotoxins, which however have a more notorious role as the cause for S. aureus food poisoning [80].
Community-associated MRSA virulence
Hospital-associated (HA) versus community-associated (CA) MRSA
Epidemiologically unassociated outbreaks of CA-MRSA have been reported throughout the world, but the epidemic caused by the clonally related USA300 strains in the United States appears to be the most serious. Notably, CA-MRSA lineages differ markedly in genotypic and phenotypic characters from traditional hospital-associated MRSA lineages [10,13,81].
Antibiotic resistance per se does not contribute to virulence
The frequent reporting in the lay press of MRSA strains as “superbugs” reflects recognition of the unusually severe and potentially fatal infections caused by CA-MRSA strains. Experimental infections in mice indicate that CA-MRSA strains indeed cause more rapidly lethal infections when compared to traditional hospital-associated MRSA strains [17]. The attenuated virulence of hospital-associated MRSA strains is due in part to the fitness cost associated with resistance to β–lactams and other antibiotics encoded by type I-III staphylococcal chromosomal cassette mec (SCCmec). A unique attribute of CA-MRSA strains is carriage of the type IV SCCmec, which is smaller in size and encodes resistance to only β-lactam class antibiotics. Notably, precise deletion of the entire resistance cassette does not impact virulence in a rabbit infection model [82]. This indicates that type IV SCCmec does not contribute to virulence and, more importantly, does not impose a biological fitness cost to CA-MRSA strains.
Virulence factors
Panton-Valentine leukocidin (PVL)
Association with CA-MRSA and PVL as a toxin
The epidemiological association between genetically diverse S. aureus strains carrying the PVL genes (lukS-PV and lukF-PV, abbreviated as lukS/F-PV) and fatal necrotizing pneumonia renewed and intensified interest in understanding the biological role of this bi-component cytolytic toxin [83]. Epidemiological data alone, however, is insufficient to establish whether PVL directly contributes to widespread dissemination of CA-MRSA clones [84,85]. It is of interest that early studies led A. M. Woodin to conclude that PVL alone is not a very toxic substance [86], as intravenous injection of PVL in rabbits resulted in granulocytopenia followed by a marked granulocytosis, and was not lethal.
Lessons learned from isogenic CA-MRSA lukS/F-PV deletion mutants
Isogenic lukS/F-PV deletion mutants and wild type parental strains of USA300 and USA400 were tested in mouse abscess and bacteremia models [16] to reproduce the most common clinical manifestations associated with CA-MRSA disease [5,6]. No significant differences between PVL-positive and PVL-negative strains were detected using these mouse models [16]. As the same mouse models have been used successfully to demonstrate the roles of other CA-MRSA virulence factors [26], the null effect of PVL in these models strongly indicates that this toxin is not a major virulence factor of USA300 and USA400 strains.
Pneumonia is a rare disease caused by CA-MRSA [5]. Using purified toxin or a laboratory strain of S. aureus that overproduced PVL, the toxin was shown to impact mouse survival in a model of pneumonia [87]. In contrast, when comparing isogenic strain pairs with and without lukS/F-PV in the USA300 and USA400 genotypes, or when over-expressing PVL in S. aureus strain Newman, no significant contribution of PVL to lethal pneumonia was found [88,89]. Additionally, passive immunization with anti-PVL immune sera failed to protect mice against challenge with USA300 in the murine pneumonia model [90], indicating that PVL is not necessary for the pathogenesis of pulmonary disease. Although future research on the biological relevance of PVL may be warranted, attributing enhanced CA-MRSA virulence to PVL alone ignores the possible contributions of numerous other determinants.
Does PVL have an impact on virulence by a gene regulatory effect?
Recently, a pronounced global gene regulatory effect was attributed to PVL [87]. In that study, PVL appeared to up-regulate production of protein A, which was thought to be fundamental in causing the overwhelming inflammation and necrosis of the mouse lungs [87,91]. However, the apparent lack of confirmatory experiments by genetic complementation analysis might have resulted in misinterpretation of the gene expression data and thus led to the model of PVL as a global regulator of gene expression [87]. Recently, the failure to replicate the regulatory effects of PVL in USA300 and USA400 indicate that PVL does not contribute to CA-MRSA virulence via a gene regulatory mechanism (DeLeo et al., 2007, Network on Antimicrobial Resistance in S. aureus (NARSA), Reston, VA).
Arginine catabolic mobile element (ACME)
The arginine catabolic mobile element (ACME) is a genetic feature of USA300 [13] found infrequently in other S. aureus strains (ACME-arcA has been detected in USA100 and multi-locus sequence types (MLST) 1 and 97 in Europe [92,93]). Deletion of ACME in USA300 attenuated pathogenicity in a rabbit bacteremia model, providing evidence that ACME contributes to pathogenesis [82]. Two gene clusters identified in ACME, arc and opp-3, may function as virulence determinants. As L-arginine is a substrate for nitric oxide production, depletion of L-arginine by the arginine deiminase system (arc) might inhibit nitric oxide production, a molecule used in both the innate and adaptive immune responses against bacterial infections [94]. L-arginine catabolism could also be important for ATP production and pH homeostasis on the acidic human skin. opp-3 belongs to the ABC transporter family, members of which have a wide variety of central physiological functions. Thus, ACME may enhance growth, survival and dissemination of USA300 during infection.
α-toxin (α-hemolysin, Hla)
α-toxin is a well-characterized pore forming cytolytic toxin that is similar in sequence and function to the leukocidins [95], albeit it does not lyse neutrophils [96]. Recently, it was shown to be an essential virulence factor during CA-MRSA pneumonia [88]. Immunization with inactivated α-toxin or passive transfer of anti-α-toxin antibodies also protected mice from lethal pneumonia [90]. Taken together, these studies indicated that α-toxin plays an essential role in pneumonia.
Phenol soluble modulin-like peptides (PSMs)
It has remained obscure which molecules are responsible for neutrophil lysis in vivo and the pronounced cytolytic activity associated with CA-MRSA. Recently, novel cytolytic peptides have been found in S. aureus, the α-type phenol-soluble modulins (PSMs), which are encoded in an operon on the genomes of all sequenced S. aureus strains [26]. The α-helical and amphipathic α-type PSMs have pronounced in vitro and in vivo leukocidal activity, in addition to pro-inflammatory and chemotactic activities [26]. However, whereas hospital-associated MRSA often lack PSM production or produce PSMs only at reduced levels, PSMs are expressed at considerable levels in CA-MRSA. Notably, over-expression of α-type PSMs in a prominent hospital-associated MRSA strain increased leukocidal activity to a level equal to that observed in CA-MRSA strains, indicating that expression of these peptides is the main cause for the extreme difference in cytolytic activity between CA-MRSA and hospital-associated MRSA and a possible major contributor to the pronounced pathogenic potential of CA-MRSA strains. In fact, a dramatic influence of the α-type PSMs on the virulence of CA-MRSA was demonstrated using mouse bacteremia and abscess models of infection [26].
Role of gene expression and regulation in CA-MRSA pathogenesis
The results obtained with the PSMs indicate that differential gene expression between CA-MRSA and HA-MRSA strains could explain differences in virulence. For example, as α-toxin and the PSMs, the only molecules described so far to impact virulence of CA-MRSA in a significant manner [26,88], are under control of the global regulator agr and there is strong expression of the agr regulatory molecule RNAIII in CA-MRSA strains, there may be a key role of this virulence regulator in CA-MRSA disease that remains to be investigated.
Should infection by toxin-producing CA-MRSA strains impact how patients are treated?
CA-MRSA strains elaborate numerous exotoxins, including PVL, α-toxin, and PSMs, that could impact disease severity. Intravenous immunoglobulin (IVIg) could improve outcome of severe staphylococcal infections when used as an adjunct to appropriate antibiotic therapy due to the presence of neutralizing anti-toxin antibodies. It remains to be determined whether PVL-specific antibodies in commercial IVIg provide any protective effect. In a mouse pneumonia model, passive transfer of anti-α-toxin antibodies—but not anti-PVL antibodies—protected against lethal pneumonia [90], providing proof-of-principle that commercial IVIg could be effective adjunct therapy. Furthermore, inappropriate treatment of CA-MRSA infections with β-lactam antibiotics or ciprofloxacin that induce bacterial SOS response in these toxin-producing strains could result in increased exotoxin production and disease severity. As such, it is reasonable to propose that clindamycin or linezolid could be beneficial in inhibiting protein synthesis and thus toxin production in CA-MRSA infections [97]. Nonetheless, the emergence of multi-drug resistant isolates of USA300 with resistance to clindamycin [13,98], and the potential to acquire even more antibiotic resistance, present a further complication in staphylococcal disease management.
Concluding comment
S. aureus has been a major cause of human infections throughout history and today remains among the most abundant causes of bacterial infections. The pathogen has evolved numerous means to avoid destruction by the human innate immune system, including those that block almost all of the key antimicrobial functions of phagocytic leukocytes. CA-MRSA is especially adept at circumventing normal neutrophil function, which could explain in part the enhanced virulence phenotype of the most prominent CA-MRSA lineages. Future studies directed to better understand the interface between innate immunity, host infection susceptibility, and S. aureus are critical for a comprehensive understanding of pathogenesis.
Acknowledgments
This work was supported by the Intramural Program of the NIAID, NIH
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, et al. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin Infect Dis. 2001;32 2:S114–S132. doi: 10.1086/320184. [DOI] [PubMed] [Google Scholar]
- 2.Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob. 2006;52 doi: 10.1186/1476-0711-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noskin GA, Rubin RJ, Schentag JJ, Kluytmans J, Hedblom EC, Jacobson C, et al. National trends in Staphylococcus aureus infection rates: impact on economic burden and mortality over a 6-year period (1998-2003) Clin Infect Dis. 2007;45(9):1132–40. doi: 10.1086/522186. [DOI] [PubMed] [Google Scholar]
- 4.Chambers HF. The changing epidemiology of Staphylococcus aureus? Emerg Infect Dis. 2001;7(2):178–82. doi: 10.3201/eid0702.010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763–71. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 6.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, et al. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med. 2006;355(7):666–74. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 7.Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle-Vavra S, et al. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279(8):593–98. doi: 10.1001/jama.279.8.593. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. Four pediatric deaths from community-acquired methicillin-resistant Staphylococcus aureus--Minnesota and North Dakota, 1997-1999. JAMA. 1999;282(12):1123–25. [PubMed] [Google Scholar]
- 9.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol. 2003;41(11):5113–20. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359(9320):1819–27. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 11.Adem PV, Montgomery CP, Husain AN, Koogler TK, Arangelovich V, Humilier M, et al. Staphylococcus aureus sepsis and the Waterhouse-Friderichsen syndrome in children. N Engl J Med. 2005;353(12):1245–51. doi: 10.1056/NEJMoa044194. [DOI] [PubMed] [Google Scholar]
- 12.King MD, Humphrey BJ, Wang YF, Kourbatova EV, Ray SM, Blumberg HM. Emergence of community-acquired methicillin-resistant Staphylococcus aureus USA 300 clone as the predominant cause of skin and soft-tissue infections. Ann Intern Med. 2006;144(5):309–17. doi: 10.7326/0003-4819-144-5-200603070-00005. [DOI] [PubMed] [Google Scholar]
- 13.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–39. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy AD, Otto M, Braughton KR, Whitney AR, Chen L, Mathema B, et al. Epidemic community-associated methicillin-resistant Staphylococcus aureus: recent clonal expansion and diversification. Proc Natl Acad Sci U S A. 2008;105(4):1327–32. doi: 10.1073/pnas.0710217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diep BA, Sensabaugh GF, Somboona NS, Carleton HA, Perdreau-Remington F. Widespread skin and soft-tissue infections due to two methicillin-resistant Staphylococcus aureus strains harboring the genes for Panton-Valentine leucocidin. J Clin Microbiol. 2004;42(5):2080–84. doi: 10.1128/JCM.42.5.2080-2084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voyich JM, Otto M, Mathema B, Braughton KR, Whitney AR, Welty D, et al. Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis. 2006;194(12):1761–70. doi: 10.1086/509506. [DOI] [PubMed] [Google Scholar]
- 17.Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol. 2005;175(6):3907–19. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- 18.Cicchetti G, Allen PG, Glogauer M. Chemotactic signaling pathways in neutrophils: from receptor to actin assembly. Crit Rev Oral Biol Med. 2002;13(3):220–28. doi: 10.1177/154411130201300302. [DOI] [PubMed] [Google Scholar]
- 19.Standiford TJ, Arenberg DA, Danforth JM, Kunkel SL, VanOtteren GM, Strieter RM. Lipoteichoic acid induces secretion of interleukin-8 from human blood monocytes: a cellular and molecular analysis. Infect Immun. 1994;62(1):119–25. doi: 10.1128/iai.62.1.119-125.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soell M. Capsular polysaccharide types 5 and 8 of Staphylococcus aureus bind specifically to human epithelial (KB) cells, endothelial cells, and monocytes and induce release of cytokines. Infect Immun. 1995;63(4):1380–86. doi: 10.1128/iai.63.4.1380-1386.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao L, Lowy FD, Berman JW. Interleukin-8 gene expression in Staphylococcus aureus-infected endothelial cells. Infect Immun. 1996;64(8):3407–09. doi: 10.1128/iai.64.8.3407-3409.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krakauer T. Interleukin-8 production by human monocytic cells in response to staphylococcal exotoxins is direct and independent of interleukin-1 and tumor necrosis factor-alpha. The journal of infectious diseases. 1998;178(2):573–77. doi: 10.1086/517477. [DOI] [PubMed] [Google Scholar]
- 23.McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, et al. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc Natl Acad Sci U S A. 2006;103(27):10408–13. doi: 10.1073/pnas.0508961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tzianabos AO, Wang JY, Lee JC. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc Natl Acad Sci U S A. 2001;98(16):9365–70. doi: 10.1073/pnas.161175598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmeling DJ, Peterson PK, Hammerschmidt DE, Kim Y, Verhoef J, Wilkinson BJ, et al. Chemotaxigenesis by cell surface components of Staphylococcus aureus. Infect Immun. 1979;26(1):57–63. doi: 10.1128/iai.26.1.57-63.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13(12):1510–14. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 27.McPhail LC, Clayton CC, Snyderman R. The NADPH oxidase of human polymorphonuclear leukocytes. Evidence for regulation by multiple signals. J Biol Chem. 1984;259(9):5768–75. [PubMed] [Google Scholar]
- 28.Kobayashi SD, Voyich JM, Burlak C, DeLeo FR. Neutrophils in the innate immune response. Arch Immunol Ther Exp (Warsz) 2005;53(6):505–17. [PubMed] [Google Scholar]
- 29.DeLeo FR, Renee J, Mccormick S, Nakamura M, Apicella M, Weiss JP, et al. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101(2):455–63. doi: 10.1172/JCI949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 31.Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, et al. Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J Immunol. 2003;170(10):5268–75. doi: 10.4049/jimmunol.170.10.5268. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood. 2003;102(7):2660–69. doi: 10.1182/blood-2003-04-1078. [DOI] [PubMed] [Google Scholar]
- 33.Lotz S, Aga E, Wilde I, van ZG, Hartung T, Solbach W, et al. Highly purified lipoteichoic acid activates neutrophil granulocytes and delays their spontaneous apoptosis via CD14 and TLR2. J Leukoc Biol. 2004;75(3):467–77. doi: 10.1189/jlb.0803360. [DOI] [PubMed] [Google Scholar]
- 34.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165(10):5392–96. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 35.Liu C, Gelius E, Liu G, Steiner H, Dziarski R. Mammalian peptidoglycan recognition protein binds peptidoglycan with high affinity, is expressed in neutrophils, and inhibits bacterial growth. J Biol Chem. 2000;275(32):24490–99. doi: 10.1074/jbc.M001239200. [DOI] [PubMed] [Google Scholar]
- 36.Liu C, Xu Z, Gupta D, Dziarski R. Peptidoglycan recognition proteins: a novel family of four human innate immunity pattern recognition molecules. J Biol Chem. 2001;276(37):34686–94. doi: 10.1074/jbc.M105566200. [DOI] [PubMed] [Google Scholar]
- 37.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27(4):549–59. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Fujita T, Matsushita M, Endo Y. The lectin-complement pathway--its role in innate immunity and evolution. Immunol Rev. 2004;198:185–202. doi: 10.1111/j.0105-2896.2004.0123.x. [DOI] [PubMed] [Google Scholar]
- 39.Quinn MT, Ammons MC, DeLeo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci (Lond) 2006;111(1):1–20. doi: 10.1042/CS20060059. [DOI] [PubMed] [Google Scholar]
- 40.Klebanoff SJ. Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J Bacteriol. 1968;95(6):2131–38. doi: 10.1128/jb.95.6.2131-2138.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosen H, Klebanoff SJ. Formation of singlet oxygen by the myeloperoxidase-mediated antimicrobial system. J Biol Chem. 1977;252(14):4803–10. [PubMed] [Google Scholar]
- 42.Marcinkiewicz J. Neutrophil chloramines: missing links between innate and acquired immunity. Immunol Today. 1997;18(12):577–80. doi: 10.1016/s0167-5699(97)01161-4. [DOI] [PubMed] [Google Scholar]
- 43.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77(5):598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 44.Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med. 2000;343(23):1703–14. doi: 10.1056/NEJM200012073432307. [DOI] [PubMed] [Google Scholar]
- 45.Hirsch JG, Cohn ZA. Degranulation of polymorphonuclear leucocytes following phagocytosis of microorganisms. J Exp Med. 1960;112:1005–14. doi: 10.1084/jem.112.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbe Infect. 2003;5(14):1317–27. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 47.Ganz T, Selsted ME, Szklarek D, Harwig SS, Daher K, Bainton DF, et al. Defensins Natural peptide antibiotics of human neutrophils. J Clin Invest. 1985;76(4):1427–35. doi: 10.1172/JCI112120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellamy W, Takase M, Yamauchi K, Wakabayashi H, Kawase K, Tomita M. Identification of the bactericidal domain of lactoferrin. Biochim Biophys Acta. 1992;1121(12):130–36. doi: 10.1016/0167-4838(92)90346-f. [DOI] [PubMed] [Google Scholar]
- 49.Femling JK, Nauseef WM, Weiss JP. Synergy between extracellular group IIA phospholipase A2 and phagocyte NADPH oxidase in digestion of phospholipids of Staphylococcus aureus ingested by human neutrophils. J Immunol. 2005;175(7):4653–61. doi: 10.4049/jimmunol.175.7.4653. [DOI] [PubMed] [Google Scholar]
- 50.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319(5865):962–65. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 51.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–35. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 52.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83(3):865–75. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Adrian UH, et al. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity. 1996;5(6):653–66. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: Cell fate is regulated at the level of gene expression. Proc Natl Acad Sci USA. 2002;99(10):6901–06. doi: 10.1073/pnas.092148299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urban CF, Lourido S, Zychlinsky A. How do microbes evade neutrophil killing? Cell Micro. 2006;8:1687–96. doi: 10.1111/j.1462-5822.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- 56.DeLeo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. 2004;9(4):399–413. doi: 10.1023/B:APPT.0000031448.64969.fa. [DOI] [PubMed] [Google Scholar]
- 57.Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. 2005;3(12):948–58. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- 58.Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 2000;164(7):3713–22. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- 59.O'Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev. 2004;17(1):218–34. doi: 10.1128/CMR.17.1.218-234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178(1):175–83. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, et al. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem. 2004;279(52):54881–86. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- 62.Vuong C, Voyich JM, Fischer ER, Braughton KR, Whitney AR, DeLeo FR, et al. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 2004;6(3):269–75. doi: 10.1046/j.1462-5822.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- 63.de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med. 2004;199(5):687–95. doi: 10.1084/jem.20031636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, et al. Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol. 2005;6(9):920–27. doi: 10.1038/ni1235. [DOI] [PubMed] [Google Scholar]
- 65.Jongerius I, Kohl J, Pandey MK, Ruyken M, Van Kessel KP, Van Strijp JA, et al. Staphylococcal complement evasion by various convertase-blocking molecules. J Exp Med. 2007;204(10):2461–71. doi: 10.1084/jem.20070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Palazzolo-Ballance AM, Reniere ML, Braughton KR, Sturdevant DE, Otto M, Kreiswirth BN, et al. Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J Immunol. 2008;180(1):500–09. doi: 10.4049/jimmunol.180.1.500. [DOI] [PubMed] [Google Scholar]
- 67.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, et al. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005;202(2):209–15. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 2004;48(12):4673–79. doi: 10.1128/AAC.48.12.4673-4679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol Microbiol. 2007;66(5):1136–47. doi: 10.1111/j.1365-2958.2007.05986.x. [DOI] [PubMed] [Google Scholar]
- 70.Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M. Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci U S A. 2007;104(22):9469–74. doi: 10.1073/pnas.0702159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med. 2001;193(9):1067–76. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 1999;274(13):8405–10. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 73.Joubert O, Voegelin J, Guillet V, Tranier S, Werner S, Colin DA, et al. Distinction between pore assembly by staphylococcal alpha-toxin versus leukotoxins. J Biomed Biotechnol. 2007 doi: 10.1155/2007/25935. 200725935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Langone JJ. Protein A of Staphylococcus aureus and related immunoglobulin receptors produced by streptococci and pneumonococci. Adv Immunol. 1982;32:157–252. [PubMed] [Google Scholar]
- 75.Forsgren A, Nordstrom K. Protein A from Staphylococcus aureus: the biological significance of its reaction with IgG. Ann N Y Acad Sci. 1974;236(0):252–66. doi: 10.1111/j.1749-6632.1974.tb41496.x. [DOI] [PubMed] [Google Scholar]
- 76.Gomez MI, Lee A, Reddy B, Muir A, Soong G, Pitt A, et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat Med. 2004;10(8):842–48. doi: 10.1038/nm1079. [DOI] [PubMed] [Google Scholar]
- 77.McCormick JK, Yarwood JM, Schlievert PM. Toxic shock syndrome and bacterial superantigens: an update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 78.Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13(1):16–34. doi: 10.1128/cmr.13.1.16-34.2000. table. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bohach GA, Fast DJ, Nelson RD, Schlievert PM. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit Rev Microbiol. 1990;17(4):251–72. doi: 10.3109/10408419009105728. [DOI] [PubMed] [Google Scholar]
- 80.Le LY, Baron F, Gautier M. Staphylococcus aureus and food poisoning. Genet Mol Res. 2003;2(1):63–76. [PubMed] [Google Scholar]
- 81.Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc Natl Acad Sci U S A. 2004;101(26):9786–91. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diep BA, Stone GG, Basuino L, Graber CJ, Miller A, des Etages SA, et al. The Arginine Catabolic Mobile Element and Staphylococcal Chromosomal Cassette mec Linkage: Convergence of Virulence and Resistance in the USA300 Clone of Methicillin-Resistant Staphylococcus aureus. J Infect Dis. 2008;197(1):1523–30. doi: 10.1086/587907. [DOI] [PubMed] [Google Scholar]
- 83.Gillet Y, Issartel B, Vanhems P, Fournet JC, Lina G, Bes M, et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet. 2002;359(9308):753–59. doi: 10.1016/S0140-6736(02)07877-7. [DOI] [PubMed] [Google Scholar]
- 84.Chambers HF. Community-associated MRSA--resistance and virulence converge. N Engl J Med. 2005;352(14):1485–87. doi: 10.1056/NEJMe058023. [DOI] [PubMed] [Google Scholar]
- 85.Rossney AS, Shore AC, Morgan PM, Fitzgibbon MM, O'Connell B, Coleman DC. The emergence and importation of diverse genotypes of methicillin-resistant Staphylococcus aureus (MRSA) harboring the Panton-Valentine leukocidin gene (pvl) reveal that pvl is a poor marker for community-acquired MRSA strains in Ireland. J Clin Microbiol. 2007;45(8):2554–63. doi: 10.1128/JCM.00245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Woodin AM. Staphylococcal leukocidin. In: Montje T, Kadis S, Ajl S, editors. Microbial toxins, (Bacterial Protein Toxins, vol 3) New York and London: Academic Press Inc.; 1970. pp. 327–55. [Google Scholar]
- 87.Labandeira-Rey M, Couzon F, Boisset S, Brown EL, Bes M, Benito Y, et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science. 2007;315(5815):1130–33. doi: 10.1126/science.1137165. [DOI] [PubMed] [Google Scholar]
- 88.Bubeck Wardenburg J, Bae T, Otto M, DeLeo FR, Schneewind O. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13(12):1405–06. doi: 10.1038/nm1207-1405. [DOI] [PubMed] [Google Scholar]
- 89.Bubeck Wardenburg J, Palazzolo-Ballance AM, Otto M, Schneewind O, DeLeo FR. Panton-Valentine leukocidin is not a virulence determinant in murine models of community-associated methicillin-resistant Staphylococcus aureus disease. J Infect Dis. 2008 doi: 10.1086/592053. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bubeck Wardenburg J, Schneewind O. Vaccine protection against Staphylococcus aureus pneumonia. J Exp Med. 2008;205(2):287–94. doi: 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kahl BC, Peters G. Microbiology. Mayhem in the lung. Science. 2007;315(5815):1082–83. doi: 10.1126/science.1139628. [DOI] [PubMed] [Google Scholar]
- 92.Goering RV, McDougal LK, Fosheim GE, Bonnstetter KK, Wolter DJ, Tenover FC. Epidemiologic distribution of the arginine catabolic mobile element among selected methicillin-resistant and methicillin-susceptible Staphylococcus aureus isolates. J Clin Microbiol. 2007;45(6):1981–84. doi: 10.1128/JCM.00273-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ellington MJ, Yearwood L, Ganner M, East C, Kearns AM. Distribution of the ACME-arcA gene among methicillin-resistant Staphylococcus aureus from England and Wales. J Antimicrob Chemother. 2008;61(1):73–77. doi: 10.1093/jac/dkm422. [DOI] [PubMed] [Google Scholar]
- 94.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329(27):2002–12. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 95.Bhakdi S, Tranum-Jensen J. Alpha-toxin of Staphylococcus aureus. Microbiol Rev. 1991;55(4):733–51. doi: 10.1128/mr.55.4.733-751.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valeva A, Walev I, Pinkernell M, Walker B, Bayley H, Palmer M, et al. Transmembrane beta-barrel of staphylococcal alpha-toxin forms in sensitive but not in resistant cells. Proc Natl Acad Sci U S A. 1997;94(21):11607–11. doi: 10.1073/pnas.94.21.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stevens DL, Ma Y, Salmi DB, McIndoo E, Wallace RJ, Bryant AE. Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J Infect Dis. 2007;195(2):202–11. doi: 10.1086/510396. [DOI] [PubMed] [Google Scholar]
- 98.Diep BA, Chambers HF, Graber CJ, Szumowski JD, Miller LG, Han LL, et al. Emergence of multidrug-resistant, community-associated, methicillin-resistant Staphylococcus aureus clone USA300 in men who have sex with men. Ann Intern Med. 2008;148(4):249–57. doi: 10.7326/0003-4819-148-4-200802190-00204. [DOI] [PubMed] [Google Scholar]