

Abstract
Aging is a risk factor for the development of adult-onset neurodegenerative diseases. Although some of the molecular pathways regulating longevity and stress resistance in lower organisms are defined (i.e., those activating the transcriptional regulators DAF-16 and HSF-1 in Caenorhabditis elegans), their relevance to mammals and disease susceptibility are unknown. We studied the signaling controlled by the mammalian homolog of DAF-16, FOXO3a, in model systems of motor neuron disease. Neuron death elicited in vitro by excitotoxic insult or the expression of mutant SOD1, mutant p150glued, or polyQ-expanded androgen receptor was abrogated by expression of nuclear-targeted FOXO3a. We identify a compound [Psammaplysene A (PA)] that increases nuclear localization of FOXO3a in vitro and in vivo and show that PA also protects against these insults in vitro. Administration of PA to invertebrate model systems of neurodegeneration similarly blocked neuron death in a DAF-16/FOXO3a-dependent manner. These results indicate that activation of the DAF-16/FOXO3a pathway, genetically or pharmacologically, confers protection against the known causes of motor neuron diseases.
Introduction
Although motor neuron diseases attributable to single gene mutations are unusual (∼10% of cases), the affected genes have been successfully used to model these diseases in experimental systems (Cleveland and Rothstein, 2001). Rare, genetic forms of motor neuron disease that arise from mutations in superoxide dismutase (SOD) or p150glued have a disease phenotype that strongly resembles sporadic amyotrophic lateral sclerosis (ALS) with lower motor neuron predominance (Pasinelli and Brown, 2006). Another predominantly lower motor neuron disease, called spinobulbar muscular atrophy (SBMA) (or Kennedy's disease) is attributable to a polyglutamine expansion in the androgen receptor (AR) (La Spada et al., 1991). Despite identification of the “disease protein,” the underlying pathogenic mechanism(s) remain incompletely understood.
In most cases of sporadic ALS, motor neuron death is triggered by the interaction of a genetic predisposition and environmental factors (Bruijn et al., 2004). Genome-wide association studies have failed to reveal consistent susceptibility loci (Dunckley et al., 2007; Kasperaviciute et al., 2007). Correlative evidence suggests that aging is a risk factor for the development of ALS as well as other adult-onset neurodegenerative disorders (Lin and Beal, 2006). Studies from a variety of experimental systems have provided insight into the genetic factors controlling aging, in particular, the insulin/insulin-like growth factor signaling pathway (Hekimi and Guarente, 2003; Longo and Finch, 2003). In Caenorhabditis elegans, hypomorphic alleles of the daf-2 gene (mammalian homolog, insulin/insulin-like growth factor receptor) and the downstream signaling molecule age-1 [mammalian homolog, phosphotidylinositol-3′-kinase (PI3′K)] promote longevity and lifespan extension. These effects require the activity of the DAF-16 transcription factor (mammalian homolog, FOXO3a) (Tatar et al., 2003).
DAF-16/FOXO3a shuttles between the cytoplasm (in which it is inactive) and the nucleus in a process that is controlled by its phosphorylation state (Brunet et al., 1999). Phosphorylation of DAF-16/FOXO3a by the PI3′K substrate kinases Akt and serum/glucocorticoid-regulated kinase (SGK) (Brunet et al., 2001) leads to the 14-3-3 protein-dependent export of nuclear DAF-16/FOXO3a and reentry into the nucleus requires dephosphorylation and release of 14-3-3 (Brunet et al., 2001; Greer and Brunet, 2005). Within the nucleus, DAF-16/FOXO3a leads to the expression of a number of genes that have context-dependent effects on cellular physiology. Expression of a constitutively nuclear FOXO3a can promote the death of purified motor neurons and cerebellar granule cells, and this has been linked to the expression of FasL (Brunet et al., 1999; Barthélémy et al., 2004). In contrast, active FOXO3a protects a variety of quiescent cells against death evoked by oxidative stress or glucose deprivation, and this has been linked to the expression of manganese superoxide dismutase (MnSOD) and catalase (Kops et al., 2002; Nemoto and Finkel, 2002). Thymocyte survival and differentiation is FOXO3a dependent (Leenders et al., 2000). Differences in beneficial versus harmful effects of FOXO3a probably relate to cell-circumstance-specific level of activation and posttranslational modifications (Birkenkamp and Coffer, 2003).
Some of the stresses that contribute to motor neuron death in ALS include excitotoxicity, reactive oxygen species, accumulation of insoluble aggregates of neurofilaments, and defects in axonal transport (Cleveland and Rothstein, 2001; Rao and Weiss, 2004; Boillée et al., 2006; Lobsiger and Cleveland, 2007). Because DAF-16/FOXO3a-dependent gene transcription, in some contexts, combats cellular stresses, we inquired whether manipulating FOXO3a signaling protected neurons from insults relevant to motor neuron diseases. We show that FOXO3a activation is neuroprotective across phyla and identify a toxicity-sparing pharmacological approach for enhancement of DAF-16/FOXO3a signaling activation.
Materials and Methods
Source of reagents.
Trophic factors [ciliary neuronotrophic factor (CNTF), brain-derived neurotrophic factor (BDNF), neurotrophin 4 (NT-4), cardiotrophin 1 (CT-1) and glial-derived neurotrophic factor (GDNF)] were obtained from Alomone Labs. Psammaplysene A (PA) was synthesized as described previously (Georgiades and Clardy, 2005). All other reagents were obtained from Sigma and were of the highest grade available.
Tissue culture.
Embryonic Sprague Dawley rat spinal cord neurons were grown on confluent monolayers of cortical astrocytes, as described previously (Hu and Kalb, 2003). The substratum was acid washed glass coverslips when imaging was performed and Primaria tissue culture plasticware (Falcon; BD Biosciences Discovery Labware) when biochemistry was performed. Culture media consisted of astrocyte-conditioned media supplemented with 10 ng/ml CNTF, BDNF, NT-4, CT-1, and GDNF, and 50% of media was replaced with fresh media every 3 d.
Male SBMA mice transgenic for the prion protein promoter-driven androgen receptor cDNA containing an expanded (112) CAG repeat tract were mated with nontransgenic C57BL/6 female mice (Chevalier-Larsen et al., 2004). Dissociated, mixed spinal cord cultures obtained from 13.5-day-old embryos were grown for 3 weeks in conditioned medium (conditioned on normal mouse astrocytes) containing charcoal-stripped serum to remove hormones. At 3 weeks, motor neurons were well differentiated and distinguishable from other neurons, including sensory neurons, by size and morphology. At this time, cultures were treated with indicated conditions for 7 d; cells were then fixed with 4% paraformaldehyde and immunostained using antibodies to neurofilament heavy chain (SMI32; Sternberger Monoclonals). Motor neurons were visualized using a Leica DMR fluorescence microscope, photographed, and analyzed using IP Labs software. Statistical analyses of results were performed using Student's t test (viral infections) or ANOVA (compound treatments; SigmaStat).
Recombinant herpes simplex virus.
cDNAs were cloned into the PrpUC amplicon plasmid to generate recombinant herpes simplex virus (HSV) as described previously (Neve et al., 1997). The titer of virus used in these studies was routinely 3–5 × 107 plaque-forming units (pfu)/ml. The sources of constructs were as follows: Michael Greenberg, Harvard University, Boston, MA [hemagglutinin (HA)-tagged wild-type (WT) and triple-mutant (TM) human FOXO3a]; David Borchelt, University of Florida, Gainesville, FL (WT and G85R mutant SOD); Erika Holzbaur, University of Pennsylvania, Philadelphia, PA (WT and mutant p150glued); and Dr. Anne Brunet, Stanford University, Stanford, CA [3x fork head response element luciferase (3xFHRE-luciferase)].
Excitotoxicity assay.
After 14 d in vitro (DIV), culture media was removed (and saved), and cells were exposed to 100 μm kainic acid (KA) for 1 h. Subsequently, they were washed three times in Locke's buffer not containing KA, and the original media were replaced and incubated for another 24 h at 37°C in 5% CO2 before fixation in 4% paraformaldehyde. Motor neurons were identified in mixed culture by immunostaining for nonphosphorylated neurofilaments and counting only labeled cells with cell body diameter of 25 μm or greater. We have previously validated this method as a means of specifically recognizing motor neurons (Mojsilovic-Petrovic et al., 2006, their Fig. 1). In experiments involving recombinant HSV, 1 μl of viral stock was added to 1 ml of culture media 24 h or more before the next manipulation. Tubes containing viruses were color coded so that the operator was blinded to the specific virus used.
Figure 1.

Triple-mutant FOXO3a is retained in the nucleus and protects against excitotoxic and proteotoxic insults. A, Mixed spinal cord neurons were transfected using Lipofectamine 2000 (Invitrogen) with HA-tagged WT FOXO3a or TM FOXO3a and, 24 h later, were fixed and immunostained with anti-HA antibody and Alexa-488 conjugated secondary antibody. In the left frame, WT FOXO3a is present in neurites and the soma cytoplasm, whereas the nucleus appears devoid of immunoreactivity. In the right frame, TM FOXO3a is seen primarily concentrated in the nucleus while soma and neurite staining is weak. Scale bar, 24 μm. B, Mixed spinal cord cultures were maintained for 14 DIV before infection with HSV–LacZ, HSV–WT-FOXO3a, or HSV–TM-FOXO3a. Twenty-four hours later, the cultures underwent a 1 h excitotoxic insult, and, after 24 h, the cultures were fixed and stained for SMI32. The number of SMI32+ motor neurons surviving each treatment is noted on the ordinate axis. A statistically significant reduction in motor neuron number is seen in all groups except the cultured treated with HSV–TM-FOXO3a. C, The 14 DIV mixed spinal cord cultures from mice engineered to express polyQ expanded AR were infected with HSV–LacZ, HSV, WT FOXO3a, or HSV–TM-FOXO3a and then treated with DHT or vehicle. After 7 d, fixed cultures were stained, and the number of motor neurons surviving was determined. A statistically significant, DHT-dependent death was found in the uninfected group and the HSV–WT-FOXO (but not HSV–TM-FOXO3a) group. D, Cultures were infected with HSV-mutant SOD (green) and HSV–LacZ (red) and subsequently stained with specific antibodies. A high degree of colocalization is evident (yellow, merge image). Scale bar, 40 μm. E, The number of motor neurons surviving each treatment was determined after infecting 14 DIV cultures with combinations of HSV–LacZ, TM FOXO3a, mutant SOD, mutant p150glued, WT SOD, or WT p150glued. Mutant SOD and mutant p150glued motor neuron death is blocked when TM FOXO3a is expressed.
Quantification of motor neurons.
The number of immunostained cells were counted in three randomly selected fields per coverslip, and the mean value was obtained. In each experiment, at least three independent coverslips were used per condition, and the results presented were obtained for at least four independent cultures and experiments.
Immunocytochemistry.
Tissue culture cells were fixed in freshly prepared 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, for 30 min before extensive washing in PBS. Overnight incubation with primary antibody was performed at room temperature, and, after washing, coverslips were incubated with Alexa fluor-conjugated secondary antibody (2–4 h). When double-labeling experiments were performed, species-specific secondary antibodies with distinct emission spectra were used. Coverslips were washed before mounting in PermaFluor (Thermo Fisher Scientific) and viewing on an Olympus FV300 Fluoview laser confocal microscope.
Western blots and quantification.
Cultures were lysed in NP-40 lysis buffer (1% NP-40, 40 mm Tris, pH 7.4, 0.15 m NaCl, 10% glycerol, 0,1% SDS, 0.1% deoxycholate plus protease inhibitors and phosphatase inhibitors) and sonicated, and particulate matter was removed by centrifugation and subjected to SDS-PAGE before transfer to nitrocellose. Equal amounts of protein (determined using BCA reagents from Pierce) were loaded in each lane. After blocking in 5% milk in PBS, membranes were incubated in primary antibody overnight, washed, incubated with secondary antibody, washed, and visualized to GE Healthcare chemiluminescent substrate according to the directions of the manufacturer. Densitometric analysis of films was obtained using TINA (Isotopenmeßgeräte) from at least four independent experiments, the results were averaged, and the mean ± SE values formed the basis of the statistical comparisons. Quantitative data on band intensity was expressed as the fold change compared with values of HSV–LacZ-infected cultures. The displayed Western blot data are representative of the results obtained from at least four independent cultures. In some experiments, the LI-COR Biosciences Odysseus system was used for visualizing and quantifying Western blot bands. Secondary antibodies were from LI-COR Biosciences (IR 800 or 680 goat anti-mouse IgG or anti-rabbit IgG). The source of primary antibodies was as follows: rabbit anti-MnSOD (Stressgen), rabbit anti- FKHRL1/FOXO3a and anti-phospho FKHRL1/FOXO3a (T32) (Upstate), anti-phosphoFOXO3a (S253) (Abcam), and rabbit anti-HA (Santa Cruz Biotechnology). Species-specific HRP-conjugated secondary antibodies were from GE Healthcare, and Alexa 488-conjugated anti-rabbit secondary antibody was from Invitrogen.
Subcellular fractionation.
Nuclear protein lysate was prepared using Sigma N-Xtract Tm kit. Briefly, cells collected from one 60 mm dish were suspended in 150 μl of hypotonic lysis buffer (in mm: 10 HEPES, pH 7.9, 1.5 MgCl2, and 10 KCl). After incubating on ice for 10 min, 10% IGEPA CA-630 was added to final concentration of 0.6%, vortexed vigorously for 10 s, and centrifuged immediately at 10,000–11,000 × g for 30 s. The supernatant was cytoplasmic fraction, and the pellet was nuclear fraction.
Luciferase reporter assay.
Human embryonic kidney 293 (HEK 293) cells were grown to ∼80% confluence and then transfected with 3xFHRE-luciferase (FHRE), the parent vector lacking the FHREs (pGL3), or pGL3 vector containing 3x retinoic acid response element (3xRARE-luciferase) (Addgene). In all cases, the Renilla luciferase reporter construct, driven by the thymidine kinase promoter, was cotransfected as an internal control for transfection efficiency and cell death. After 24 h, the cells were trypsinized and replated into media containing the appropriate concentration of PA or vehicle. After 48 h, the cells were trypsinized again and replated into a 96-well plate in fresh media containing PA or vehicle (12 replicates per condition). Twenty-four hours later, the plates were analyzed using the Dual-Glo Luciferase Assay System (Promega) on the Xenogen In Vitro Imaging System. The ratio of luciferase signal to Renilla signal for each well was calculated, and the various treatment groups were compared with vehicle.
Bioinformatics.
To identify genes that were differentially expressed in ALS, we accessed the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database and searched the available datasets for ALS-related microarray data. Five datasets were identified, and these were analyzed using the two-tailed t test analysis with a 0.050 significance level, as provided on the GEO site, to compare the control group with the ALS group in each of the datasets. The data from these differentially expressed genes were then downloaded into a spreadsheet and searched for genes that were previously determined to be controlled by FOXO (Ramaswamy et al., 2002). Genes that were present in both lists were added to the supplemental table (available at www.jneurosci.org as supplemental material), and their direction of regulation was indicated.
Study of mice.
B6SJL-Tg(SOD1–G93A)1Gur/J mice were obtained from The Jackson Laboratory and were bred to the F1 generation of C57BL/6 × SJL mice. The offspring of this cross was used for all of the biochemical studies (n = 4 in each experimental group). Lumbar spinal cord was obtained at postnatal day 87, frozen on dry ice until use. The spinal cord segments were homogenized with a Dounce in NP-40 buffer as above (10:1 v/w). All other manipulation of the lysates was as described for tissue culture cells.
Study of Drosophila.
Drosophila stocks were crossed on standard cornmeal agar media at 29°C. Food was supplemented with DHT (Steraloids) and Psammaplysene A once it had cooled to <50°C, to final concentrations of 1 and 0.5 mm, respectively. Eye phenotypes of female flies of each condition were examined and blindly scored according to the criteria described with one scoring modification to increase sensitivity to differences in affected eye areas (Pandey et al., 2007).
Flies haploinsufficient for Drosophila FOXO (dFOXO) were generated by mating GMR–GAL4, AR52Q flies to the following fly stock lines: foxoBG01018 (Bloomington Stock Center stock 12530); foxo21, foxo25 (Jünger et al., 2003), and foxoc01841 (Exelixis).
Study of C. elegans.
The following C. elegans strains were obtained from the C. elegans Genetic Center (biosci.umn.edu/CGC) or constructed by us: Δglt-3, ZB1096 glt-3(bz34) IV; nuIs5, KP742[glr-1::gfp; glr-1::G∝s(Q227L) V; lin-15(+)]; Δglt-3; nuIs5, ZB1102 glt-3(bz34) IV; nuIs5 V. For age-1; Δglt-3; nuIs5, strain ZB1102 glt-3(bz34) IV; nuIs5 V was crossed with strain BE13 sqt-1(sc13)II. sqt-1 was then kicked out and replaced by age-1 using strain TJ1052 age-1(hx546) II. For Pmyo-3:daf-16::gfp, strain ZB2283 was produced by Jian Xue and Carolina Ibanez-Ventoso (both from Rutgers University, Piscataway, NJ) by making transgenic animals carrying an extrachromosomal array containing Pmyo-3:daf-16:gfp.
For PA treatment, we soaked the diluted drug into standard nematode growth medium culture plates. We transferred freshly growing nematode cultures to these plates and allowed them to grow for 2 d before assessing the effect. To determine the ratio of nuclear versus cytoplasmic DAF-16 levels, we used a strain transgenic for Pmyo-3:daf-16::gfp, in which the DAF-16::GFP reporter is expressed in body wall muscle cells. We compared the intensity of green fluorescent protein (GFP) labeling in the nucleus and adjacent cytoplasm using NIH Image. We monitored the effect of PA or age-1 on nematode excitotoxicity by measuring the extent of neurodegeneration in Δglt-3; nuIs5 animals with or without PA or with or without age-1 mutation. We observed free-moving animals with an inverted scope under Nomarski differential interference contrast optics with no anesthetics. Swollen cells in the nerve-ring region were counted as head necrotic figures indicative of neurodegeneration.
Statistics.
Pairwise comparisons used two-tailed Student's t test, and, when three or more groups were compared, we used ANOVA and post hoc analysis with significance set at p < 0.05.
Results
In fibroblasts, expression of FOXO3a with alanine substitutions at three phosphorylation sites [threonine 32 (T32), serine 253 (S253), and serine 315 (S315)] leads to nuclear retention of the transcriptionally active protein (Brunet et al., 2001). Cultures of rat spinal cord neurons were infected with recombinant HSV engineered to express the TM or WT FOXO3a. Immunostaining for the transgene demonstrated that the WT FOXO3a is restricted to the neuronal cytoplasm and the TM FOXO3a is primarily nuclear (Fig. 1A). The transgene products were detectable in all neurons for >5 d without any apparent toxicity. To determine whether these transgenes influenced the susceptibility of motor neurons to excitotoxic insult, 14 DIV mixed spinal cord cultures were infected with HSV–WT-FOXO3a, HSV–TM-FOXO3a, HSV–LacZ, or no virus, the following day were exposed to an excitotoxic challenge (100 μm KA or vehicle for 1 h), and the number of surviving motor neurons determined 24 h later (Fryer et al., 1999, 2000). Although ∼45% of motor neurons were killed by KA in the HSV–LacZ- and HSV–WT-FOXO3a-infected cultures, no KA-induced motor neuron death occurred in the HSV–TM-FOXO3a-infected cultures (Fig. 1B). ANOVA using transgene expression as the between-group factor and survival as the within-group factor demonstrated a significant difference between groups (F(5,12) = 15.68; p < 0.001, ANOVA). The post hoc comparisons between groups using Scheffé's F test with significance set at p < 0.05 showed that significant motor neuron death only occurred in the cultures expressing the WT FOXO3a or LacZ. Thus, expression of the TM FOXO3a protects cultured motor neurons from excitotoxic insult.
Next we explored the capacity of TM FOXO3a to protect motor neurons in vitro from a variety of proteotoxic insults relevant to motor neuron diseases. In spinobulbar muscular atrophy, a polyglutamine expansion in the AR leads to testosterone-dependent motor neuron death (Chevalier-Larsen et al., 2004). We made cultures from spinal cord of mutant AR-expressing mice and found that a 7 d treatment with DHT led to the loss of ∼25% of motor neurons compared with the vehicle-treated cultures (p < 0.05) (Fig. 1C). We observed an equivalent rate of DHT-dependent motor neuron death in cultures treated with HSV–WT-FOXO3a (p < 0.05). Infection of cultures with HSV–TM-FOXO3a, however, completely blocked DHT-dependent death of motor neurons, with all motor neurons surviving in the presence of DHT (Fig. 1C).
Proteotoxic motor neuron death can also be precipitated by expression of mutant forms of human superoxide dismutase or p150glued (Mojsilovic-Petrovic et al., 2006). We asked whether treating cultures expressing these mutant proteins with TM FOXO3a affected motor neuron death. We began these studies by determining concentrations of viruses that led to coexpression of both transgenes in neurons but did not lead to toxicity because of the viral burden. We established that infection of spinal cord neurons with ∼8 × 104 pfu of HSV-mutant SOD per milliliter of culture media reliably induced 50% motor neuron loss 7 d after infection. Addition of ∼8 × 104 more pfu of HSV–LacZ to these wells did not exacerbate motor neuron death. Immunocytological localization studies revealed that 97 ± 2% of motor neurons expressing mutant SOD also expressed β-galactosidase (Fig. 1D). In all subsequent studies, we confirmed a >95% coexpression of the toxicity-inducing mutant protein (SOD or p150glued) and LacZ or TM FOXO3a. We also established that coexpression of LacZ with either WT SOD or WT p150glued had no adverse effect on motor neuron survival. We have shown previously that this concentration of HSV-mutant SOD infects all motor neurons at this plating density (Hu and Kalb, 2003; Mojsilovic-Petrovic et al., 2006).
We compared next the outcome of cultures infected with HSV-mutant SOD plus HSV–LacZ or HSV–TM-FOXO3a as well as cultures infected with HSV-mutant p150glued plus HSV–LacZ or HSV–TM-FOXO3a (Fig. 1E). The ANOVA indicates that statistically significant differences between groups existed (F(5,12) = 70.14; p < 0.0001). Infection of cultures with HSV-mutant SOD plus HSV–LacZ led to significantly lower motor neuron numbers when compared with cultures infected with HSV-mutant SOD plus HSV–TM-FOXO3a (35.0 ± 4.0 vs 69.0 ± 2.0; p < 0.05 in post hoc analysis). Similarly, motor neuron numbers from cultures infected with HSV-mutant p150glued plus HSV–LacZ were significantly lower than motor neuron numbers from cultures infected with mutant p150glued plus HSV–TM-FOXO3a (36.0 ± 4.0 vs 68.0 ± 3.0; p < 0.05 in post hoc analysis) (Fig. 1E). Thus, the toxicity of three different mutant proteins that cause motor neuron disease can be blocked by expression of a version of FOXO3a that constitutively resides in the nucleus.
Inhibition of PI3′K should lead to accumulation of FOXO3a dephosphorylated at T32, S253, and S315 and enhanced partitioning of FOXO3a into the nucleus. Given this, we wondered whether PI3′K inhibition would confer neuroprotection. We began these experiments by determining that application of the PI3′K inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one] (final concentration, 20 μm) every other day for 6 d did not adversely affect motor neuron survival under our culture conditions (data not shown). Next we examined the biochemical effects of LY294002. For this set of experiments, the culture media was supplemented with CNTF, CT-1, NT-4, and GDNF but lacked BDNF. Neurons were pretreated with LY294002 or vehicle for 30 min before acute stimulation with BDNF (50 ng/ml) or vehicle, and then lysates were prepared 30 min later (Fig. 2A). In the absence of LY294002, BDNF evoked an increase in the phosphorylation of AKT, mitogen-activated protein kinase (MAPK), and T32 and S253 of FOXO3a. No changes in the abundance of the nonphosphorylated species were noted. In cultures pretreated with LY294002, application of BDNF evoked an increase in MAPK phosphorylation, but there were no changes in the phosphorylation of AKT and T32 and S253 of FOXO3a. No changes in the abundance of the nonphosphorylated species were noted. These results suggest that LY294002 selectively inhibit the PI3′K signaling pathway and phosphorylation of FOXO3a. Next we investigated the effects of chronic LY294002 application to cultures supplemented with all five of the above trophic factors. Twenty-four hour incubation with LY294002 compared with vehicle-treated cultures led to a decrease in the abundance of phospho-AKT and phosphorylation at the S253 site on FOXO3a (Fig. 2B). No other changes were noted in the phosphorylated proteins studied, and no changes were detected in the abundance of the nonphosphorylated species. Next we looked at the effects of 6 d incubation with LY294002 (fresh drug applied every other day) (Fig. 2C). We found that incubation with LY294002 compared with vehicle-treated cultures led to a decrease in the abundance of phosphor-AKT, phosphor-MAPK, and phosphorylation of T32 and S253 of FOXO3a. In summary, these experiments indicate that inhibition of PI3′K has the desired effect of decreasing the abundance of phosphorylated FOXO3a.
Figure 2.

Inhibition of PI3′K reduces FOXO3a phosphorylation but does not protect against mutant SOD toxicity. Mixed spinal cord cultures were grown in the absence of BDNF and pretreated with LY294002 or vehicle before acute stimulation with BDNF. Thirty minutes after application of BDNF, lysates were prepared for Western blot analysis. A shows that LY294002 blocks the BDNF stimulated increase in phospho-AKT, phospho-MAPK, and phosphorylation of FOXO3a on T32 and S253. B shows the effect of a 24 h treatment with LY294002 of cultures chronically grown in five trophic factors, including BDNF. By Western blot analysis, LY294002 suppresses phospho-AKT and phosphorylation of FOXO3a on S253. C shows the effect of a 6 d treatment with LY294002 of cultures chronically grown in five trophic factors, including BDNF. By Western blot analysis, LY294002 suppresses phospho-AKT, phospho-MAPK, and phosphorylation of FOXO3a on T32 and S253. D shows the number of motor neurons surviving 6 d of infection with HSV–WT-SOD or HSV-mutant SOD grown in the presence or absence of chronic LY294002. Mutant SOD led to ∼50% motor neuron loss, and LY294002 did not abrogate the process.
To determine whether LY294002 was neuroprotective, cultures were infected with either WT or mutant SOD-expressing viruses, and LY294002 or vehicle was added to the culture media every other day for 6 d (Fig. 2D). In the presence of WT SOD, equivalent levels of motor neuron survival was found in the presence or absence of LY294002. In addition, in the presence of mutant SOD, equivalent levels of motor neuron death were found in the presence or absence of LY294002. These results indicate that LY294002 is not toxic on its own and does not protect motor neurons from mutant SOD toxicity. Because LY294002 reduces FOXO3a phosphorylation but is not neuroprotective, it may be that phosphorylation of other PI3′K targets in mammalian cells offset any potential beneficial actions of dephosphorylated FOXO3a.
A chemical-genetic screen recently reported the identification of a series of compounds that can inhibit FOXO1a nuclear export. Compounds fell into two classes: (1) inhibitors of general nuclear export machinery and (2) inhibitors specific to the FOXO1a pathway (Kau et al., 2003). We inquired whether compounds in the second class would also block the nuclear export of FOXO3a because they would be predicted (based on the results above) to display neuroprotective activity. We focused on PA, which was isolated from a marine sponge, because it was the most potent of the class 2 inhibitors (Fig. 3A) (Schroeder et al., 2005). In addition, PA was reported to have no effect on AKT phosphorylation, suggesting that it acted either downstream of AKT or in a synergistic pathway.
Figure 3.

PA drives FOXO3a into the neuronal nucleus, promotes FOXO-dependent transcription, and protects against the proteotoxicity of mutant SOD and mutant p150glued. A, The chemical structure of PA. B, Subcellular fractionation of 14 DIV spinal cord culture lysates treated with PA or vehicle. Nuclear fraction (enriched for the nuclear marker lamin) contains more FOXO3a in PA- versus vehicle-treated cultures. The abundance of total FOXO3a, lamin, or actin is the same under both conditions. Quantification of band intensities show that PA treatment leads to a statistically significant increase in nuclear FOXO3a/nuclear lamin compared with vehicle-treated cultures. C, PA promotes transcription from a 3xFHRE-luciferase reporter in a dose-dependent manner. PA affected neither transcription from the parent vector (pGL3 that lacks the FHREs) nor a 3xRARE-luciferase reporter. There was a statistically significant difference between vehicle treatment of 3xFHRE versus PA concentrations 10 nm (*p < 0.05), 50 nm (**p < 0.01), and 100 nm (***p < 0.001). D, Pretreatment of mixed spinal cord cultures with PA (+PA) protected motor neurons from excitotoxic insult (left bar graph) and DHT-dependent death of polyQ expanded AR toxicity (right bar graph). E, One set of 14 DIV mixed spinal cord cultures was infected with HSV–LacZ, HSV–WT-SOD, or HSV-mutant SOD, and a second set of cultures was infected with HSV–LacZ, HSV–WT-p150glued, or HSV-mutant p150glued. Survival of SMI32-stained motor neurons was determined 4 d later. Infected cultures were either treated with vehicle (−PA) or PA (+PA). Mutant SOD led to a statistically significant reduction in motor neuron number (by ANOVA; see Results for details), and this was prevented in PA-treated cultures. Similarly, mutant p150glued led to a statistically significant reduction in motor neuron number, and this was prevented in PA-treated cultures. *p < 0.05.
We began by looking at the effect of a synthetic sample of PA (for synthesis, see Georgiades and Clardy, 2005) on the distribution of FOXO3a endogenously expressed by neurons. We biochemically isolated nuclei from spinal cord cultures treated with PA or vehicle (Fig. 3B). Based on the distribution of the nuclear envelope protein lamin, it is clear that our subcellular fractionation procedure greatly enriched nuclei. There was an ∼2.5-fold increase in the nuclear FOXO3a/nuclear lamin ratio in the PA-treated cultures compared with vehicle-treated cultures. Total FOXO3a and lamin levels were unaffected by drug treatment. These observations indicate that PA promotes the sequestration of FOXO3a into nuclei.
We next tested the ability of PA to promote FOXO-dependent transcription. HEK 293 cells were transfected with a plasmid containing a minimal promoter containing 3xFHRE driving luciferase expression and then treated with various concentrations of PA or vehicle for 72 h. As a control for a nonspecific action of PA on transcription, a parallel set of cells were transfected with the parent plasmid (pGL3) that lacked the 3xFHRE. In all experiments, an internal control plasmid expressing Renilla luciferase was cotransfected, and all results are expressed as the ratio luciferase/Renilla fluorescence. PA treatment led to a dose-dependent increase in luciferase signal in 3xFHRE-expressing but not pGL3-expressing cells (Fig. 3C). As an added control for specificity, we looked at the ability of PA to enhance transcription from a 3xRARE luciferase construct and found that PA had no effect on this promoter. These results indicate that PA can promote FOXO transcriptional activity.
To determine whether PA had neuroprotective activity, spinal cord cultures were treated with the drug (10 nm) for 2 d and then subjected to an excitotoxic challenge (Fig. 3D). The percentage of KA-induced cell death was ∼50% in vehicle-treated cultures (65 ± 2 vs 35 ± 3 for vehicle vs KA; p < 0.01, Student's t test) and ∼3% in PA-treated cultures (67 ± 3 vs 65 ± 2 for vehicle vs KA; p < 0.6, Student's t test), indicating that PA protected motor neurons from excitotoxic challenge. Next we looked at mutant AR proteotoxicity (Fig. 3D). Significant differences between groups (F(2,6) = 18.84, ANOVA) were found in the three-way comparison of (1) no DHT, (2) DHT plus vehicle, and (3) DHT plus PA). The post hoc analysis demonstrated a DHT-dependent ∼25% loss of motor neurons in vehicle-treated cultures (p < 0.01) and neuroprotection in the PA-treated cultures (p > 0.05 in the comparison of no DHT vs DHT plus PA).
We followed up these observations by asking whether PA blocked the proteotoxicity of SOD or p150glued. Spinal cord cultures were infected with HSV engineered to express the WT or mutant forms of SOD or the WT or mutant forms of p150glued and received PA (or vehicle) every other day for 4 d. The drug treatment had no effect on transgene expression (data not shown). After 4 d, the cultures were fixed, and motor neuron number was determined. ANOVA revealed statistically significant differences between groups in LacZ versus WT SOD versus mutant SOD (with or without PA) comparisons (F(5,12) = 18.41; p < 0.001) as well as LacZ versus WT p150glued versus mutant 150glued (with or without PA) comparisons (F(5,12) = 19.26; p < 0.001) (Fig. 3E). The post hoc analysis revealed that statistically significant protection against the toxicity of mutant SOD or p150glued was conferred by PA treatment on motor neuron survival. PA had no adverse effect on survival of motor neurons expressing LacZ or wild-type versions of SOD or p150glued. Thus, PA is nontoxic on its own but can protect against four different insults in vitro that are directly relevant to motor neuron diseases.
Although the direct molecular target of PA is unknown, we examined the effect of PA on some candidate biochemical and cell biological processes that have been implicated previously in neuron death. Neurotrophins (such as BDNF) can promote neuronal survival by activating its receptor, TrkB, both during development and after insult (Koliatsos et al., 1993; Ernfors et al., 1994; Klöcker et al., 1998) (but see Koh et al., 1995; Mojsilovic-Petrovic et al., 2006), and so we wondered whether PA had demonstrable effects on this signaling pathway. Spinal cord cultures grown for 14 DIV were infected with HSV-mutant SOD and then PA or vehicle was added to the cultures. Under these conditions, in the absence of BDNF, the level of active, phosphor-Trk receptor is very low (Fig. 4A). Similarly, downstream signaling involving AKT and MAPK are only modestly active. In response to BDNF addition to the media, there is a rapid and robust activation of the Trk receptor (as monitored by assaying for the phosphorylated form of the receptor) as well as phosphor-AKT and phosphor-MAPK. The temporal pattern of receptor activation and downstream signaling in our cultures conforms to previous observations (Fryer et al., 2000; Hu and Kalb, 2003) (Fig. 4A). Identical results were obtained in cultures uninfected with viruses (data not shown). Thus, we find no evidence that pretreatment of cultures with PA has any effect on the magnitude or duration of BDNF–TrkB signaling.
Figure 4.

Effects of PA on the distribution of WT and mutant SOD in vitro and biochemical consequences of WT and mutant SOD in vitro and in vivo. A, Mixed spinal cord cultures infected with HSV–WT-SOD or HSV–G87R–SOD were treated with PA or vehicle overnight before stimulation with 50 ng/ml BDNF. Lysates of unstimulated cells or stimulated cells at the given time points were subjected to Western blot analysis. The kinetics and degree of TrkB pathway stimulation is the same in PA- versus vehicle-treated cells. B, Mixed spinal cord cultures infected with HSV–WT-SOD or HSV–G85R–SOD were treated with PA or vehicle for 3 d before fixation and immunostaining for human SOD. WT SOD is homogenously distributed throughout the cytoplasm in the neuronal soma and processes in the absence of PA (−PA) and in the presence of PA (+PA). In contrast, G87R–SOD assumes a punctate pattern of expression indicative of aggregates of the protein in both the absence of PA (−PA) and the presence of PA (+PA). Scale bar, 20 μm. Below, using cultures infected with HSV–G85R–SOD, motor neurons are stained with SMI32 (left) and anti-human SOD (middle). Aggregated mutant SOD is localized within motor neurons (yellow in right). Scale bar, 15 μm. C, In the absence of PA (−PA) treatment, spinal cord cultures expressing mutant SOD or WT SOD have the same abundance of phosphor-FOXO3a (T32) and the nonphosphorylated species. In addition, mutant SOD-expressing cultures have decreased abundance of MnSOD. In the presence of PA (+PA), there is no difference in the abundance of phospho-FOXO3 or nonphosphorylated FOXO3a in cultures expressing WT or mutant SOD. In addition, the level of expression of MnSOD is no longer suppressed by mutant SOD expression in PA-treated cultures. D, Western blots of lumbar spinal cord lysates from two wild-type (labeled 1 and 2) and G93A mutant SOD mice (labeled 1 and 2). Representative Western blots show an increase in the abundance of T32 phospho-FOXO3a in the lysates from the mutant mice as well as a decrease in the abundance of MnSOD. Quantification of these results are shown in the histograms below.
Insoluble aggregates of mutant SOD are detectable within cells from transgenic mice engineered to express mutant SOD (Bruijn et al., 1997; Watanabe et al., 2001). We wondered whether WT or mutant SOD aggregated in neurons in vitro and whether treating cultures with PA influenced the cellular distribution of human transgene SOD. Immunocytological location of human SOD in cultures infected with HSV–WT-SOD revealed that the protein is homogeneously distributed throughout the cytoplasm and extends centrifugally for >100 μm into axons and dendrites (Fig. 4B). In contrast, in cultures infected with HSV–mutant SOD immunoreactivity is concentrated into puncta (the cytological signature of insoluble aggregated proteins) in the soma cytoplasm and neurites. Double-labeling studies reveal that mutant SOD puncta are present in motor neurons (identified by SMI32 immunoreactivity) as well as nonmotor neurons in our cultures (Fig. 4B). Treatment of cultures with PA had no effect on the subcellular distribution of human SOD in cultures infected with either of the recombinant HSVs. Although the pathophysiological significance of aggregated protein is controversial, these results indicate that the neuroprotective action of PA is dissociable from the accumulation of aggregated mutant SOD. A similar observation has been made in C. elegans wherein DAF-16 protects against β-amyloid Aβ1–42 toxicity but does not influence the accumulation of protein aggregates (Cohen et al., 2006).
Because the neuroprotection conferred by PA does not seem to be linked to alterations in trophic factor signaling or the generation of macroscopic mutant SOD aggregates, perhaps PA action is linked to its capacity to promote nuclear localization of FOXO3a. This would be consistent with the observations that PA causes nuclear partitioning of FOXO3a in neurons and constitutive localization of FOXO3a in the nucleus is broadly neuroprotective. We began with biochemical studies of spinal cord neurons in vitro expressing WT SOD or mutant SOD. In the absence of PA, the abundance of phosphorylated FOXO3a at T32 (or its ratio to the nonphosphorylated species) is the same in mutant versus WT SOD-expressing cultures (Fig. 4C). The same was true when we assayed phosphorylation of S253 (data not shown). Conversely, one transcriptional target of FOXO3a is MnSOD, and the abundance of this protein is markedly depressed in mutant SOD compared with WT SOD-expressing cultures. Expression of neither mutant nor WT SOD led to measurable changes in total FOXO3a or actin. In the presence of PA treatment, the abundance of phosphorylated FOXO3a (or its ratio to the nonphosphorylated species) is the same in mutant versus WT SOD-expressing cultures (Fig. 4C). Interestingly, PA treatment rescued the mutant SOD effect on the level of MnSOD such that MnSOD abundance was equal in the mutant versus WT SOD-expressing cultures. PA had no effect on the abundance of total FOXO3a or actin. These findings suggest that mutant SOD proteotoxic stress in vitro is not associated with a change in the state of phosphorylation of FOXO3a at sites that regulate its subcellular distribution. However, mutant SOD expression results in a reduction in the abundance of a FOXO3a transcriptional target (MnSOD), raising the possibility that proteotoxic stress might influence the FOXO3a signaling pathway. How PA treatment reverses the mutant SOD-dependent effect on MnSOD expression, and whether it is linked to FOXO3a, will require additional study.
Given these in vitro observations, we wondered whether a similar phenomenon occurs in mutant SOD mice. We examined lysates from spinal cords of mice expressing the G93A mutant form of human SOD or wild-type controls for the expression of phosphor-FOXO3a and target genes. The mice were 87 d old, a time when they are asymptomatic in terms of weakness but do manifest other subtle abnormalities (Mourelatos et al., 1996; Frey et al., 2000; Pun et al., 2006). A consistent increase in the phosphorylation of T32 of FOXO3a (and the ratio of phosphor-FOXO3a to the nonphosphorylated species) was seen in the G93A mice compared with the wild-type animals (n = 4 in each experimental group) (Fig. 4D). This was associated with a reduction in MnSOD in the G93A mice compared with the wild-type animals. No differences were noted in the abundance of actin in the mutant versus wild-type animals. Thus, in both in vitro and in vivo experiments, mutant SOD expression is associated with a reduction in MnSOD expression. The effect of mutant SOD expression on FOXO3a phosphorylation differed in the in vitro versus in vivo paradigms.
To further explore this issue, we used bioinformatics tools to query existing microarray profiles for a potential connection between motor neuron disease and FOXO3a-dependent transcription. We identified five microarray datasets located in the NCBI GEO server that are relevant to motor neuron disease. These datasets come from studies of postmortem tissue from ALS patients and mutant SOD mouse or rat tissues at various stages of disease. To our knowledge, the only description of the FOXO3a transcriptome comes from a study of phosphatase and tensin homolog (PTEN) null 786-O renal carcinoma cells (Ramaswamy et al., 2002). This study identified 198 transcripts whose level of expression is changed by at least twofold when a DNA binding-competent, constitutively nuclear (triple-mutant) FOXO3a, is expressed in these cells. When we asked how many genes in the motor neuron disease dataset were also components of the FOXO3a transcriptome, we identified 22 transcripts in both datasets (supplemental table, available at www.jneurosci.org as supplemental material). Even given the disparate experimental platforms used in these studies, the presence of any overlapping dataset is intriguing. A better-controlled prospective study is required to obtain a more full understanding of the potential importance of FOXO3a in motor neuron disease pathogenesis.
In light of the neuroprotective effect of PA in vitro, we examined the effects of the drug in two in vivo model systems of neuronal degeneration. Expression of polyglutamine-expanded AR in the Drosophila eye leads to DHT-dependent degeneration (Takeyama et al., 2002; Pandey et al., 2007). We found that flies reared on food supplemented with 0.5 mm PA had a reduced degenerative phenotype when compared with vehicle-treated flies (Fig. 5a,b). These in vivo results complement the observations made in spinal cord cultures from mice expressing polyglutamine-expanded AR wherein we found expression of TM FOXO or treatment with PA blunts DHT-dependent mutant AR toxicity.
Figure 5.
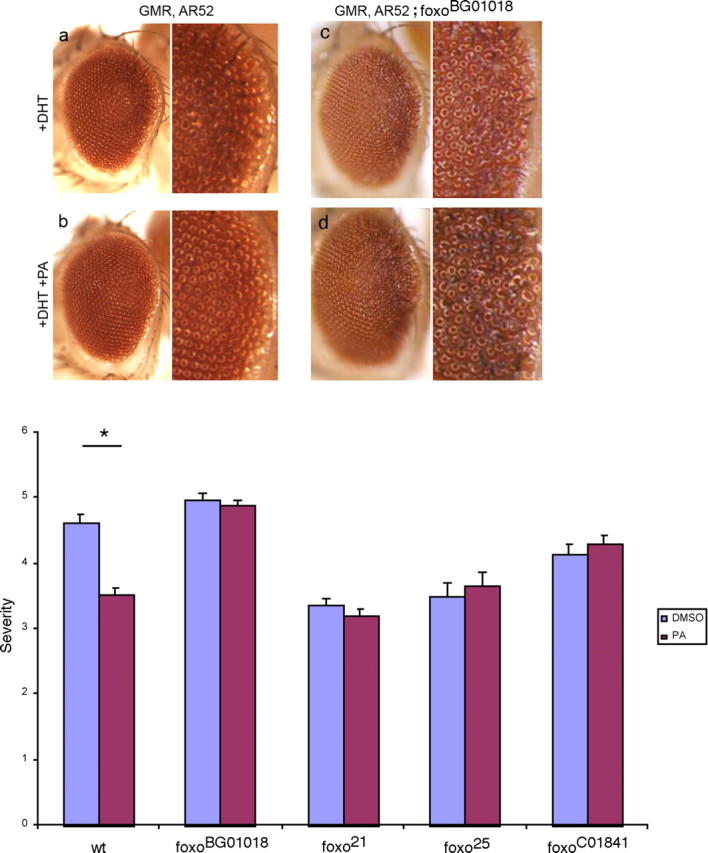
PA suppresses degeneration in a Drosophila model of SBMA in a dFOXO-dependent manner. Right panel of each diptych shows higher magnification of the posterior margin of the eye, in which degeneration is concentrated. a, Flies expressing polyglutamine-expanded human androgen receptor (GMR, AR52Q) show degeneration when reared on food containing 1 mm dihydrotestosterone (+DHT). b, This degeneration is suppressed when food is supplemented with 0.5 mm PA. c, d, PA fails to rescue AR52Q-mediated degeneration in a dFOXO-deficient background. Bar graph below, Quantification of the degenerative eye phenotype (scored according to Pandey et al., 2007) shows that the neuroprotective effect of PA is lost when flies are haploinsufficient for dFOXO. Numbers of eyes scored are as follows: GMR, AR52: DMSO, n = 34, PA, n = 41; GMR, AR52; foxoBG01018: DMSO, n = 37, PA, n = 47; GMR, AR52; foxo21: DMSO, n = 20, PA, n = 16; GMR, AR52; foxo25: DMSO, n = 8, PA, n = 14; GMR, AR52; foxoC01841: DMSO, n = 28, PA, n = 20. Mean values are indicated with SE. p value was determined using Student's t test. *p < 0.05.
We next asked whether the neuroprotective action of PA depended on FOXO. To this end, we examined the efficacy of PA in flies that both expressed the polyglutamine-expanded AR and were haploinsufficient for dFOXO (Fig. 5c,d). In a dFOXO-deficient background (foxoBG01018 allele) (Dionne et al., 2006), PA lost its ability to protect against DHT-dependent degeneration. To corroborate these observations, we studied two additional dFOXO loss-of-function alleles (foxo21 and foxo25) and one predicted loss-of function allele (foxoC01841) (Jünger et al., 2003; Thibault et al., 2004). As above, on all of these dFOXO-deficient backgrounds, PA lost its ability to protect against DHT-dependent degeneration. Using a quantitative rating score, we found that PA led to a statistically significant mitigation of polyglutamine-expanded AR degeneration, but this was lost in the dFOXO haploinsufficient flies (Fig. 5, bar graph). Variation in the baseline (no PA provided) level of neurodegeneration phenotype among the studied fly lines is likely attributable to differences in the background genotype. These observations indicate that PA confers protection against mutant AR proteotoxicity in a FOXO-dependent manner.
We developed a C. elegans model system of neurodegeneration by combining a null mutation in the glutamate transporter GLT-3 (Δglt-3) with a transgenic strain [nuIs5 (Berger et al., 1998)] in which the glr-1 promoter drives expression of an activated form of G∝s (abbreviated G∝s*) and GFP in glutamatergic neurons (Mano and Driscoll, 2009). The Δglt-3; nuIs5 double mutants exhibit necrotic neuron death at all stages of postembryonic development, with the strongest effect seen in developmental stage L3. PA had a dose-dependent neuroprotective effect at the L3 stage, with complete rescue from death using 10 nm PA (Fig. 6A). This concentration of PA had no adverse effect on WT nematodes. We asked whether the effect of PA is mediated by changing the timing of neurodegeneration or by reducing it throughout development (Fig. 6B). To examine this, we studied the effect of PA on the Δglt-3; nuIs5 double mutants as a function of larval stage, and we found that neuronal death was reduced in all developmental stages, with the strongest effect observed in the developmental stages most prone to excitotoxicity. Neuron death was reduced in a statistically significant manner in larval stages L2 (3.4 ± 0.2 vs 2.5 ± 0.2 dying neurons per animals; n = 44 vs n = 49, vehicle vs PA; p = 0.006) and L3 (4.2 ± 0.2 vs 2.2 ± 0.1 dying neurons per animals; n = 63 vs n = 65, vehicle vs PA; p < 0.001) (Fig. 6B).
Figure 6.

PA treatment or reducing the activity of IGFR signaling pathway alleviates nematode excitotoxicity. A, PA has a dose-dependent neuroprotective effect on nematode excitotoxicity: Δglt-3; nuIs5 double-mutant nematodes were cultured in the presence of increasing concentrations of PA in the medium. The number of swollen, degenerating head neurons was scored under Nomarski differential interference contrast optics in live animals at the L3 developmental stage. PA shows a statistically significant, dose-dependent neuroprotective effect. B, The neuroprotective effect of PA is mediated by reducing the extent of neurodegeneration throughout development. C, age-1(hx546), a mutation that reduces IGFR signaling pathway without altering developmental decisions, protects C. elegans from excitotoxicity. Degenerating head neurons were scored in live animals as above. D, PA causes preferential nuclear localization of DAF-16, the nematode homolog of FOXO3a. Animals express a transgene (Pmyo-3:daf-16::gfp) that directs the expression of DAF-16::GFP fusion protein in body-wall muscle cells. In control animals (i.), DAF-16::GFP is abundantly present in the cytoplasm. Animals grown in the presence of 10 nm PA (ii.) show preferential (although not uniform) accumulation of DAF-16::GFP in the nucleus.
In C. elegans, stress resistance and longevity are promoted by a reduction in the activity of insulin growth factor receptor (IGFR) signaling pathway (i.e., hypomorphic alleles of IGFR, daf-2, and PI3′K, age-1), and this requires daf-16, the nematode homolog of FOXO3a. This led us to wonder whether reducing activity in the IGFR signaling pathway would alleviate nematode excitotoxicity. To that end, we crossed the Δglt-3; nuIs5 double mutants with an age-1 mutant that carries the hx546 allele. This allele has a specific anti-aging effect but does not affect development (Friedman and Johnson, 1988). We generated triple-mutant nematodes (age-1; Δglt-3; nuIs5) and found a robust neuroprotective effect of age-1 at larval stages L1–L3 (all p values <0.001) (Fig. 6C).
Finally, although we showed that PA leads to accumulation of FOXO3a in mammalian neuronal nuclei, we wanted to determine whether the same was true in C. elegans. To this end, we studied nematodes in which a DAF-16::GFP fusion protein was expressed in body wall muscles. Addition of PA, but not vehicle, to the growth substrate led to nuclear localization of the fusion protein and quantification of the nucleus/cytoplasm ratio of DAF-16::GFP revealed a statistically significant effect of PA (1.46 ± 0.05 vs 3.44 ± 0.55; n = 36 vs n = 27, vehicle vs PA; p < 0.0001) (Fig. 6D). This result indicates that PA has an evolutionarily conserved capacity to promote nuclear localization of the DAF16/FOXO3a transcription factor, and this is associated with resistance to necrotic neuron death. Another method for promoting nuclear localization of DAF-16 (i.e., mutation of age-1) has a neuroprotective effect and further reinforces the notion that manipulation of the IGFR signaling pathway could have promise as a neuroprotective strategy.
Discussion
Biochemical pathways that regulate longevity in yeast, C. elegans, Drosophila melanogaster, and mice also play a fundamental role in resistance to stresses such as UV radiation, oxidative conditions, heat shock, and misfolded and aggregation-prone proteins (Finkel and Holbrook, 2000). Two transcription factors, heat-shock factor 1 and DAF-16/FOXO3a, are essential mediators of this longevity/stress resistance program in nematodes (Hsu et al., 2003; Morley and Morimoto, 2004). Here we show that genetic and pharmacological maneuvers that enhance nuclear localization of FOXO3a protect mammalian motor neurons in vitro from four insults directly relevant to motor neuron diseases and abrogate neurodegeneration in two in vivo invertebrate model systems. The broad neuroprotective action of PA suggests that it acts on a phylogenetically conserved, core stress resistance pathway.
Nuclear FOXO3a and PA can evoke a neuroprotective program
We have used several complementary experimental platforms (each with its own unique advantage) to investigate the biological actions of PA. In summary, the experimental result suggest that the neuroprotective actions of PA are mediated, at least in part, by FOXO3a/DAF-16. First, in both rat neuron cultures and C. elegans, application of PA promotes nuclear accumulation of FOXO3a or DAF-16. Second, PA evokes a dose-dependent increase in FOXO-dependent transcription. Third, constitutive nuclear localization of FOXO3a mimics the neuroprotective action of PA. Fourth, PA-mediated protection against mutant AR-evoked degeneration is dFOXO dependent. Even without knowledge of the direct molecular target of PA, this evidence favors the view that PA acts, at least in part, in a FOXO3a transcription-dependent manner.
During nuclear localization, members of the FOXO family of transcription factors can control the expression of messages that impact glucose metabolism, tumor suppression (through effects on cell cycle progression and apoptotic responses), stress resistance, and longevity (Ramaswamy et al., 2002; Greer and Brunet, 2005). The specific effects of FOXOs are both cell-type and context dependent, and specific posttranslational modifications (i.e., phosphorylation, ubiquitination, acetylation) play a key role in controlling the transcriptional readout (Brunet et al., 1999, 2004; Hu et al., 2004; Berdichevsky et al., 2006). For example, work from the Brunet laboratory has demonstrated that AMP-activated protein kinase (AMPK) phosphorylates FOXO3a at six sites, and the state of phosphorylation at these sites does not affect nuclear localization (Greer et al., 2007a). Instead, these sites are vital for expression of genes involved in oxidative stress management and energy metabolism. Our finding that induction of MnSOD by PA in cultures expressing mutant SOD parallels the observation of Greer et al. (2007b) that AMPK induction of sod-3 expression in nematodes is FOXO/DAF-16 dependent. Our preliminary studies suggest that expression of mutant SOD with or without PA does not influence the state of phosphorylation of FOXO3a at AMPK sites S413 or S588 (our unpublished observations). Additional work is required to determine whether FOXO3a undergoes posttranslational modifications in cells expressing mutant SOD, whether PA influences such a change, and whether this has functional consequences.
It is noteworthy that expression of mutant SOD in neurons in vivo, before neuron loss, is associated with an increase in FOXO3a phosphorylation at T32 (one of the three key sites controlling nuclear/cytoplasmic partitioning) and a reduction in the abundance of MnSOD. These observations may indicate that, as a consequence of expressing mutant SOD, neurons upregulate the activity of FOXO3a kinases (and/or decrease the activity of FOXO3a phosphatases), and this results in a reduction in the expression of FOXO3a-regulated transcripts that combat proteotoxicity. In fact, there is a 2.5-fold increase in PI3′K activity and protein in spinal cords of ALS patients [the activator of the FOXO3a kinases AKT and SGK (Wagey et al., 1998)] as well as a 3.6-fold increase in phosphor-AKT itself in mutant SOD mice (Hu et al., 2003). These observations may indicate that a putatively beneficial response to stress, such as increased expression and activity of an antiapoptotic pathway, may have an unintended adverse additional action on FOXO3a transcription.
Although we found that constitutive nuclear localization of FOXO3a is neuroprotective, other groups obtained the opposite result. Heterologous expression of TM FOXO3a led to death of purified motor neurons and cerebellar granule neurons (Brunet et al., 1999; Barthélémy et al., 2004) as well as PTEN-deficient tumor cells (Nakamura et al., 2000). One factor that may contribute to these disparate observations is experimental platform. A second contributory factor may be the level of transgene expression. It has been suggested that the level of nuclear FOXO3a dictates the cellular readout (Nemoto and Finkel, 2002), and the promoter in the HSV-based system we use generally evokes moderate level of transgene expression (Neve et al., 1997).
Invertebrate models support PA acts via a conserved mechanism
Given the potent neuroprotective activity of PA in vitro, we sought to examine its effects in vivo. The two model systems we chose have features that are reminiscent of human motor neuron diseases. Neurons in the Δglt-3: nuIs5 nematode die an excitotoxic cell death, and there is abundant evidence implicating excitotoxic mechanisms in ALS (Rao and Weiss, 2004). Spinobulbar muscular atrophy is caused by expression of the proteotoxic polyQ expanded AR, and transgenic expression of this mutant in the fly eye similarly causes degeneration (Pandey et al., 2007). The neuroprotective activity of PA in these model systems complements the in vitro studies with mammalian motor neurons. A simple explanation for these observations is that PA operates on a biochemical pathway common in all these experimental systems that can overcome a diversity of noxious insults.
Extending these observations to the mutant SOD mouse model of ALS at the present time is hindered by the lack of evidence that PA can cross the blood–brain barrier. In preliminary studies, we have not detected PA in brain homogenates after intraperitoneal administration. Administration using an intrathecal catheter/osmotic minipump may be a viable option (Storkebaum et al., 2005), although the long-term stability of PA at body temperatures is not known. It may be possible to synthesize derivatives of PA that retain the essential biological activity and cross the blood–brain barrier (Georgiades and Clardy, 2006).
Healthful aging and the FOXO3a signaling pathway
The onset of many neurodegenerative diseases in adulthood raises the possibility that the causes of aging are specific contributors to disease pathogenesis. To the extent that this is true, applying our understanding of the molecular biology of longevity may lead to new types of therapy for neurodegenerative diseases. The present work on FOXO3a is an example of how manipulation of a phylogenetically conserved, longevity-promoting signaling pathway can effectively block neurodegeneration. This is particularly interesting in light of a recent population-based study of human longevity showing a strong association of FOXO3a and healthy aging (Willcox et al., 2008). Agents that trigger neurodegeneration may act in part by subverting the normally healthful actions of an aging pathway.
Footnotes
This work was supported by National Institutes of Health Grants NS34435 (M.D.), CA24487 (J.C.), NS32214 (D.M.), NS053825 (J.P.T.), and NS52325 (R.G.K.) and the ALS and Muscular Dystrophy Associations. We thank Dr. David Borchelt for the gift of the anti-human SOD rabbit serum and Ernst Hafen and Julia Lüdke (Institute of Molecular Systems Biology, Zurich, Switzerland) for the Foxo21 and Foxo25 fly lines.
The authors declare no competing financial interests.
References
- Barthélémy C, Henderson CE, Pettmann B. Foxo3a induces motoneuron death through the Fas pathway in cooperation with JNK. BMC Neurosci. 2004;5:48. doi: 10.1186/1471-2202-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell. 2006;125:1165–1177. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- Berger AJ, Hart AC, Kaplan JM. Gαs-induced neurodegeneration in Caenorhabditis elegans. J Neurosci. 1998;18:2871–2880. doi: 10.1523/JNEUROSCI.18-08-02871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenkamp KU, Coffer PJ. Regulation of cell survival and proliferation by the FOXO (Forkhead box, class O) subfamily of Forkhead transcription factors. Biochem Soc Trans. 2003;31:292–297. doi: 10.1042/bst0310292. [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the Forkhead Transcription Facotr FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, O'Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Curr Biol. 2006;16:1977–1985. doi: 10.1016/j.cub.2006.08.052. [DOI] [PubMed] [Google Scholar]
- Dunckley T, Huentelman MJ, Craig DW, Pearson JV, Szelinger S, Joshipura K, Halperin RF, Stamper C, Jensen KR, Letizia D, Hesterlee SE, Pestronk A, Levine T, Bertorini T, Graves MC, Mozaffar T, Jackson CE, Bosch P, McVey A, Dick A, et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N Engl J Med. 2007;357:775–788. doi: 10.1056/NEJMoa070174. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer HJ, Knox RJ, Strittmatter SM, Kalb RG. Excitotoxic death of a subset of embryonic rat motor neurons in vitro. J Neurochem. 1999;72:500–513. doi: 10.1046/j.1471-4159.1999.0720500.x. [DOI] [PubMed] [Google Scholar]
- Fryer HJ, Wolf DH, Knox RJ, Strittmatter SM, Pennica D, O'Leary RM, Russell DS, Kalb RG. Brain-derived neurotrophic factor induces excitotoxic sensitivity in cultured embryonic rat spinal motor neurons through activation of the phosphatidylinositol 3-kinase pathway. J Neurochem. 2000;74:582–595. doi: 10.1046/j.1471-4159.2000.740582.x. [DOI] [PubMed] [Google Scholar]
- Georgiades SN, Clardy J. Total synthesis of psammaplysenes A and B, naturally occurring inhibitors of FOXO1a nuclear export. Org Lett. 2005;7:4091–4094. doi: 10.1021/ol0513286. [DOI] [PubMed] [Google Scholar]
- Georgiades SN, Clardy J. Preparation of a psammaplysene-based library. Org Lett. 2006;8:4251–4254. doi: 10.1021/ol061599w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007a;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007b;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekimi S, Guarente L. Genetics and the specificity of the aging process. Science. 2003;299:1351–1354. doi: 10.1126/science.1082358. [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Hu JH, Chernoff K, Pelech S, Krieger C. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem. 2003;85:422–431. doi: 10.1046/j.1471-4159.2003.01669.x. [DOI] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Hu P, Kalb RG. BDNF heightens the sensitivity of motor neurons to excitotoxic insults through activation of TrkB. J Neurochem. 2003;84:1421–1430. doi: 10.1046/j.1471-4159.2003.01599.x. [DOI] [PubMed] [Google Scholar]
- Jünger MA, Rintelen F, Stocker H, Wasserman JD, Végh M, Radimerski T, Greenberg ME, Hafen E. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol. 2003;2:20. doi: 10.1186/1475-4924-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasperaviciute D, Weale ME, Shianna KV, Banks GT, Simpson CL, Hansen VK, Turner MR, Shaw CE, Al-Chalabi A, Pall HS, Goodall EF, Morrison KE, Orrell RW, Beck M, Jablonka S, Sendtner M, Brockington A, Ince PG, Hartley J, Nixon H, Shaw PJ, Schiavo G, Wood NW, Goldstein DB, Fisher EM. Large-scale pathways-based association study in amyotrophic lateral sclerosis. Brain. 2007;130:2292–2301. doi: 10.1093/brain/awm055. [DOI] [PubMed] [Google Scholar]
- Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463–476. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- Klöcker N, Cellerino A, Bähr M. Free radical scavenging and inhibition of nitric oxide synthase potentiates the neurotrophic effects of brain-derived neurotrophic factor on axotomized retinal ganglion cells in vivo. J Neurosci. 1998;18:1038–1046. doi: 10.1523/JNEUROSCI.18-03-01038.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Leenders H, Whiffield S, Benoist C, Mathis D. Role of the forkhead transcription family member, FKHR, in thymocyte differentiation. Eur J Immunol. 2000;30:2980–2990. doi: 10.1002/1521-4141(200010)30:10<2980::AID-IMMU2980>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007;10:1355–1360. doi: 10.1038/nn1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians? Science. 2003;299:1342–1346. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- Mano I, Driscoll M. Caenorhabditis elegans glutamate transporter deletion induces AMPA-receptor/adenylyl cyclase 9-dependent excitotoxicity. J Neurochem. 2009;108:1373–1384. doi: 10.1111/j.1471-4159.2008.05804.x. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Jeong GB, Crocker A, Arneja A, David S, Russell DS, Russell D, Kalb RG. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci. 2006;26:9250–9263. doi: 10.1523/JNEUROSCI.1856-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci U S A. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- Neve RL, Howe JR, Hong S, Kalb RG. Introduction of the glutamate receptor subunit 1 into motor neurons in vitro and in vivo using a recombinant herpes simplex virus. Neuroscience. 1997;79:435–447. doi: 10.1016/s0306-4522(96)00645-8. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- Rao SD, Weiss JH. Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci. 2004;27:17–23. doi: 10.1016/j.tins.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Schroeder FC, Kau TR, Silver PA, Clardy J. The psammaplysenes, specific inhibitors of FOXO1a nuclear export. J Nat Prod. 2005;68:574–576. doi: 10.1021/np049624z. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, Singh CM, Buchholz R, Demsky M, Fawcett R, Francis-Lang HL, Ryner L, Cheung LM, Chong A, Erickson C, Fisher WW, Greer K, Hartouni SR, Howie E, Jakkula L, Joo D, et al. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- Wagey R, Pelech SL, Duronio V, Krieger C. Phosphatidylinositol 3-kinase: increased activity and protein level in amyotrophic lateral sclerosis. J Neurochem. 1998;71:716–722. doi: 10.1046/j.1471-4159.1998.71020716.x. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]